Abstract
Cell cycle inhibitors, such as the cyclin-dependent kinase (Cdk) inhibitor proteins and retinoblastoma (Rb) family members, control exit from the cell cycle during the development of a variety of terminally differentiated tissues. It is unclear whether sustained expression of these proteins is required to prevent cell cycle re-entry in quiescent and terminally differentiated cells. The organ of Corti (cochlear sensory epithelium) and pars intermedia (intermediate lobe of the pituitary) are two tissues that share the characteristic of ongoing cell division in mice lacking either the p27Kip1 Cdk inhibitor, Ink4 proteins, or Rb. Here, we use tamoxifen-inducible mouse models to delete p27Kip1 in postnatal animals and show this is sufficient to induce proliferation in both the organ of Corti and pars intermedia. Thus, these tissues remain sensitive to the presence of p27Kip1 even after their developmental exit from the cell cycle. The neonatal cochlea displayed heightened sensitivity to changes in p27Kip1 expression, with a proliferative response higher than that of constitutive null mice. In adults, the proliferative response was reduced but was accompanied by increased cell survival. In contrast, re-establishment of normal p27Kip1 expression in animals with established pituitary tumors, in an inducible “knock-on” model, led to cessation of pituitary tumor growth, indicating the cells had maintained their susceptibility to p27-mediated growth suppression. Although restoration of p27Kip1 did not induce apoptosis, it did lead to resolution of pathological features and normalization of gene expression. Our data underscore the importance of p27Kip1 expression in the maintenance of cellular quiescence and terminal differentiation.
Introduction
Mice with constitutive deletion of the p27Kip1 gene (Cdkn1b) exhibit ongoing abnormal proliferation in two adult tissues: the auditory sensory epithelium of the cochlea, known as the organ of Corti, and in melanotroph cells, which comprise the pituitary pars intermedia.Citation1–Citation6 Both of these tissues proliferate during development and then exit the cell cycle at a defined time point. In the case of the organ of Corti, the tissue is considered terminally differentiated after embryonic day 12–14, as no proliferation occurs under physiological or pathological conditions.Citation7,Citation8 Ordinarily, a wave of cell cycle withdrawal proceeds from the apex to base that coincides with the expression of the cyclin-kinase inhibitor p27Kip1.Citation1,Citation2,Citation7 Likewise, pituitary melanotrophs proliferate in neonatal animals until the onset of postnatal dopaminergic innervation.Citation9,Citation10
p27Kip1 is a member of the Cip/Kip class of cyclin-dependent kinase (Cdk) inhibitors and, as such, can inhibit multiple Cdk protein complexes, including those containing either cyclins D, E or A.Citation11 In contrast, a second class of Cdk inhibitors, the Ink4 proteins, are obligate inhibitors of cyclin D, and thereby prevent phosphorylation of the retinoblastoma proteins. Mice lacking the p19Ink4d Cdk inhibitor show proliferation in the postnatal organ of Corti.Citation12 In comparison to p27 null mice, which mostly show proliferation in organ of Corti support cells (SCs), p19Ink4d knockout mice have increased proliferation in the mechanoreceptor hair cells (HCs), which then undergo apoptosis. Co-deletion of p21Cip1 with p19Ink4d enhanced proliferation in the early postnatal period, indicating cooperation of the Cip/Kip and Ink4 family Cdk inhibitors.Citation13 In the pituitary, deletion of a different Ink4 family member, p18Ink4c, leads to persistent melanotroph proliferation, analogous to that seen in p27Kip1 knockout mice.Citation14 Co-deletion of p18Ink4c and p27Kip1 increased the incidence and histological grade of pituitary tumors.Citation14,Citation15 Interestingly, deletion of p57Kip2, a member of the Kip/Cip family like p21 and p27, prevented cell cycle exit of immature anterior pituitary cells during embryonic development, suggesting tissue-specific differences in the action of the respective Cdk inhibitors.Citation16 Complete elimination of Rb, in constitutive knockout mice, is lethal due to effects on extra-embryonic tissues.Citation17 However, SC-specific deletion of a floxed Rb allele in newborn (P0-P1) mice resulted in proliferation of two types of SCs, pillar and Deiters' cells, but no new HC addition.Citation18 However, HC-specific deletion of a floxed Rb allele caused aberrant proliferation and HC death.Citation19 Similarly, inducible deletion of Rb in HCs at birth (P0) led to a synchronous wave of HC proliferation and then apoptosis.Citation19 But, inducible deletion of Rb in SCs and HCs in adults failed to induce proliferation or apoptosis, demonstrating an age-dependent effect of Rb deletion in cell cycle re-entry and suggesting additional mechanisms are involved in the mechanism of quiescence in the adult inner ear.Citation20 In the pituitary, tissue-specific deletion of Rb in melanotrophs resulted in massive pars intermedia growth.Citation21,Citation22 In contrast to the organ of Corti, proliferation of pars intermedia melanotrophs was not immediately followed by apoptosis, but massive enlargement of the organ is eventually accompanied by ischemic necrosis, apparently due to inadequate angiogenesis.Citation21 In summary, animal models demonstrate that cell cycle inhibition by the Rb pathway is essential to prevent ongoing proliferation in both the organ of Corti and the pituitary pars intermedia. However, because these models induce gene deletion at early developmental stages, it is less clear whether the ongoing cell proliferation in these two tissues depends on the absence of Rb pathway inhibition at a critical stage of development or whether there is a dependence on cell cycle inhibitors in adult tissues. In the case of p27Kip1, it is unknown whether it acts chiefly as a developmental trigger to induce cell cycle withdrawal, or whether it is also needed to prevent aberrant cell cycle re-entry at later stages.
Cellular proliferation in the organ of Corti is of tremendous interest, because sensory HC loss is responsible for the majority of hearing disorders and because mammalian auditory HCs do not regenerate. In contrast, non-mammalian vertebrates regenerate sensory HCs via two mechanisms: mitotic regeneration, where SCs undergo asymmetric cell division to replace both damaged HCs and differentiated SCs or direct transformation of SCs into HCs.Citation23,Citation24 Thus, a major goal of hearing research is to understand the molecular constraints on cellular proliferation in the mature organ of Corti. We addressed this issue for p27Kip1, using an inducible model of p27Kip1 deletion. The aim was to determine the dependence of post-mitotic inner ear sensory epithelial cells on the presence of p27Kip1 at varying ages. With similar methodology, we sought to determine its ability to induce proliferation in the mature pituitary pars intermedia. In the pituitary, we addressed a second important question regarding the temporal requirement for p27Kip1. That is, whether the ongoing growth of pituitary tumors renders them unsusceptible to the inhibitory effect of p27, e.g., through the acquisition of other growth promoting mutations. We addressed this question through the use of a second, “knock-on” animal model, in which p27Kip1 is not expressed until it is induced with tamoxifen.
Results
Efficiency of tamoxifen-induced gene recombination in the cochlea.
To determine the efficiency and temporal specificity of tamoxifen-induced gene deletion in inner ear sensory epithelia, we inter-crossed CreER transgenics to a reporter mouse (Z/EG) which activates GFP in response to Cre.Citation26,Citation27 The frequency of GFP activation in mice treated both as neonates and as adults (G1 and G6 paradigms, ) was evaluated. As listed in Table 2, negative control animals (given vehicle alone) showed no GFP expression as neonates, indicating no spontaneous (leaky) Cre activity. In neonates, tamoxifen induced GFP in multiple cell types (), including inner and outer hair cells (5% and 8% GFP+, respectively, ), whereas the percentage of GFP-positive Deiters' cells was smaller (1%). In adult cochleas, the CreER transgene exhibits a low level of leakiness in untreated controls, particularly in the inner hair cells (4% GFP+ IHCs, and and H). Nonetheless, tamoxifen induced a 3-fold higher Cre-mediated GFP expression in the adult organ of Corti compared to neonates (p ≤ 0.01, mean adults = 1,888 GFP+ OC cells/mm2 SE, mean neonates = 663 GFP+ OC cells/mm2 SE, ).
Induced knockout of p27 gene resulted in proliferation in neonatal organ of Corti cells.
In order to study the effect of p27Kip1 loss in postnatal animals, we induced deletion of p27Kip1 by administering tamoxifen and BrdU to neonatal p27L+/L+;CreER+ mice (group L1, ). When observed on postnatal day 7, a high level of proliferation was noted both in sensory (organ of Corti) and non-sensory cells of the cochlea ( and and F). Mitotic cells were seen in the organ of Corti (Sup. Fig. 2) and the GER (). Patterns of condensed chromosomes indicated the presence of cells in M phase at the time of tissue harvest. Negative control animals consisted both of p27L+/L+;CreER+ mice given vehicle instead of tamoxifen, and p27L+/L+ mice (no Cre) given tamoxifen. In both of these types of negative controls no proliferation was detectible in the organ of Corti, but there were BrdU+ cells in the non-sensory Claudius cells that abut the organ of Corti ( and Sup. Table 1). The lack of proliferation in the neonatal organ of Corti in the control animals confirms that sensory epithelial cells of p27LoxP mice, like wild-type mice, are in a post-mitotic state. It is also consistent with the GFP-reporter data that indicate that, in the neonatal cochlea, there is no spontaneous Cre activity in CreER transgenics in the absence of tamoxifen. Thus, loss of p27Kip1 in early postnatal animals is sufficient to induce proliferation in both the organ of Corti and in non-sensory cells of the cochlea.
Survival of newly divided cells in the neonatal cochlea.
To determine whether proliferating cells in the neonatal animals survive and are capable of differentiating into new hair cells, an additional set of animals were administered tamoxifen and BrdU during week 1, but were not sacrificed until week 4 (group L1–4, ). As shown in , BrdU+ organ of Corti cells in tamoxifen-treated neonatal conditionals persist at 4 weeks. However, numbers of BrdU-labeled cells were significantly decreased relative to that seen at week 1. Overall, roughly 10% of the BrdU+ organ of Corti cells are retained at week 4 (, group L1–4 mean = 539 BrdU+ OC cells per mm2 SE, group L1 mean = 5,666 BrdU+ OC cells per mm2 SE). However, we observed no retention of BrdU amongst Deiters' support cells, nor did we observe the appearance of BrdU-label within the inner or outer hair cell populations. The decreased number of BrdU+ cells at 4 weeks probably is due to death of a large fraction of the newly divided cells. A less likely explanation is that ongoing cell proliferation diluted the BrdU down to the point that it was no longer detectible. Arguing against the latter, we did not observe reduced BrdU staining intensities in the organ of Corti at this late time point. Furthermore, mitotic cells are not seen at week 4, as they were at week 1. These data indicate that, although induced loss of p27Kip1 induces a marked wave of proliferation in the neonatal cochlea, most of these newly divided cells soon disappear, most likely via apoptosis.
Proliferation and fate of new cells in the adult organ of Corti.
Despite being post-mitotic, the neonatal mouse organ of Corti is immature and undergoes significant morphological and functional differentiation prior to adulthood. In order to determine whether induced loss of p27Kip1 is capable of triggering cell cycle reentry in the post-mitotic cells of the mature cochlea, we administered the combination of tamoxifen and BrdU to 6 week-old mice. These animals were then sacrificed either immediately after their course of treatment (group L6) or 6 weeks later (group L6–12). Control p27L+/L+ mice without Cre exhibited a complete lack of proliferation in the organ of Corti ( and Sup. Table 1), again indicating that the sensory epithelium has undergone normal terminal differentiation. In contrast, animals with p27L+/L+;CreER+ which were treated with vehicle instead of tamoxifen exhibited a significant number of BrdU-positive cells in the sensory epithelium ( and Sup. Table 1). This indicates that, in the adult cochlea, the CreER transgene exhibits leakiness and thus lacks temporal specificity. Leakiness was also seen in the Z/EG;CreER+ reporter mice (), yet the level of proliferation seen in untreated p27L+/L+;CreER+ adults was even higher. Indeed, the level of proliferation in vehicle-treated p27L+/L+;CreER+ animals was virtually identical to their tamoxifen-treated, experimental counterparts. Because there was no spontaneous proliferation in p27L+/L+;CreER+ neonates, we conclude that the spontaneous Cre activity occurred at some point after the neonatal period, at some time between 1 and 6 weeks of age. Deletion of p27Kip1 in adults also resulted in a different pattern of proliferation compared to animals treated as neonates. The level of proliferation was 18-fold lower in adult animals (317 BrdU+ cells/mm2 SE in adults vs. 5,666 cells/mm2 SE in neonates). Interestingly, the percent survival of BrdU+ cells in adults was significantly higher in adult animals compared to neonates (). In Deiters' cells, for example, the BrdU+ cell density was relatively low in adult animals (18 BrdU+ cells/mm2 SE) ( and D) but was nearly as high at 12 weeks (14 cells/mm2 SE). Furthermore, although there were no labeled inner or outer hair cells noted at the earlier time points, rare BrdU+/Sox2− cells in the inner and outer hair cell nuclear layers were noted in adult mice after 6 weeks of recovery (Sup. Fig. 3 and ). Interestingly, attempts to label several of these cells with the HC-specific marker, myosin6, did not reveal detectable myosin6 expression (not shown). Thus, despite their localization adjacent to support cells, we lack positive confirmation of their cellular identity. One possibility is that these represent a transitional stage between SCs and HCs. In this case they may have been Sox2-positive during the BrdU labeling, but that they lost Sox2 expression and have not yet gained myosin6 expression. Alternative explanations are that these are new HCs that are undergoing cell death and have lost myosin6 labeling, infiltrating inflammatory cells or non-sensory cells that have become mislocated due to progressive anatomical derangements in the p27 null cochlea ( and F).
Vestibular support cells re-enter S phase after p27 elimination.
As in the organ of Corti, we examined the ability of tamoxifen and CreER to induce GFP in the utricular macula (vestibular sensory epithelium of the utricle) of Z/EG reporter mice, and to induce proliferation in the macula of p27LoxP mice. Numerous GFP+ cells were present in the macula of animals treated either as neonates or adults (not shown). Cell counts indicated that a sizable fraction of the vestibular hair cells (26%; mean 3,182 GFP+ HCs/mm2 SE ± 531 SEM, n = 4 animals), but only a small fraction of support cells (0.4%; mean 107 GFP+ SCs/mm2 SE ± 27 SEM), are GFP-positive. Spontaneous Cre activity was not observed in SCs in the utricular macula of untreated Z/ EG;CreER+ reporter mice but was seen in a few HCs (1%, 159 GFP+ HCs/mm2 SE ± 39 SEM, n = 4 animals). Induced deletion of p27Kip1 in neonates led to 5-fold increased BrdU-labeling in neonatal p27L+/L+;CreER+ vestibular macula relative to negative controls (mean p27L+/L+;CreER+ = 1,948 BrdU+ cells/mm2 SE, mean negative controls = 375 BrdU+ cells/mm2 SE, Sup. Table 3, p < 0.05, n = 4–5 animals/paradigm). It was also increased relative to constitutive p27 null mice (2-fold increased, p < 0.05). The increase was primarily due to increased numbers of BrdU-labeled SCs (Sup. Table 3). Roughly 7% as many BrdU+ cells are seen in the sensory epithelium after 6 weeks (group L6–12) compared to the immediate observation (group L6) (group L6–12 mean = 144 BrdU+ cells/mm2 SE ± 26 SEM; group L6 mean = 1,948 BrdU+ cells/mm2 SE ± 361 SEM, n = 4 animals/paradigm).
Tamoxifen-induced deletion or reactivation of p27Kip1 in the pituitary pars intermedia.
To determine whether p27Kip1 is also required to maintain quiescence in the pars intermedia, we treated p27L+/L+;CreER+ mice with tamoxifen at 15 weeks of age (group L15-30, n = 6). Control mice either lacked Cre or were given vehicle and had normal appearing pituitaries at 30 weeks ( and n = 11). In contrast, p27L+/L+;CreER+ mice treated with tamoxifen developed typical pars intermedia pituitary tumors, histologically similar to those in p27L−/L− or p27S−/S− mice. Despite the fact that the pars intermedia normally accounts for only a fraction of the pituitary, the mass of whole pituitaries from mice with induced loss of p27 was triple that of control animals at 30 weeks of age ( and p ≤ 0.001, t-test). Immunostaining for p27 confirmed the expression of p27 in control pituitaries and nearly complete elimination of p27 in tamoxifen-treated animals ( and D). The tumors often had small intraparenchymal hemorrhages and extensive, old hemorrhages in the space between the pars intermedia and pars distalis (Rathke's pouch). These data indicate that adult mice remain dependent on p27 to prevent hemorrhagic pituitary tumor growth.
To determine whether established pars intermedia tumors remain sensitive to p27Kip1 growth inhibition, we reactivated the gene by administering tamoxifen to p27S−/S−;CreER+ animals. Animals given tamoxifen at 6 weeks of age and then observed at 15 weeks of age (group S6-15, n = 4) showed no pathological pars intermedia hyperplasia. Control animals, treated with vehicle or lacking the Cre recombinase (n = 8), developed early pathological enlargement of their pituitaries as demonstrated in . Tamoxifen treatment at this early age prevented pathological changes (). Immunostaining for p27Kip1 showed that, following tamoxifen treatment, p27Kip1 expression was restored in the pars intermedia and pars distalis lobes of the pituitary, whereas it was undetectable in untreated controls ( and D). When treatment was delayed to an older age (15 weeks), inducible knock-on animals (p27S−/S−;CreER+) had already developed early pars intermedia tumors (not shown). We examined pituitaries in animals at 30 weeks of age, 15 weeks after treatment. In this case, p27S−/S−;CreER+ mice treated with tamoxifen had very small pituitary tumors () comparable to untreated 15 week-old animals. These pituitaries were significantly smaller (), than those of sham-treated p27S−/S−;CreER+ mice or of p27 null (p27S−/S−, Cre−) mice. The gross and histological appearance of these pituitaries was notable for resolution of the hemorrhagic areas. In Rathke's pouch, hemorrhage was often replaced with hemosiderin-laden macrophages, fibroblasts and lipid crystals, all consistent with a repair process in response to prior hemorrhage ( and H).
Pars intermedia proliferation and apoptosis in response to induced p27Kip1 mutation.
We measured the immediate effect of tamoxifen-induced p27Kip1 deletion on the proliferation in the pars intermedia of 6 week-old animals (group L6). Immunofluorescence staining showed a marked increase in BrdU+ cell counts in p27L+/L+;CreER+ mice (), whereas control animals had little detectable BrdU immunoreactivity. These data indicate that, despite having withdrawn from the cell cycle, pituitary melanotrophs remain dependent on the presence of p27Kip1 to prevent cell cycle entry. In contrast, p27stop mice, like constitutive nulls, displayed high levels of BrdU labeling and restoration of p27Kip1 expression led to a sharp decline of proliferation down to normal background levels. TUNEL staining (not shown) in these animals remained low (range 0.4%–0.6%) regardless of p27 induction status. This indicates that the pars intermedia tumors, despite the development of adenomatous features, remain susceptible to p27Kip1 mediated cell cycle arrest without undergoing apoptosis. In a separate group of p27S−/S−;CreER+ mice, we administered IdU one day prior to, and CldU immediately after, tamoxifen administration (). In this case, we observed very few CldU labeled cells, which indicates that the effect of p27Kip1 on the cessation of cell growth was very rapid. In contrast, untreated mice showed single-labeled IdU and CldU, but only rare double-labeled cells. This indicates that proliferation within the tumor is widespread and sporadic, rather than being confined to a small subset of continuously cycling cells.
Normalization of gene expression following p27 reactivation in pituitary tumors.
We previously characterized abnormal gene expression in pituitary-specific p27Kip1 and Rb knockout tumors.Citation21 Here, we examined RNA expression of several genes involved with cell cycle control and angiogenesis () with RT-QPCR. Mice with pituitary tumors in which p27Kip1 was reactivated (p27S−/S−;CreER+) by tamoxifen showed normalization of the cell cycle regulators cyclin E2 and p18Ink4c. The tumors also regained normal expression of Timp2 and Bnip3, which have been implicated in angiogenesis in the cellular response to hypoxia.Citation32–Citation34 In summary, our data indicate that restoration of p27Kip1 expression in pars intermedia pituitary tumors normalized gene expression resulting in cell cycle arrest. Resolution of the vascular lesions underlying tumor parenchymal hemorrhage was accompanied by normalized expression of specific angiogenic factors.
Discussion
These studies indicate that two tissues, namely the cochlea's organ of Corti and the pituitary's pars intermedia, remain dependent on p27Kip1 expression to maintain their proliferative quiescence even after they have exited the cell cycle as part of normal development. Induced loss of p27Kip1 in neonatal mice did not simply convert them to a null phenotype. Instead, a marked increase in proliferation was seen in the organ of Corti compared to neonatal p27 null animals ( and Sup. Table 2). This indicates that neonatal organ of Corti cells, despite being postmitotic under physiological conditions, remain primed and ready to enter the cell cycle upon loss of Cdk inhibition. In order to determine what fraction of cells enter the cell cycle upon elimination of p27, it is necessary to estimate the fraction of cells that tamoxifen was able to induce Cre/LoxP-mediated DNA excision. We therefore used a GFP reporter (Z/EG) as a marker for Cre-mediated excision. In this system, both the CreER and Z/EG transgenes are driven by the constitutive chicken actin promoter, which minimizes reporter promoter bias. Overall, tamoxifen and CreER led to significant levels of GFP expression from the Z/EG reporter transgene in neonatal animals, with no spontaneous Cre activation (leakiness). However, induction of GFP was variable amongst different cell types and was less than 100% complete, indicating that tamoxifen was probably only partially effective in inducing p27 deletion in p27L+/L+;CreER+ mice. Thus, complete deletion of p27Kip1 may induce a higher level of proliferation than we measured. However, the Z/EG reporter was an imperfect predictor of CreER's effectiveness in p27LoxP mice. We can deduce this from the observation that a large fraction of Hensen cells proliferated in response to induced loss of p27Kip1 () whereas, in reporter mice, tamoxifen induced GFP in only 1% of Hensen cells. This was a surprising observation, considering the widespread use of reporter mice to judge the effectiveness of Cre transgenes. Despite this caveat, there also appears to be cell-type specificity to p27's effect, since the pattern of proliferation does not mirror the relative effectiveness of tamoxifen-induced reporter activity. Among the SC subtypes, the highest level of proliferation was seen in pillar (neonate) and Hensen's (adult) cells. In neonatal mice, p27 is strongly expressed in all SC subtypes and its expression in Deiters' cells decreases with age such that it is weakly expressed in these cells in adulthood.Citation35 In contrast, strong p27 expression is maintained in all other SC subtypes in adulthood. The different abilities of SC subtypes to re-enter the cell cycle upon p27 loss is not simply explained by p27 expression patterns, suggesting SC quiescence is regulated by additional mechanisms.
The level of proliferation in the inner ear induced by p27Kip1 deletion was reduced in adult animals compared to neonates. This was probably not from age-related changes in CreER activity in adult animals, because tamoxifen was equally or more efficient at inducing the GFP in Z/EG reporter mice in adults vs. neonates. Adult reporter mice (Z/EG;CreER+) again underestimated the effectiveness of tamoxifen in p27L+/L+;CreER+ cochleas. This is evident by the fact that extensive BrdU labeling was seen in p27L+/L+;CreER+ mice given vehicle (without tamoxifen), whereas only a small percentage of cells showed ‘leaky’ Cre activity in Z/EG reporter mice. Because of this spontaneous deletion of p27 by CreER in postnatal animals, we cannot pinpoint the time point at which the majority of p27 was deleted. Indeed, the level of proliferation in the organ of Corti of adult p27L+/L+;CreER+ animals was comparable to constitutive p27−/− null animals regardless of whether they were given tamoxifen. Therefore, the relative reduction in proliferation in adult animals, compared to neonates, may partly be due the lack of synchronization of p27 deletion and BrdU labeling in adult mice in this model system.
GFP activation was commonly seen in the mechanoreceptor hair cells of tamoxifen-treated Z/EG;CreER+ reporter mice. Hair cells in neonatal and adult mice do not normally express detectable levels of p27, and the absence of proliferation seen in these cells with the short-term survival time following p27Kip1 deletion (groups L1 and G1) is consistent with this finding.Citation35,Citation36 The rare examples of BrdU+/Sox2− cells located in the HC layer that we observed occurred in adults 5 weeks following BrdU administration. These cells were not labeled by the HC-specific marker myosin6. We cannot rule out the possibility that they are leukocytes, but their nuclear shape and size are consistent with those of organ of Corti cells. The proliferation induced by p27Kip1 deletion likely occurred in an underlying SC that with the passage of time lost their Sox2 expression and began to differentiate as HCs. At time of tissue harvest, these rare cells may not have yet fully differentiated into a mature HC phenotype, or they may have stalled and failed to fully differentiate. However, proof of this point would require a larger number of events, in conjunction with additional very early markers of differentiating HCs. It is likely that the large numbers of native (resident) HCs present in the tissue inhibited the majority of proliferating cells from differentiating into HCs. We anticipate that greater numbers of BrdU+ HCs would be seen if p27Kip1 were to be conditionally deleted in damaged ears missing significant numbers of HCs. Nonetheless, the initiation of SC proliferation alone in the in situ adult organ of Corti is of considerable importance for the field of HC regeneration, as manipulation of p27 levels could be combined with additional mechanisms facilitating the differentiation of proliferating cells into new HCs (e.g., by manipulations of the Notch signaling pathway) and used therapeutically. Further, while transdifferentiation-based therapeutic strategies to restore hearing and balance deficits in mammals may be useful (where differentiated SCs are converted into HCs without an intervening cell division, e.g., via overexpression of atoh1, a transcription factor necessary for HC development), these therapies will likely require the mitotic production of new SCs for full restoration of function. Transdifferentiation depletes SC numbers, potentially leading to significant changes in the cellular organization of hearing and balance organs and loss of trophic support for the HCs.
Markers of apoptosis, such as TUNEL or caspase staining, are commonly employed to quantify cell death. Although these assays give an accurate snapshot of the numbers of dying cells at a given time-point, they may not reflect the overall percentage of cells that will live or die in the long run. In the case of p19Ink4d knockouts and Rb deletion, increased proliferation in the organ of Corti was accompanied, or followed soon after, by cellular apoptosis.Citation13,Citation19 As an alternative to measuring cell death, we estimated the survival of cells that had undergone cell division. We compared densities of BrdU immunostaining, in specific cell types, at early and late time points after the BrdU administration. Strictly speaking, this measure of BrdU “retention” may not be exactly equal to cell survival. Another way that BrdU labeling could be lost would be if the labeled cell were ejected from the organ but, in the organ of Corti, this is tantamount to cell death. Another possibility is that the cell continued to divide so many times that the BrdU was diluted to the point of being nondetectable. Tamoxifen-induced deletion of p27 in neonatal mice led to a high level of proliferation in the organ of Corti as discussed above, but only 10% of these cells persisted to 4-weeks of age. Indeed, in the Deiters' cell subtype, there was no persistent BrdU labeling at 4-weeks. However, the outcome was markedly different when p27 was deleted after the neonatal time-period. Although the level of proliferation was markedly reduced from that seen in neonates, the percentage of cell survival was much higher and averaged 75% (including Deiters' cells).
In the absence of Cre, there is no proliferation in the organ of Corti of neonatal or adult p27L+/L+ mice. However, deletion of p27 in the neonatal time period and beyond leads to varying levels of cellular proliferation. This observation disproves the hypothesis that the ongoing cellular proliferation in the organ of Corti of constitutive p27 knockout mice is due to abnormal persistence of developmentally immature precursor cells. In mature animals, organ of Corti cells are capable of both cell division and prolonged survival following loss of p27Kip1. In the field of regenerative medicine, there is considerable interest in the use of embryonic stem cells and induced progenitor cells for the repair of damaged post-mitotic tissues.Citation37,Citation38 Our data indicate that the use of immature progenitor cells may not be the only route to the regeneration of post-mitotic tissues since, at least in the organ of Corti, manipulation of cell cycle inhibitors can over come cell cycle arrest in post-mitotic cells. Furthermore, the relatively weak effect of tamoxifen of inducing gene deletion of the Z/ EG;CreER+ reporter in the organ of Corti, suggests that increased levels of proliferation may be achieved with a more robust system for inducing p27 loss. We also anticipate that increased levels of proliferation will be seen simply with increased BrdU exposure. Our BrdU-injection paradigm, a single daily injection of BrdU for 4 or 7 consecutive days, probably underestimated the total number of dividing cells during the interval due to the short half life of BrdU, as evidenced by the presence of numerous unlabeled mitotic cells (e.g., Sup. Fig. 2 and 2G). An alternative explanation for unlabeled mitotic figures, is that neonatal cochlea have a population of cells that are arrested in G2 and thus do not need to pass through S phase before reaching M. Together, these data add to a growing body of literature that substantiates the requirement of active inhibition to prevent cell cycle re-entry in specific terminally differentiated tissues. Our data are consistent with prior work demonstrating that knocking down p27Kip1 expression, in neonatal cochlea explants, can induce SC proliferation.Citation39 However, in the current work we demonstrate that this also holds true in live animals and in mature tissues, and we demonstrate the heterogeneity of the cellular response to p27 loss. The organ of Corti has an exquisite three-dimensional structure and, rather than growth inhibition being entirely cell autonomous, there may be additional signals from neighboring cells. This is supported by the observation that constitutive p27−/− mice show increased levels of proliferation in the organ of Corti following exposure to the ototoxic drug, amikacin.Citation40
Like the organ of Corti, the melanotroph cells of the rodent pars intermedia normally exit the cell cycle at a developmentally determined time point. At 10 days of age, proliferation ceases in the pars intermedia, coinciding with the onset of dopaminergic innervation and the expression of pro-opiomelanocortin.Citation41 In theory, it is possible that the establishment of quiescence in adult melanotroph cells would render them insensitive to loss of p27Kip1. For example, if melanotroph cells lose expression of activated cyclin/Cdk enzyme complexes, then removing the p27Kip1 inhibitor protein may have no effect on cell proliferation. However, our results show that the mature pars intermedia remains dependent on the presence of p27Kip1 to maintain cellular quiescence and, ultimately, to prevent tumor formation. As they progressed, pars intermedia tumors developed progressive cellular atypia and intraparenchymal hemorrhages, which is similar to the pituitary tumors in p27−/− constitutive knockouts. That the tumors were less massive than those of constitutive knockouts is to be expected given their delayed onset in this experimental system.
Regeneration of endocrine cells may be of interest for endocrine deficiency states, but in oncology the interest is in halting abnormal proliferation in neoplasms. Our results using tamoxifen induction of CreER in p27stop (knock-on) mice indicate that, pars intermedia tumors remain sensitive to the growth inhibitory effects of p27Kip1. Furthermore, our system re-established p27 expression under the control of its endogenous promoter. This indicates that normal levels of p27 were sufficient to halt tumor growth; it was not necessary to overexpress the protein. Not only did p27 halt abnormal proliferation, but it also normalized patterns of gene expression. Likewise, intraparenchymal hemorrhages, as a secondary feature of pars intermedia tumors, completely resolved. In comparison to pars intermedia tumors arising in p27 null mice, pars intermedia tumors in Rb knockouts exhibit more extreme cellular atypia. In addition, the tumors arise as clonal neoplasms, even in experimental systems that delete both copies of Rb, suggesting that additional mutations may occur.Citation21 Thus, it remains to be seen whether re-establishment of Rb expression can halt the growth of pars intermedia tumors caused by Rb mutations.
Germline mutation of p27Kip1 has been shown to be associated with a multiple endocrine neoplasia syndrome both in a rat model and in a human cancer family.Citation42 In addition, mice harboring combinations of p27Kip1, p18Ink4c and Rb develop a variety of endocrine neoplasias. The finding that restoration of p27Kip1 expression in pituitary tumors induces growth arrest without inducing apoptosis suggests that a similar effect would be seen for small molecule Cdk inhibitors. However, the ability of Cdk inhibition to induce apoptosis may also be determined by the status of Rb; mice lacking Rb are more susceptible to bromocryptine-induced pituitary tumor cell apoptosis if p27 is intact.Citation43 The first generation of Cdk inhibitors that were used in clinical trials, as cancer therapeutic agents, were not highly specific and exhibited dose-limiting toxicities through off-target effects.Citation44 Our data suggests that optimized p27Kip1 mimetics would have cytostatic, but not necessarily cytotoxic, effects on tumor growth. Nonetheless, recent clinical experience with kinase inhibitors for the treatment of solid tumors indicates that cytostatic agents may have improved efficacy and lower side effects than conventional chemotherapy agents.Citation45 In humans, both mimetics or antagonists of p27Kip1 may be therapeutic for either tissue regeneration or tumor growth inhibition, in the appropriate clinical setting.
Materials and Methods
Mouse models.
The p27loxP knockout mouse strain harbors a conditional knockout allele (p27L+) which normally expresses p27 until it is deleted by Cre recombinase, resulting in a null allele (p27L−).Citation25 A second mouse model, p27stop, contains a ‘knock-on’ allele (p27S−) which, at baseline, does not express p27 due a transcriptional stop cassette in the promoter region.Citation25 In this strain, Cre recombinase reactivates p27 expression, resulting in an active allele (p27S+). Mice with either p27L+ or p27S− were bred to CAGG-CreER transgenics (CreER+, courtesy of Andy McMahon, Harvard University) after having been backcrossed to 129S4 for six generations and then intercrossed to produce homozygous alleles of p27.Citation26 CreER is a fusion protein that is activated by tamoxifen and is expressed by the chicken beta-actin promoter. Constitutive p27 knockout mice, co-isogenic to the 129S4 strain, were used for comparison.Citation5 In order to assess the efficiency of tamoxifen-induced recombination, we intercrossed CreER transgenics to Z/EG reporter mice. These mice express GFP under control of the chicken actin promoter following Cremediated excision of a LoxP transcriptional stop cassette.Citation27
Tamoxifen and BrdU administration.
To activate the Cre recombinase, mice were injected with tamoxifen (30 µg/g body weight/day) at 5–10 mg/ml i.p. in canola oil. Age-matched littermate controls received a comparable volume of vehicle (canola oil). Tamoxifen was administered starting at one of three different time points: at 1 week (postnatal day 3), 6 weeks or at 15 weeks of age, as outlined in . The drug was given daily for 4 days for neonatal animals, or for 7 days for adults, concurrently with the nucleoside analog, BrdU (50 µg/g/day). Tissues were harvested 24 hours after the final dose or 3 to 15 weeks later (as in ). Each experimental group was accompanied by two sets of negative controls: animals given oil instead of tamoxifen and animals without Cre. Likewise, the p27 wild-type and constitutive p27 knockout mice were used for comparisons. The rate of ongoing proliferation in the pituitary was measured with in vivo labeling by two different nucleoside analogs in separate group of 6 week-old p27S+/S+;CreER+ mice (n = 6). In these animals, IdU (57 µg/g) was given ×7 days, tamoxifen −1 week, then CldU (44 µg/g) ×7 days and then tissue was harvested the following day.
Tissue preparation.
Inner ear tissues were fixed with cold 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4 for 4 hours, washed in buffer and prepared as whole-mount preparations as described previously in reference Citation28. Pituitaries were photographed in situ on the floor of the cranium, then were removed and weighed intact. Whole pituitaries were fixed in 4% paraformaldehyde in PBS and paraffin embedded for histology. Alternatively, the pars intermedia and pars nervosa were peeled away from the pars distalis under a stereomicroscope, along the tissue plane formed by the remnant's of Rathke's pouch and then used for expression studies.
Immunostaining inner ear end organs.
Whole-mount organ of Corti and utricular macula preparations from neonatal and adult mice were immunostained for Sox2 using a goat polyclonal antibody (1:500, Santa Cruz, SC-17320), as previously described in reference Citation28. The tissues labeled with BrdU-specific antibody (monoclonal rat anti-BrdU, 1:300, Accurate Chemical Scientific Corporation; Clone BU1/75) were incubated in 2N HCl in 0.1% Triton-X/PBS buffer for 30 minutes to denature the DNA prior to application of blocking solution. A rabbit anti-myosin6 polyclonal antibody (1:500, Proteus Biosciences 25-6791) was used to label hair cells (HCs) as previously described in references Citation28–Citation30. Labeling with multiple primary antibodies was done sequentially. Secondary antibodies with fluorescent labels (Alexa 488, 568, 594) from Invitrogen were used at 1:200–1:500 dilutions per the manufacturer. Nuclear counterstaining was performed for 15 minutes with 1 µg/ml DAPI (4′,6-diamidino-2-phenylindole), followed by a PBS rinse and glass coverslipping with Vectashield (Vector Labs) mounting medium. Antibody specificity were checked by substituting non-immune sera for the primary antibody. In double-labeling experiments, antibodies raised in different species were used to avoid cross-reactivity among secondary antibodies.
Inner ear imaging and cell quantitation.
Cells were quantified using a non-biased sampling method. In summary, halfturn preparations of cochlear whole-mounts were viewed at 60x with a laser scanning confocal microscope in various sub-divided regions (described in Sup. Fig. 1). Each region (area 0.045 mm2) was recorded as vertical stack of confocal images (Z-series, at 1 µm increments) through the entire depth of the sensory epithelium. Utricules were treated similarly, except regions were not further subdivided because of the small size of the macula. Labeled cell counts and locations were recorded using the Cell Counter plug-in for ImageJ (NIH). Sox2 labeling was used to identify SCs (Sox2+), whereas HCs were identified as (Sox2−) cells, with large round nuclei, located immediately above the SC layer. Myosin6 and Sox2 labeling were both used in the studies of the fate of BrdU-labeled cells (L1–4, L6–12). Similar strategies were used for the vestibular tissues, except that Sox2 also labels type II HCs in this tissue.Citation28 The architecture of the organ of Corti allowed us to determine support cell subtypes based on their nuclear morphology, patterning, and location relative to inner and outer hair cell nuclei in confocal image stacks. Cell densities were expressed as a function of sensory epithelium area (per mm2 SE) for cell types located within the sensory receptor epithelium. Percentages of GFP-positive cells in each region were calculated as the ratio of GFP+ cells to the total number of cells x100. Significance values were determined using ANOVAs or Student's t-tests (Prism, GraphPad Software). p ≤ 0.05 was considered statistically significant.
Pituitary immunohistochemistry.
Following antigen retrieval (10 mM sodium citrate, pH 6.0, 15 min. 100°C), slides with 5 µm sections were blocked with 10% goat or horse serum for 30 minutes and then stained with primary antibody at 4°C overnight. Cy3 anti-rabbit (Chemicon, AP182C) or streptavidin-Alexa 555 (Invitrogen) labeled secondary antibodies were applied at room temperature for one hour. Sections were then DAPI counterstained (Vecta-Shield, Vector Labs). For BrdU staining, sections were pretreated with 2 N HCl for 15 minutes. Primary antibodies were: rabbit anti-mouse p27, rat anti-CD34 (BD, clone 1H6) and biotinylated anti-BrdU (Invitrogen, clone ZBU-30).Citation25 To detect apoptosis, sections were TUNEL stained (FragEL kit, Calbiochem). IdU and CldU double immunofluorescent staining was performed as previously described in reference Citation31. Cell counts were obtained under a fluorescent microscope from three random 60x high power fields (0.075 mm2) from each of three pituitaries.
Gene expression.
RNA from the pars intermedia was prepared with TriZol (Invitrogen). Pituitaries were harvested from p27S−/S−;CreER+ mice (group S15) treated with either tamoxifen (n = 4) or canola oil (n = 4) and 30 week-old wild-type controls (n = 10). Total RNA was reverse transcribed and subjected to QPCR using primer sets for S16, cyclin E2, cyclin D2, p130Rb2, p18Ink4c, Timp2, p19Arf, Bnip3 and Usp1 Power SYBR-Green, as described in reference Citation21. Gene expression was calculated in terms of PCR cycle number adjusted according to S16 levels and expressed as the difference compared to control mice (−ΔΔCt).
Abbreviations
| i.p. | = | intraperitoneal |
| SEM | = | standard error of the mean |
| SE | = | sensory epithelium |
| HC | = | hair cell |
| SC | = | support cell |
| OC | = | organ of Corti |
| GER | = | greater epithelial ridge |
| IS | = | inner sulcus |
| DAPI | = | 4′,6-diamidino-2-phenylindole |
| BrdU | = | 5-bromo-2′-deoxyuridine |
| CldU | = | 5-chloro-2′-deoxyuridine |
| IdU | = | 5-iodo-2′-deoxyuridine |
| Cdk | = | cyclin-dependent kinase |
| Rb | = | retinoblastoma |
Figures and Tables
Figure 1 Visualization of CreER-induced gene recombination in adult cochlea with a GFP reporter. GFP expression is shown in confocal images from whole-mount preparations of apical turn organ of Corti from 6 week-old tamoxifen-injected (group G6 animal 1, A–C; animal 2, D–F) or vehicle only (oil, G and H) animals. Tissues are labeled with Sox2 antibody (red) that specifically labels support cells and GFP (green), plus a nuclear counterstain, DAPI (blue).Citation28 Panels show a brightest-point projection of a confocal Z-series spanning the sensory epithelium. GFP label alone, in grayscale, is shown in the middle panels (B, E and H) to illustrate the morphology of the GFP-positive cells. Only Sox2 and DAPI are shown in the right panels, demonstrating 3 rows of Sox2-negative outer hair cell nuclei (OHC1–3) and 1 row of inner hair cell nuclei (IHC). Many GFP-positive support cells and hair cells are present in the CreER+ animals treated with tamoxifen (A–F). (G and H) A rare GFP-positive IHC (arrow) present in a vehicle (oil) treated adult control mouse. Scale bar = 10 µm (A–C) or 20 µm (D–H).
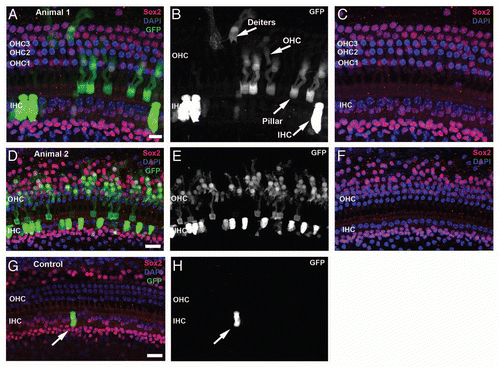
Figure 2 Proliferation of neonatal organ of Corti cells after induced deletion of p27Kip1. Confocal images from cochlea of neonatal mice given BrdU (group L1) are shown at the level of HC (left column) or Deiters/pillar nuclei (right column). Control animals, including those given oil (without tamoxifen, A and B) and tamoxifen-treated p27 wild-type mice (C and D), show Sox2 staining (red) in the greater epithelial ridge (GER) and in SCs cells. One row of inner (IHC) and 3 rows of outer (OHC) hair cells are Sox2- and seen with DAPI nuclear staining (blue). BrdU staining (green) is not seen in the organ of Corti or GER but is present in the Sox2- Claudius cells (arrow, D) and in the underlying stroma (B). In the brightest-point projection shown in (B), the BrdU labeling that appears in the GER is actually in underlying BrdU-labeled stromal cells; the GER cells are unlabeled. (E and F) Experimental animals (p27L+/L+;CreER+) given tamoxifen and BrdU show numerous Sox2+/BrdU+ cells in both the organ of Corti and GER. (G and H) Constitutive p27 knockout animal (positive control) treated with BrdU. BrdU+/Sox2+ cells are seen in the Hensen (G), pillar/tunnel (H), border cell (G and H) and GER (G and H) regions. Multiple Sox2+ mitotic figures (arrows) are present in the GER (G) and two additional mitotic cells (labeled 1 and 2) are shown at higher magnification in the insets. (H) Numerous BrdU+ Claudius cells are detectable. (C, Claudius; D, Deiters; H, Hensen; T, pillar/tunnel; GER, Greater epithelial ridge; WT, wild type). The scale bar in (B) (20 µm) applies to all parts.
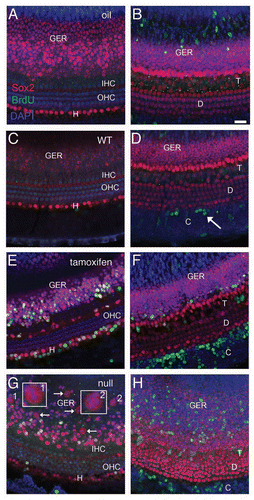
Figure 3 Proliferation in the organ of Corti after induced deletion of p27Kip1 in adult animals. Adult mice were given BrdU ± tamoxifen and observed immediately afterwards (group L6, A–D) or 6 weeks later (group L6-12, E and F). Tissues were immunostained for Sox2 (red) and BrdU (green), with DAPI staining nuclei (blue). Shown are brightest-point projections from a confocal Z-series spanning the sensory epithelium (A, B, E and F) or Deiters' cell nuclear region (C and D, inset for F). No BrdU-labeled cells are detectable in a p27 wild-type (WT) mouse (A), but BrdU+ cells are present in a vehicle-treated adult p27L+/L+;CreER+ animal (B) in the organ of Corti and in the Claudius cell region. (C and D) A tamoxifen-treated p27L+/L+;CreER+ mouse (Group L6) shows two BrdU-labeled Deiters' cells (arrows). The BrdU channel is shown in gray scale in (D). (E and F) Tamoxifen-treated p27L+/L+;CreER+ mouse euthanized 6 weeks later (group L6–12) showing BrdU+ Claudius, Hensen and inner sulcus cells (asterisk). A labeled Deiters' cell (arrow) is also shown in (F) (BrdU channel only in gray scale) and as an inset in a narrower Z-series spanning only the Deiters' nuclei region. All scale bars equal 20 µm. OHC, outer hair cell; IHC, inner hair cell; IS, inner sulcus region; C, Claudius cell region, D, Deiters' cell.
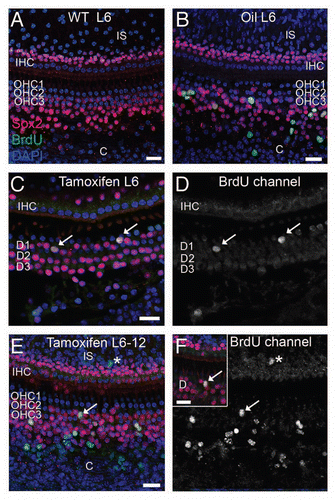
Figure 4 Deletion of p27Kip1 induced in adult p27L+/L+;CreER mice. Dorsal view of pituitaries from p271L+/L+;CreER+ (inducible knockout) mice treated at 15 weeks of age with vehicle (A) or tamoxifen (B) and then examined at 30 weeks of age. Immunostaining for p27Kip1 (red) is apparent in vehicle-treated controls (C), but is eliminated following tamoxifen treatment (D, blue = DAPI nuclear stain). Black scale bar = 1 mm, white scale bar = 100 µm. pd, pars distalis; pi, pars intermedia.
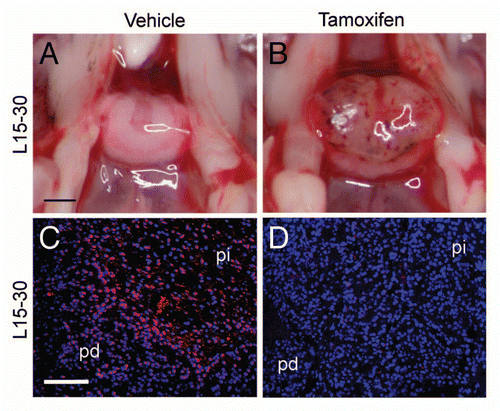
Figure 5 Reactivation of p27Kip1 expression in p27S−/S−;CreER mice. Dorsal view of pituitary (A) from p27S−/S−;CreER+ (inducible knock-on) mice treated with vehicle (A) or tamoxifen (B) at 6 weeks of age and harvested at age 15 weeks (group S6–15). Immunostaining shows absence of p27 in vehicle-treated control pituitary (C), compared to tamoxifen-treated animals (D). Pituitaries from p27S−/S−;CreER+ mice are pathologically enlarged in mice treated with vehicle at 15 weeks of age (E) and observed at 30 weeks of age (group S15–30). Comparable treatment with tamoxifen (F) led to resolution of hemorrhage. An H&E stained section (G) of a tamoxifen-treated mouse pituitary (S15–30 treatment group) shows resolution of hemorrhage and the infiltration of macrophages (m) into Rathke's pouch, which (H) stain positive for CD34 immunoperoxidase (hematoxylin counterstain). Black scale bar = 1 mm (applies to parts A, B, E and F); white scale bar = 100 µm (applies to C, D, G and H). pd, pars distalis; pi, pars intermedia; pn, pars nervosa.
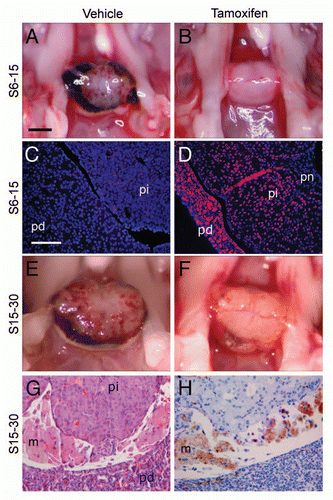
Figure 6 Proliferation and gene expression following loss or reactivation of p27Kip1. Mass of whole pituitaries (A) of p27L+/L+ mice (group L15–30, mean ± SD) treated with tamoxifen (Tam+) and compared to control animals without Cre (CreER−) or treated with vehicle (Tam−). (B) p27S−/S− mice treated at 6 weeks of age (group S6–15) or 15 weeks of age (S15–30) are shown. (C) Cell counts of BrdU-positive cells per high-power field (0.075 mm2, with a mean of 142 ± 12 DAPI+ nuclei) following one week of tamoxifen administration in 6 week-old mice. Homozygous p27loxP mice (L+), p27stop mice (S−) and wild-type (+) mice were examined with or without CreER and tamoxifen. (D) Pars intermedia from p27S−/S−;CreER+ mice treated with IdU and tamoxifen (Tam+) or vehicle (Tam−) followed by CldU and scored for the percentage of IdU, CldU or double-positive cells. (E) RNA expression levels by RT-QPCR are expressed as change in PCR cycle number (−ΔΔCt, mean change ± SEM) for selected genes altered in p27 null pituitary tumors from p27S−/S−;CreER+ mice treated with vehicle vs. tamoxifen. (*p value = 0.01, **p value < 0.001, t-test).
Table 1 Treatment schedules
Table 2 GFP expression in neonatal and adult auditory sensory epithelia in Z/EG; CreER+ reporter miceTable Footnote1
Table 3 BrdU-labeled cell density and retention in neonatal and adult cochleasTable Footnote1
Additional material
Download Zip (882 KB)Acknowledgements
We thank Andrew McMahon for providing CreER mice, Kendra Garrison for mouse colony assistance, Glen MacDonald for confocal imaging, Julie Randolph-Habecker for histopathology, and Ed Rubel for sharing facilities and helpful discussion. This research was supported by grants from the NIDCD: R01-DC03944 (EO), P30-DC04661 and by the NICHHD: P30-HD002274. It was also supported by the National Cancer Institute: R01-CA100053 (W.M.C., M.F.).
References
- Chen P, Segil N. p27(Kip1) links cell proliferation to morphogenesis in the developing organ of Corti. Development 1999; 126:1581 - 1590
- Löwenheim H, Furness DN, Kil J, Zinn C, Gültig K, Fero ML, et al. Gene disruption of p27(Kip1) allows cell proliferation in the postnatal and adult organ of Corti. Proc Natl Acad Sci USA 1999; 96:4084 - 4088
- Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, et al. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia and pituitary tumors. Cell 1996; 85:707 - 720
- Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, et al. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell 1996; 85:721 - 732
- Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis and female sterility in p27(Kip1)-deficient mice. Cell 1996; 85:733 - 744
- Kanzaki S, Beyer LA, Swiderski DL, Izumikawa M, Stöver T, Kawamoto K, et al. p27(Kip1) deficiency causes organ of Corti pathology and hearing loss. Hear Res 2006; 214:28 - 36
- Lee YS, Liu F, Segil N. A morphogenetic wave of p27Kip1 transcription directs cell cycle exit during organ of Corti development. Development 2006; 133:2817 - 2826
- Ruben RJ. Development of the inner ear of the mouse: A radioautographic study of terminal mitoses. Acta Otolaryngol 1967; 220:1 - 44
- Davis MD, Lichtensteiger W, Schlumpf M, Bruinink A. Early postnatal development of pituitary intermediate lobe control in the rat by dopamine neurons. Neuroendocrinology 1984; 39:1 - 12
- Yamaguchi H, Aiba A, Nakamura K, Nakao K, Sakagami H, Goto K, et al. Dopamine D2 receptor plays a critical role in cell proliferation and proopiomelanocortin expression in the pituitary. Genes Cells 1996; 1:253 - 268
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev 1995; 9:1149 - 1163
- Chen P, Zindy F, Abdala C, Liu F, Li X, Roussel MF, et al. Progressive hearing loss in mice lacking the cyclin-dependent kinase inhibitor Ink4d. Nat Cell Biol 2003; 5:422 - 426
- Laine H, Doetzlhofer A, Mantela J, Ylikoski J, Laiho M, Roussel MF, et al. p19(Ink4d) and p21(Cip1) collaborate to maintain the postmitotic state of auditory hair cells, their codeletion leading to DNA damage and p53-mediated apoptosis. J Neurosci 2007; 27:1434 - 1444
- Franklin DS, Godfrey VL, Lee H, Kovalev GI, Schoonhoven R, Chen-Kiang S, et al. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev 1998; 12:2899 - 2911
- Franklin DS, Godfrey VL, O'Brien DA, Deng C, Xiong Y. Functional collaboration between different cyclin-dependent kinase inhibitors suppresses tumor growth with distinct tissue specificity. Mol Cell Biol 2000; 20:6147 - 6158
- Bilodeau S, Roussel-Gervais A, Drouin J. Distinct developmental roles of cell cycle inhibitors p57Kip2 and p27Kip1 distinguish pituitary progenitor cell cycle exit from cell cycle reentry of differentiated cells. Mol Cell Biol 2009; 29:1895 - 1908
- Wu L, de Bruin A, Saavedra HI, Starovic M, Trimboli A, Yang Y, et al. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature 2003; 421:942 - 947
- Yu Y, Weber T, Yamashita T, Liu Z, Valentine MB, Cox BC, et al. In vivo proliferation of postmitotic cochlear supporting cells by acute ablation of the retinoblastoma protein in neonatal mice. J Neurosci 2010; 30:5927 - 5936
- Weber T, Corbett MK, Chow LM, Valentine MB, Baker SJ, Zuo J. Rapid cell cycle reentry and cell death after acute inactivation of the retinoblastoma gene product in postnatal cochlear hair cells. Proc Natl Acad Sci USA 2008; 105:781 - 785
- Huang M, Sage C, Tang Y, Lee SG, Petrillo M, Hinds PW, et al. Overlapping and distinct pRb pathways in the mammalian auditory and vestibular organs. Cell Cycle 2011; 10:337 - 351
- Chien WM, Garrison K, Caufield E, Orthel J, Dill J, Fero ML. Differential gene expression of p27Kip1 and Rb knockout pituitary tumors associated with altered growth and angiogenesis. Cell Cycle 2007; 6:750 - 757
- Vooijs M, van der Valk M, te Riele H, Berns A. Flpmediated tissue-specific inactivation of the retinoblastoma tumor suppressor gene in the mouse. Oncogene 1998; 17:1 - 12
- Oesterle EC, Stone JS. Salvi RJ, Popper AN, Fay RR. Hair Cell Regeneration, Repair and Protection. Hair cell regeneration 2008; New York, NY Springer Verlag 141 - 197
- Stone JS, Cotanche DA. Hair cell regeneration in the avian auditory epithelium. Int J Dev Biol 2007; 51:633 - 647
- Chien WM, Rabin S, Macias E, Miliani de Marval PL, Garrison K, Orthel J, et al. Genetic mosaics reveal both cell-autonomous and cell-nonautonomous function of murine p27Kip1. Proc Natl Acad Sci USA 2006; 103:4122 - 4127
- Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: A tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 2002; 244:305 - 318
- Novak A, Guo C, Yang W, Nagy A, Lobe CG. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis 2000; 28:147 - 155
- Oesterle EC, Campbell S, Taylor RR, Forge A, Hume CR. Sox2 and Jagged1 expression in normal and drug-damaged adult mouse inner ear. J Assoc Res Otolaryngol 2008; 9:65 - 89
- Hasson T, Mooseker MS. Porcine myosin-VI: Characterization of a new mammalian unconventional myosin. J Cell Biol 1994; 127:425 - 440
- Hume CR, Bratt DL, Oesterle EC. Expression of LHX3 and SOX2 during mouse inner ear development. Gene Expr Patterns 2007; 7:798 - 807
- Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell 2007; 12:817 - 826
- Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, Macleod KF. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol 2007; 27:6229 - 6242
- Seo DW, Li H, Guedez L, Wingfield PT, Diaz T, Salloum R, et al. TIMP-2 mediated inhibition of angiogenesis: An MMP-independent mechanism. Cell 2003; 114:171 - 180
- Stetler-Stevenson WG, Seo DW. TIMP-2: An endogenous inhibitor of angiogenesis. Trends Mol Med 2005; 11:97 - 103
- Laine H, Sulg M, Kirjavainen A, Pirvola U. Cell cycle regulation in the inner ear sensory epithelia: Role of cyclin D1 and cyclin-dependent kinase inhibitors. Dev Biol 2010; 337:134 - 146
- Endo T, Nakagawa T, Lee JE, Dong Y, Kim TS, Iguchi F, et al. Alteration in expression of p27 in auditory epithelia and neurons of mice during degeneration. Neurosci Lett 2002; 334:173 - 176
- Lengner CJ. iPS cell technology in regenerative medicine. Ann NY Acad Sci 2010; 1192:38 - 44
- Kesser BW, Lalwani AK. Gene therapy and stem cell transplantation: Strategies for hearing restoration. Adv Otorhinolaryngol 2009; 66:64 - 86
- Ono K, Nakagawa T, Kojima K, Matsumoto M, Kawauchi T, Hoshino M, et al. Silencing p27 reverses post-mitotic state of supporting cells in neonatal mouse cochleae. Mol Cell Neurosci 2009; 42:391 - 398
- Kil J, Gu R, Fero M, Lynch ED. The Open Otorhinolaryngology. Journal 2011; In press
- Gary KA, Chronwall BM. The onset of dopaminergic innervation during ontogeny decreases melanotrope proliferation in the intermediate lobe of the rat pituitary. Int J Dev Neurosci 1992; 10:131 - 142
- Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Höfler H, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci USA 2006; 103:15558 - 15563
- Carneiro C, Jiao MS, Hu M, Shaffer D, Park M, Pandolfi PP, et al. p27 deficiency desensitizes Rb−/− cells to signals that trigger apoptosis during pituitary tumor development. Oncogene 2003; 22:361 - 369
- Benson C, White J, De Bono J, O'Donnell A, Raynaud F, Cruickshank C, et al. A phase I trial of the selective oral cyclin-dependent kinase inhibitor seliciclib (CYC202; R-Roscovitine), administered twice daily for 7 days every 21 days. Br J Cancer 2007; 96:29 - 37
- Hahn O, Stadler W. Sorafenib. Curr Opin Oncol 2006; 18:615 - 621