Abstract
Hypoxia inducible factor (HIF) is the major transcription factor involved in the regulation of the cellular response to hypoxia, or low oxygen tensions. Even though HIF-1 function is mostly studied following hypoxic stress, well oxygenated areas of several diseased tissues have detectable levels of this transcription factor. Therefore, it is surprising how little is known about the function of HIF in normoxia. This study seeks to fill this gap. Using transient HIF-1α knockdown, as well as, stable cell lines generated using short hairpin RNAs (shRNA), we have further characterized the role of HIF-1α in normoxia. Our data reveals that knockdown of HIF-1α results in a significant increase in cells in the G1 phase of the cell cycle. We find that HIF-1α depletion increases the protein and mRNA of both p21 and p27. p21 is induced via, at least in part, p53-independent but SP1-dependent mechanisms. Interestingly, HIF-1α knockdown also alters the cellular response to chemotherapeutic agents. These data have important implications in not only for the further understanding of HIF-1α, a major transcription factor, but also for the use of HIF-targeted and combination therapies in cancer treatment.
Introduction
HIF-1α, the major transcription factor responsible for oxygen homeostasis in mammalian cells, is involved in many aspects of cancer biology including tumorigenesis, metabolism, differentiation, proliferation, angiogenesis, metastasis and responsiveness to therapy.Citation1 Importantly, levels of HIF-1α are increased in many human tumors and this expression correlates with a poor patient outcome in many of these cancers including colorectal, pancreatic, breast, ovarian, head and neck, gastric, cervical, bladder and osteosarcoma.Citation2–Citation10 Moreover, the activation of HIF in tumor cells has also been shown to mediate resistance to both chemotherapy and radiation therapyCitation11,Citation12 and is expressed in both hypoxic and normoxic regions of solid tumors.Citation1,Citation13–Citation15
Typically, in normoxia, HIF-1α is negatively regulated by the oxygen-dependent activity of a class of prolyl hydroxylase domain enzymes (PHDs) and the factor inhibiting HIF (FIH). The asparginyl hydroxylase activity of FIH prevents HIF from interacting with transcriptional coactivators such as CBP/p300, effectively suppressing HIF-1α's transcriptional activity. While, the PHD-dependent hydroxylation of the HIF-1α subunit facilitates the binding of HIF-1α to the E3 ligase complex, Von-Hippel-Lindau (VHL), targeting HIF-1α for proteasomal degradation. Importantly, both the PHD enzymes and FIH require the availability of oxygen in addition to iron and 2-oxoglutarate for full activity.Citation16,Citation17 Therefore, it is perhaps not surprising that, HIF-1α activity has been best characterized in response to hypoxia, when this negative regulation is abrogated, resulting in the rapid accumulation of HIF-1α. However, despite the negative regulation at the protein level, HIF-1α is constitutively transcribed and translated under normoxic conditions and maintains a low level of transcriptional activity.Citation16,Citation17 Moreover, it is stabilized in an oxygen-independent manner in response to a variety of other stimuli.Citation18–Citation20
HIF-1α levels are elevated in both hypoxic and normoxic regions of tumors.Citation1,Citation13–Citation15 Normoxic activation of HIF has been attributed to genetic alterations within the oxygen sensing pathway, such as VHL inactivation and activation of cell signaling pathways such as the PI-3 kinase/AKT pathway.Citation1 In addition, several cytokines and growth factors including transforming growth factor-β, platelet-derived growth factor, thrombin, epidermal growth factor, insulin, insulin like growth factor and interleukin-1β can readily stabilize HIF-1α under normoxic conditions.Citation19,Citation21–Citation28
In this study, we investigate the effect of HIF-1α knockdown on cell cycle, cell cycle regulators, and responsiveness to chemotherapeutic agents in normoxia. Our data reveals that knockdown of HIF-1α results in a significant increase in cells in the G1 phase of the cell cycle and in the protein and mRNA expression of both p21 and p27. This accumulation occurs in normal and transformed cell types. Moreover, the accumulation of p21 in response to HIF-1α depletion is partly independent of p53. Importantly, concomitant knockdown of SP1 and HIF abrogates p21 induction, as well as, partially rescues the HIF-1α induced G1 accumulation. In addition to altering cell cycle and p21 transcription, HIF-1α knockdown also has functional consequences to the cell by altering cellular responsiveness to chemotherapeutic agents. HIF-1α depletion impairs the autophagy response while apoptosis is slightly enhanced. These data not only further the understanding of HIF-1α in normoxia, but also have implications on the use of HIF-1α as a therapeutic target in cancer treatment.
Results
HIF-1α knockdown results in G1 arrest and p21 induction while inhibiting retinoblastoma protein phosphorylation.
To investigate the effect of HIF-1α knockdown on the cell cycle, the human osteosarcoma cell line, U2OS, was used. U2OS cells were chosen since they have been used extensively to study HIF-1α biology and therefore made an attractive model system for our initial study.Citation29,Citation30
We used siRNA to transiently knockdown HIF-1α in U2OS cells in normoxic conditions and analyzed cells cell cycle distribution using FACS. As shown in , siRNA knockdown of HIF-1α was extremely effective in our model system. After 2 h of the hypoxia mimetic drug Desferroxamine (DFX) treatment virtually no HIF-1α protein was detectable by immunoblot, in the HIF-1α depleted cells, as compared to non-targeted siRNA control. Interestingly, HIF-1α knockdown resulted in a significant (p ≤ 0.01) accumulation of cells in the G1 phase of the cell cycle and a concomitant significant (p ≤ 0.01) reduction in cells in the S phase of the cell cycle ().
To determine the mechanism behind the G1-cell cycle arrest, whole cell lysates (WCL) from control and HIF-1α depleted cells were analyzed for the levels of key markers of cell cycle progression (). Western blot analysis revealed that HIF-1α depletion caused increases in both Cyclin D1 and Cyclin E and a decrease in Cyclin B1. Importantly, despite the changes in the levels of Cyclin D1 and E, two critical regulators of the G1-S transition, we observed a decrease in phosphorylation of the retinoblastoma (Rb) protein indicating that cells are not transitioning into S phase.Citation31
The cyclin-dependent kinase inhibitors p21 and p27 are important negative regulators of the cyclin-dependent kinases Cyclin D and Cyclin E. These inhibitors abrogate the activity of both cyclin D and E/CDK containing complexes and thereby inhibit phosphorylation of the Rb protein. This loss of phosphorylation subsequently prevents transition into S phase of the cell cycle.Citation31 Therefore we determined if p21 and p27 protein levels were altered during HIF-1α knockdown. Western blot analysis of WCL obtained for non-targeted control and HIF-1α siRNA treated cells () shows a dramatic increase in p21 protein levels, as well as, an increase in p27 levels when HIF-1α is depleted.
To determine the mechanism behind the induction of p21 and p27 proteins, we investigated if depletion of HIF-1α was increasing these proteins at the transcriptional level. Quantitative RT-PCR (qRT-PCR) was performed using RNA from control and HIF-1α depleted cells and the levels of p21, p27 and GLUT3, a well characterized HIF target gene were analyzed (). HIF-1α mRNA and the levels of the known HIF-1α target gene GLUT3 were significantly reduced (). Interestingly, we could detect a significant increase in both p21 (p ≤ 0.01) and p27 (p ≤ 0.05) mRNA levels. These data demonstrates that HIF-1α is transcriptionally active under normoxic conditions, as is evident by the significant (p ≤ 0.01) decrease in GLUT3 mRNA levels during HIF-1α depletion in normoxia. Moreover, these data demonstrate that, p21 and p27 protein increases observed following HIF-1α knockdown, are mediated by increased mRNA levels.
G1 accumulation and p21 induction occurs in p53-impaired HeLa cells.
One of the major transcription factors responsible for the regulation of the p21 promoter is p53.Citation4 Therefore, we wanted to determine if the p21 induction observed in response to HIF-1α depletion was mediated through p53. As HeLa cells have an impaired p53 due to the HPV protein E6, we used this cell line to evaluate the effect of HIF-1α knockdown on cell cycle and p21 expression levels. Transient depletion of HIF-1α in HeLa cells was extremely effective (). Importantly, HIF-1α knockdown resulted in a significant (p ≤ 0.01) accumulation of cells in the G1 phase of the cell cycle and a concomitant significant (p ≤ 0.01) reduction in cells in the S phase of the cell cycle (). As shown in , western blot analysis of WCL revealed that this G1 accumulation in HIF-1α depleted HeLa cells was accompanied by the same alterations in specific cell cycle regulators we had observed in the U2OS cells, with increases in both Cyclin D1 and Cyclin E and a decrease in Cyclin B1. Moreover, just as observed in the U2OS cell line, Rb protein does not become phosphorylated in HIF-1α knockdown cells. Similarly, we observed an increase in p21 protein levels. However, p27 protein was only slightly induced in HeLa cells (). Importantly, HIF-1α depletion also resulted in increased mRNA levels of p21 and, to a lesser extent, of p27 in HeLa cells (). Similar results were also obtained in MDA-MB-231 cells that possess mutant p53 (data not shown). These data demonstrate that a p53-independent effect is occurring during HIF-1α depletion resulting in G1 accumulation and the observed increase in p21 at both the protein level and at the mRNA level.
HIF-2α does not alter p21 or p27 message levels.
As the HIF-1α and HIF-2α transcription factors have overlapping functions and target genes,Citation32 we wanted to determine if HIF-2α depletion would have the same effect on p21 and p27 message levels as we observed in response to HIF-1α knockdown. We used a specific siRNA oligonucleotide against HIF-2α and then analyzed by qRT-PCR mRNA levels of p21, p27 and GLUT3. HeLa cells were chosen to determine the effect of HIF-2α knockdown because they express readily detectable levels of HIF-2α while U2OS cells express only very low levels. Knockdown of HIF-2α was confirmed by analysis of HIF-2α mRNA levels. As shown in , HIF-2α reduction has no significant effect on p21 or p27 levels. Therefore, the effects we have observed are unique to HIF-1α depletion.
Simultaneous knockdown of HIF-1α and SP1 abrogates p21 induction and partially rescues G1 accumulation.
Another major transcription factor controlling the expression of p21 in response to a broad range of stimuli is SP1. In fact, there are a total of six SP1 binding sites on the p21 promoter.Citation31 Therefore, we sought to determine if SP1 was involved in the upregulation of p21 we observed in response to HIF-1α knockdown. U2OS cells were transfected with control, HIF-1α, SP1 or both SP1 and HIF-1α siRNA. The mRNA levels for p21, HIF-1α and SP1 were then analyzed by qRT-PCR. Knockdown of both SP1 and HIF-1α was confirmed by analysis of specific mRNA. As shown in , a significant induction of p21 (p ≤ 0.01) was again observed in response to HIF-1α depletion, while SP1 knockdown alone had no significant effects on p21 levels. Importantly, simultaneous knockdown of both SP1 and HIF-1α significantly (p ≤ 0.01) reduced p21 induction, as compared to HIF-1α alone. Interestingly, HIF-1α reduction alone resulted in an increase in SP1 message level (). Similar results were also observed at the protein levels ().
To firmly establish the role of SP1 in the control of p21 gene, we determined if HIF-1α depletion resulted in changes of SP1 recruitment to the p21 promoter using chromatin immunoprecipitation (ChIP). Analysis of the p21 promoter revealed that HIF-1α knockdown results in enhanced levels of SP1 present at the p21 promoter (). Importantly, this enhancement was restricted to the SP1 binding sites present at the p21 promoter, as no recruitment could be measured when a control region of the promoter was used (). Furthermore and in agreement with our expression data, HIF-1α depletion results in enhanced levels of RNA polymerase II present at the p21 promoter and indication of active transcription (). These data demonstrate that SP1 is required for p21 induction in response to HIF-1α knockdown.
To determine the consequences for cell cycle progression, U2OS cells were transfected with control, HIF-1α, SP1 or both SP1 and HIF-1α siRNA and the cells harvested for cell cycle analysis. A significant accumulation (p ≤ 0.01) of cells in the G1 phase of the cell cycle was again observed in response to HIF-1α knockdown, while SP1 knockdown alone had no significant effects on the number of G1 phase cells (). Importantly, simultaneous knockdown of both SP1 and HIF-1α significantly (p ≤ 0.01) reduced the number of cells in the G1 phase of the cell cycle, as compared to HIF-1α knockdown alone (). Taken together these data demonstrate that not only does the simultaneous reduction of both SP1 and HIF-1α abrogate p21 induction, but it also significantly (p ≤ 0.01) rescues the accumulation of cells in the G1 phase of the cell cycle that occurs in response to HIF-1α depletion alone. Therefore, SP1 is indeed involved in the G1 arrest and p21 induction observed in response to HIF-1α knockdown.
p21 induction and G1 accumulation also occurs in HIF-1α stable knockdown cells.
To rule out the possibility that transient transfection of HIF-1α siRNAs was causing an off-target cell cycle arrest, we created HIF-1α stable knockdown cells using two different shRNA sequences Sequence 1 (S1) and Sequence 2 (S2). WCL were obtained from stable HIF-1α depleted cells and compared with that of nontargeted control cells. demonstrates the high level of HIF-1α knockdown observed with both sequences following 2 h of DFX treatment, with very little HIF-1α stabilization observed in either of the stable HIF-1α stable knockdown cell lines. Importantly, as shown in , p21 protein is increased in both sets of HIF-1α depleted cell lines as compared to the nontargeted control. Moreover, when samples were harvested and analyzed for cell cycle analysis both sequences had a significant [(S1 p ≤ 0.05) (S2 p ≤ 0.01)] increase in cells in the G1 phases of the cell cycle with a concomitant reduction in cells in the S phase of the cell cycle (). These data demonstrate that stable depletion of HIF-1α, just like transient depletion, causes an increase in p21 protein levels as well as an accumulation of cells in the G1 phase of the cell cycle.
HIF-1α knockdown in non-transformed MCF10A and human foreskin fibroblast cells results in p21 mRNA induction.
In order to evaluate if the effects of HIF-1α knockdown on p21 were restricted to transformed cell lines we used the breast epithelial cell line MCF10A and human foreskin fibroblast (HFF). Knockdown of HIF-1α was confirmed in both cell lines. Importantly, the results in all of the nontransformed cell lines tested, mirror those we had observed in both HeLa and U2OS cells. Both p21 (p ≤ 0.05) and p27 (p ≤ 0.01) mRNA levels were significantly induced with HIF-1α depletion ( and E). These data demonstrate that HIF-1α reduction not only increases p21 and p27 message in transformed cell lines, but also, in non-transformed cell lines.
HIF-1α depletion protects against autophagy in chemotherapy treated U2OS cells.
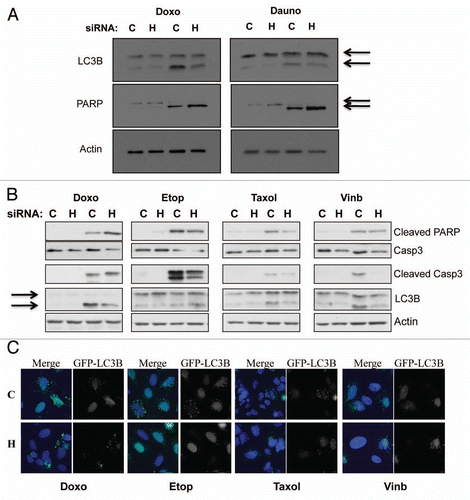
Given that HIF-1α is a viable cancer therapy target, we wanted to determine how HIF-1α knockdown would alter responsiveness to treatment with traditional chemotherapeutic drugs, as combination therapies tend to be more effective. This question became even more pertinent when coupled with our new findings that HIF-1α depletion induces p21 levels. p21 is a well known modulator of both apoptosis and autophagy in response to a variety of stimuli.Citation8,Citation33–Citation36 Therefore, to address this question U2OS cells were transfected using nontargeted or HIF-1α siRNA for 48 h prior to a 24 h treatment with the chemotherapeutic drugs doxorubicin or daunorubicin. Following treatment WCL were harvested and samples were analyzed by western blot using specific antibodies for LC3B and PARP. Our data, demonstrates that when HIF-1α is knockdown LC3B cleavage, a well characterized marker for autophagy, is reduced in response to doxorubicin and daunorubicin (). In response to doxorubicin and daunorubicin, PARP cleavage is also slightly enhanced in the HIF-1α knockdown cells as compared to the nontargeted control (). To extend and confirm these observations, we analyzed the cell responses to additional chemotherapeutic drugs such as etoposide, taxol and vinblastine in the presence or absence of HIF-1α ( and C). We could confirm that when HIF-1α is depleted, doxorubicin mainly induced apoptosis, assessed by PARP-cleavage and activation of caspase-3 (). However, we did not detect any increased levels of apoptosis markers in HIF-1α depleted cells when we used etoposide, taxol or vinblastine (). Interestingly, HIF-1α depletion resulted in lowering of LC3 cleavage in all treatments used ( and C). Taken together these data providing intriguing evidence that HIF-1α depletion inhibits the autophagy response, while slightly enhancing the apoptosis pathway in response to specific chemotherapy treatments.
Discussion
HIF-1α is involved in many aspects of cancer biology and its expression correlates with poor patient outcome in many cancers, as well as, resistance to both chemotherapy and radiation therapy.Citation2–Citation12 Moreover, HIF-1α is expressed in both hypoxic and normoxic regions of solid tumors.Citation1,Citation13–Citation15 However, the role of HIF-1α in normoxia has not been elucidated thus far. In this study, we investigated the effect of HIF-1α knockdown on cell cycle, cell cycle regulators and responsiveness to chemotherapeutic agents in normoxia. Our data reveals that knockdown of HIF-1α not only significantly increases cells arrested in the G1 phase of the cell cycle (p ≤ 0.01), but also, results in a significant increase in the protein and mRNA expression of both p21 (p ≤ 0.01) and p27 (p ≤ 0.05) ( and ). Importantly the accumulation of p21 in response to HIF-1α reduction occurs in transformed ( and ) and normal () cell types, as well as, in stable knockdown cells (). However, p21 is not induced in response to HIF-2α depletion (), thus indicating a specific function for HIF-1α. Interestingly, HIF-1α knockdown induces an increase in p21 in both p53 wild type U2OS cells () and p53 impaired HeLa cells ().
The cyclin-dependent kinase inhibitor, p21, is a major checkpoint in the cell cycle and can inhibit cyclin-dependent kinases at the G1/S boundary by direct interaction and thereby, inhibit cell cycle progression by preventing phosphorylation of the Rb protein due to the resulting blockade of the E2F pathway.Citation33,Citation37–Citation39 This is, to our knowledge, the first time the effects of HIF-1α depletion on the p21 checkpoint have been investigated in normoxia and our data clearly demonstrates that HIF-1α reduction results in a significant increase in p21 message and protein expression, loss of Rb phosphorylation and subsequent G1 arrest ( and ).
p21 expression is regulated by p53-dependent and p53-independent pathways.Citation40 Because our data indicated that the accumulation of p21 and the subsequent G1 arrest in response to HIF-1α knockdown was, at least in part, independent of p53 (), we began to explore p53-independent activation of the p21 promoter. The major p53-independent regulator of p21 is the transcription factor SP1. Indeed, six SP1 binding sites exist on the human p21 gene in the region between −119 bp and the start of transcription.Citation41 Importantly, these SP1 binding sites not only contribute to the basal expression of p21,Citation34 but also, have been shown to be responsible for the p21 inductions observed in response to a variety of stimuli and cellular contexts including the tumor suppressor BRCA, PMA, okadaic acid, TGFβ, calcium, butyrate, lovastatin, trichostatin and nerve growth factor to name a few.Citation41–Citation47 Importantly, the SP1-mediated activation of the p21 promoter in several of these cellular contexts also occurs during G1 arrest.Citation42,Citation44 This is important to note because SP1 is most active during the G1 phase of the cell cycle, indicating that G1 cell cycle arrest may, in itself, contribute to increased SP1 activity.Citation48
Therefore, it is perhaps not surprising that the concomitant knockdown of SP1 and HIF-1α very significantly (p ≤ 0.01) abrogates p21 induction and partially, but significantly (p ≤ 0.01), rescued the G1 accumulation induced by HIF-1α depletion (). We did note that while the reduction in p21 levels with simultaneous HIF-1α and SP1 knockdown was nearly complete ( and B), as compared to HIF-1α depletion alone; the G1 arrest, while very significantly (p ≤ 0.01), was not completely rescued (). This result is logical as p27, another cyclin-dependent kinase inhibitor that results in G1 arrest, is also induced during HIF-1α knockdown ( and ). Therefore, the effects of p27 and p21 are likely additive with regard to the G1 arrest observed.
Taken together these data indicate that SP1 is a p53-independent regulator of p21 induction in response to HIF-1α depletion, as both p21 message and subsequent G1 arrest are very significantly reduced (p ≤ 0.01) with double knockdown of SP1 and HIF-1α. We also found that HIF-1α depletion induced an increase in the recruitment of SP1 and RNA polymerase II to the p21 promoter ( and D). These data further demonstrating the importance of SP1 in the control of p21 expression when HIF-1α is reduced.
Because HIF-1α is an important target for cancer therapeutics and because combination therapies are of particular interest in cancer treatments we wanted to assess the effect HIF-1α knockdown would have on the cellular responsiveness to chemotherapeutic drugs. Our data demonstrate, for the first time, that HIF-1α knockdown in normoxia has functional consequences to the cell in response to chemotherapeutic treatment. During HIF-1α depletion the autophagy response is reduced while apoptosis is slightly enhanced (). Accumulating evidence has shown that in addition to its well known function as a cyclin kinase inhibitor, p21 also modulates the cell death pathways of both autophagy and apoptosis. While both positive and negative regulation of apoptosis have been reported, depending on cell type and context, to our knowledge only negative regulation by p21 of autophagy has been demonstrated.Citation35,Citation36
Importantly, a 2008 study by Fujiwara and colleagues clearly demonstrates that in response to ceramide and γ-Irradiation, which in turn generates ceramide, autophagy is reduced and apoptosis enhanced but only in p21 expressing cells.Citation35 The knockdown of p21 reverses this effect and shunts cell death in response to ceramide towards autophagy and away from apoptosis. Moreover, that study demonstrated that overexpression of p21 alone increased apoptosis and decreased autophagy.Citation35,Citation49 The published data demonstrates that p21 is responsible for shunting cellular fate towards apoptosis and away from autophagy.Citation35 These findings are especially applicable to our data as not only do we observe a significant increase in p21 in response to HIF-1α knockdown, but also, doxorubicin and daunorubicin result in the generation of ceramide.Citation50–Citation52 Interestingly, we did not observe any increased in apoptosis markers in HIF-1α depleted cells treated with etoposide, taxol and vinblastine ( and C). Taxol and Vinblastine are microtubule-targeting agents that have been shown to induce p21 and alter the effects of chemotherapeutic drugs.Citation53 In addition, microtubule-disrupting drugs also lead to inhibition of HIF-1α levels via translation repression.Citation54 As such, our results are in agreement with the published work, demonstrating that further increases in p21 provided by HIF-1α depletion, do not result in increases in apoptosis markers. Our data demonstrate, that HIF-1α knockdown has functional consequences to the cell in response to specific chemotherapeutic agents and may have important implications to cancer treatment and therapies.
Initial studies suggested that the additive cell death effect of autophagy and apoptosis was beneficial to treatments, however, the preponderance of the evidence now indicates that autophagy is in fact an adaptive mechanism of the tumor cell. Autophagy contributes to cell survival by facilitating resistance to chemotherapeutic and radiation treatments. Indeed, many clinical trials are currently underway to develop inhibitors of autophagy that can be used in conjunction with traditional chemotherapeutics and radiation therapies to prevent the generation of these resistant cells.Citation55–Citation57
Taken together our data demonstrates for the first time that reduction of HIF-1α in normoxic conditions results in the induction of p21 and p27 and G1 cell cycle arrest. This is a HIF-1α specific response, as depletion of HIF-2α had no significant effect on p21 or p27 message levels. Mechanistically, this is dependent on the SP1 transcription factor. Importantly, we have demonstrated that HIF-1α knockdown alters cellular responsiveness to chemotherapeutic agents resulting in a decreased autophagy response and a slightly enhanced apoptosis response. These data offer valuable insight into the potential effectiveness of HIF-1α based therapies in conjunction with traditional treatments, while demonstrating the importance of further understanding of the role of HIF-1α in normoxia.
Materials and Methods
Cells.
Human Osteosarcoma cells, U2OS and Human Cervical Epithelial cells, HeLa, were maintained at 5% CO2 in Dulbecco's modified Eagle's medium (Lonza) supplemented with 10% fetal bovine serum (Invitrogen), 1% penicillin-streptomycin (Lonza) and 1% L-glutamine (Lonza). Human Foreskin Fibroblasts (HFF) were maintained at 5% CO2 in Dulbecco's modified Eagle's medium (Lonza) supplemented with 15% fetal bovine serum (Invitrogen), 1% penicillin-streptomycin (Lonza) and 1% L-glutamine (Lonza). U2OS, HFF and HeLa cells were obtained from the European Collection of Cell Cultures (ECACC). U2OS cells containing stably transfected HIF-1α or nontargeting shRNAs were created using the pSilencer vectors as described previously in reference Citation58. Human Normal Breast Epithelial cells MCF10A were maintained at 5% CO2 in DMEM/Hans nutrient mixture F12 with 5% FBS (Invitrogen), 1% penicillin-streptomycin (Lonza) and 1% L-glutamine (Lonza), supplemented with 10 µg/ml insulin (Sigma), 20 ng/ml EGF (Sigma) and 5 µg/ml hydrocortisone (Sigma). MCF10A cells were a kind gift from Dr. A. Schulze (CRUK, London). U2OS-GFP-LC3B cells were maintained in maintained at 5% CO2 in Dulbecco's modified Eagle's medium (Lonza) supplemented with 10% fetal bovine serum (Invitrogen), 1% penicillin-streptomycin (Lonza) and 1% L-glutamine (Lonza) with addition 200 µg/mL G418.
Cell cycle analysis by flow cytometry.
Cells were prepared for flow cytometry analysis as described in reference Citation58. Cells with DNA content between 2N and 4N were designated as being in the G1, S or G2/M phase of the cell cycle. The number of cells in each phase of the cell cycle was expressed as a percentage of the total number of cells present after gating to eliminate doublets and determined using CellQuest software.
Antibodies.
Antibodies used were: anti-HIF-1α (MAB1536, R&D Systems), anti-β-actin (A5441, Sigma), anti-cyclin D1 (2926, Cell Signaling), anti-Cyclin B1 (SC-752, Santa Cruz), anti-cyclin E (4129, Cell Signaling), anti-phospho-RB780 (9307, Cell Signaling), anti-phospho-RB795 (9301, Cell Signaling), anti-phospho-RB807/811 (9308, Cell Signaling), anti-Rb (9309, Cell Signaling), anti-p21 (554228, Pharmingen), anti-p27 (1641, Cell Signaling), anti-LC3B (2775, Cell Signaling), anti-PARP (9542, Cell Signaling), anti-Casp-3 (9662, Cell Signalling), anti-Cleaved PARP (Cell Signalling, 9541), anti-SP1 (07-645, Millipore), anti-RNA Polymerase II (sc-47701, Santa Cruz).
Western blot.
Western blots were performed essentially as described previously in reference Citation59. Assays analyzed whole cell lysates and used 20–30 µg of protein. Whole cell protein extracts were performed as previously described in reference Citation59.
Hypoxia mimetic treatments.
For hypoxia mimetic conditions, cells were treated with 100 µM of Desferoxamine (Sigma) for 2 h immediately following 48 h siRNA treatments and harvested for western blot analysis as previously described.
siRNA.
siRNA duplex oligonucleotides were synthesized by MWG and transfected using Interferin (Polyplus) as per manufacturer's instruction. In brief, cells were plated the day before transfection at the concentration of 1.5 × 105 cells per well in 6.well plates. The following day, cells were transfected with the final concentration of 5 nM of siRNA oligonucleotides in fresh media, final volume of 2.2 mL. Cells were incubated for additional 48 h prior to harvesting.
Quantitative RT-PCR analysis.
Total RNA was extracted with the Peqgold RNA extraction kit (Peqlab), according to the manufacturer's directions. RNA was converted to cDNA using the Quantitect Reverse Transcription Kit (Qiagen). cDNA was amplified using specific primer sets and the Stratagene Brilliant II SYBR green qPCR mix according to the manufacturer instructions. Amplification and detection were performed using a Stratagene Mx3005P detection system. Sample values obtained with specific primer sets were normalized to β-actin primer set values.
Oligonucleotide sequences.
qRT-PCR. Actin For-CTG GGA GTG GGT GGA GGC Rev-TCA ACT GGT CTC AAG TCA GTG HIF-1α For-CAC TGA GGC AGT GGA GAC AG Rev-TGC AGT CCC AGC TAC TTG TG p21 For-GTC CAC TGG GCC GAA GAG Rev-TGC GTT CAC AGG TGT TTC TG p27 For-GTG GAC CCA AAG ACT GAT Rev-GGA ACC GTC TGA AAC ATT GLUT3 For-CAA TGC TCC TGA GAA GAT CAT AA Rev-AAA GCG GTT GAC GAA GAG T HIF-2α For-TTT GAT GTG GAA ACG GAT GA Rev-GGA ACC TGC TCT TGC TGT TC SP1 For-ACC AGG CTG AGC TCC ATG AT Rev-CCT CAG TGC ATT GGG TAC TTC HIF-2α For-TTT GAT GTG GAA ACG GAT GA Rev-GGA ACC TGC TCT TGC TGT TC.
siRNA. Nontargeted-CAG UCG CGU UUG CGA CUG G HIF-1α-CUG AUG ACC AGC AAC UU SP1-CCU GGA GUG AUG CCU AAU ATT HIF-2α-CAG CAU CUU UGA CAG UTT.
shRNA. Control and HIF-1α sequences were previously described in reference Citation60 and Citation61.
ChIP. SP1 proximal promoter
For-TTG TAT ATC AGG GCC GCG CT Rev-CGA ATC CGC GCC CAG CTC SP1 control region For-TGG CCC CTC TGT GAA AAC AT Rev-TTC CTG TTC CTG GCT CTA ACA AC.
Chromatin immunoprecipitation (ChIP).
Cells were grown to 70% confluency and cross-linked with 1% formaldehyde at room temperature for 10 minutes (m). Glycine was added to a final concentration of 0.125 M for 5 m at room temperature. Cells were washed twice with 10 ml of ice-cold phosphate-buffered saline and then scraped into 2 mls ice cold harvest buffer (PBS, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 µg/ml leupeptin, 1 µg/ml aprotinin) before being centrifuged at 1,000 rpm in an Avanti benchtop centrifuge at 4°C for 10 m. The supernatant was removed and the pellet was resuspended in 0.5 ml of lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1, 1 mM PMSF, 1 µg/ml leupeptin, 1 µg/ml aprotinin) and left on ice for 10 m. Samples were then sonicated at 4°C seven times. Each sonication was for 20 seconds (s) with a 1 m gap between each sonication. Supernatants were recovered by centrifugation at 12,000 rpm in an eppendorf microfuge for 10 m at 4°C before being diluted 10-fold in dilution buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl, pH 8.1). Samples were then pre-cleared for 2 hours (h) at 4°C with 2 µg of sheared salmon sperm DNA and 20 µl of protein A-Sepharose (50% slurry). At this stage, 10% of the material was kept and stored at −20°C as Input material. Immunoprecipitations were performed overnight with specific antibodies (2 µg), with the addition of BRIJ-35 detergent to a final concentration of 0.1%. The immune complexes were captured by incubation with 30 µl of protein A-Sepharose (50% slurry) and 2 µg salmon sperm DNA for 1 h at 4°C. The immunoprecipitates were washed sequentially for 5 m each at 4°C in Wash Buffer 1 (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCL, pH 8.1, 150 mM NaCl), Wash Buffer 2 (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl) and Wash Buffer 3 (0.25 M LiCl, 1% Nonidet P-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris-HCl, pH 8.1). Beads were washed twice with Tris-EDTA (TE) buffer and eluted with 100 µl of elution buffer (1% SDS, 0.1 M NaHCO3). Elutes were purified using a PCR purification kit (NBS).
Chemotherapeutic drug treatments.
For chemotherapeutic drug treatments all chemicals were obtained from Sigma and prepared according to the manufacturer's directions. Cells were treated with a final concentration per well of 2 µm Doxorubicin (Enzo Life Sci.), 2 µM Daunorubicin (Enzo Life Sci.), 25 µM Etoposide (Enzo Life Sci.), 50 ng/mL Taxol (Enzo Life Sci.), 25 ng/mL (Enzo Life Sci.), for 24 h immediately following the 48 h siRNA protocol and harvested for western blot analysis as previously described.
Fluorescence microscopy.
For immunofluorescence, cells were grown on coverslips and treated as indicated prior to fixation by incubation in 3.7% formaldehyde/PBS (pH 6.8) for 15 minutes. Cells were permeabilized in PBS-0.1% Triton X-100 for 15 minutes and then blocked in PBS-0.05% Tween-20 supplemented with 1% normal donkey serum for 30 minutes. Cells were analyzed and images were acquired using a DeltaVision microscope. Images were deconvolved and analyzed using OMERO client software (Open Microscopy Environment).
Statistical analysis.
Student's t-tests were performed using the means and p values were calculated. *p ≤ 0.050; **p ≤ 0.01 using Graphpad PRISM software.
Abbreviations
| HIF | = | hypoxia inducible factor |
| siRNA | = | small interfering RNA |
| shRNA | = | short hairpin RNA |
| SP1 | = | specificity protein 1 |
| PHD | = | prolyl-hydroxylase |
| FIH | = | factor inhibiting HIF |
| Rb | = | retinoblastoma protein |
| CDK | = | cyclin-dependent inase |
| WCL | = | whole cell lysates |
| DFX | = | deferroxamine |
| qRT-PCR | = | quantitative reverse transcriptase PCR |
| ChIP | = | chromatin immunoprecipitation |
Figures and Tables
Figure 1 HIF-1α knockdown results in G1 accumulation and p21 induction in U2OS cells. (A) Confirmation of HIF-1α knockdown using U2OS whole cell lysates (WCL) for western blot analysis. Samples were harvested following siRNA protocol and 2 h of DFX treatment. (B) U2OS cells were transfected with nontargeted or HIF-1α siRNA oligonucleotides prior to harvesting for cell cycle analysis using the propidium iodide staining protocol. Student's t-tests (two tailed) were performed and p-values calculated. *p ≤ 0.05 and **p ≤ 0.01. (C and D) U2OS cells were transfected using nontargeted or HIF-1α siRNA prior to harvest of WCL. Samples were analyzed by western blot using the specific antibodies indicated. (E) U2OS cells were transfected as in (B) and mRNA extracted. HIF-1α, p21, p27 and GLUT3 mRNA were analyzed using quantitative PCR. Graphs depict HIF-1α, p21, p27 and GLUT3 mRNA normalized to actin mRNA and compared to untreated samples. Student t-tests (two tailed) were performed and p-values calculated. *p ≤ 0.05 and **p ≤ 0.01. Data refers to a minimum of three independent experiments.
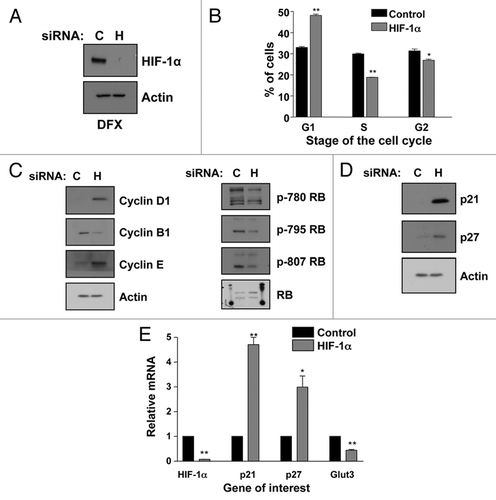
Figure 2 HIF-1α knockdown results in G1 accumulation and p21 induction in HeLa cells. (A) Confirmation of HIF-1α knockdown using HeLa whole cell lysates (WCL) for western blot analysis. Samples were harvested following siRNA protocol and 2 h of DFX treatment. (B) HeLa cells were transfected with nontargeted or HIF-1α siRNA oligonucleotides prior to harvesting for cell cycle analysis using the propidium iodide staining protocol. (C) HeLa cells were transfected using nontargeted or HIF-1α siRNA prior to harvest of WCL. Samples were analyzed by western blot using the specific antibodies indicated. (D) HeLa cells were transfected as in (B) but mRNA was extracted. HIF-1α, p21, p27 and GLUT3 mRNA were analyzed using quantitative PCR. Graph depicts mRNA levels normalized to actin. Student's t-tests (two tailed) were performed and p-values calculated. *p ≤ 0.05 and **p ≤ 0.01. (E) HeLa cells were transfected with control and HIF-2α siRNA oligonucleotides and mRNA extracted. HIF-1α, HIF-2α, p21, p27 and GLUT3 mRNA were analyzed using quantitative PCR. Graph depicts mRNA levels normalized to actin. Student's t-tests (two tailed) were performed and p-values calculated. *p ≤ 0.05 and **p ≤ 0.01. Data refers to a minimum of three independent experiments.
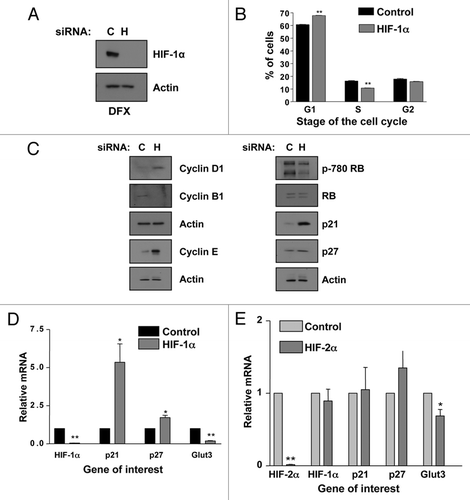
Figure 3 Simultaneous knockdown of HIF-1α and SP1 abrogates p21 induction and partially rescues G1 accumulation. (A) U2OS cells were transfected using nontargeted, HIF-1α, SP1 or both SP1 and HIF-1α siRNA prior to extraction of mRNA. Subsequently, p21, HIF-1α and SP1 mRNA were analyzed using quantitative PCR. Graph depicts mRNA levels normalized to actin. Student t-tests (two tailed) were performed and p-values calculated. *p ≤ 0.05 and **p ≤ 0.01. (B) U2OS cells were transfected as in A prior to lysis. WCL were analyzed by western blot using the indicated antibodies. (C) U2OS were transfected with control and HIF-1α siRNA oligonucleotides prior to fixation and lysis. ChIP were performed using SP1 and control IgG antibodies. p21 promoter regions were amplified using specific primers and levels of SP1 recruitment were analyzed by qPCR. (D) U2OS cells were treated and processed as in (C), but levels of RNA Polymerase II present at the p21 promoter were analyzed. (E) U2OS cells were transfected as in (A) prior to harvesting for cell cycle analysis using the propidium iodide staining protocol. Student's t-tests (two tailed) were performed and p-values calculated. *p ≤ 0.05 and **p ≤ 0.01. Data refers to a minimum of three independent experiments.
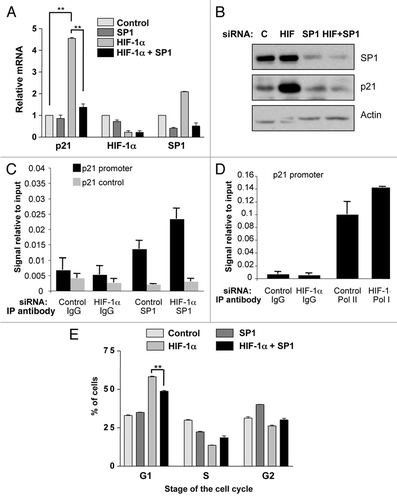
Figure 4 p21 induction and G1 accumulation also occurs in HIF-1α stable knockdowns and non-transformed cells. (A) Two sequences (1 and 2) of shRNA were used to generate U2OS-HIF-1α stable knockdown cells as reported previously in reference Citation61. Confirmation of HIF-1α knockdown in HIF-1α stable cells was performed using western blot analysis. WCL were harvested following 2 h of DFX treatment from non-targeted and HIF-1α stable cell lines. (B) WCLs from U2OS-HIF-1α stable knockdown cell lines were analyzed by western blot using the specific antibodies indicated. (C) U2OS-HIF-1α stable knockdown were harvested for cell cycle analysis using propidium iodide staining protocol. Student's t-tests (two tailed) were performed and p-values calculated. *p ≤ 0.05 and **p ≤ 0.01. (D) MCF10A cells and (E) HFF cells were transfected with non-targeted or HIF-1α siRNA oligonucleotides prior to mRNA extraction. HIF-1α, p21, p27, SP1 and GLUT3 mRNA were analyzed using quantitative PCR. Graphs depict mRNA levels normalized to actin. Student's t-tests (two tailed) were performed and p-values calculated. *p ≤ 0.05 and **p ≤ 0.01. Data refers to a minimum of three independent experiments.
Figure 5 HIF-1α knockdown protects against genotoxic drug induced autophagy, while increasing apoptosis-mediated cell death. (A) U2OS cells were transfected using non-targeted or HIF-1α siRNA prior to 24 h treatment with the genotoxic drugs doxorubicin or daunorubicin as described in the materials and methods. Following treatment WCL were harvested and samples were analyzed by western blot using the specific antibodies indicated. (B) U2OS cells were transfected using non-targeted or HIF-1α siRNA prior to 24 h treatment with the chemotherapeutic drugs doxorubicin, etoposide, taxol and vinblastine as described in the materials and methods. Following treatment WCL were harvested and samples were analyzed by western blot using the specific antibodies indicated. (C) U2OS-GFP-LC3B cells grown on coverslips were transfected using non-targeted or HIF-1α siRNA prior to 24 h treatment with the chemotherapeutic drugs doxorubicin, etoposide, taxol and vinblastine as described in the materials and methods. Cells were fixed, stained with DAPI and analyzed by microscopy for GFP-LC3B foci.
Acknowledgements
We are thankful to all members of the Rocha laboratory for comments on the manuscript. We are very grateful to Prof. N. Perkins for providing reagents and valuable comments. We want to thank Prof. T. Yoshimori (Osaka, Japan) for providing the GFP-LC3B plasmid. This study was mainly funded by a New Investigator Research grant from the Medical Research Council (C.C., S.M., S.R.). A.M. is funded by the Wellcome Trust. S.R. is funded by a Research Council UK fellowship and the University of Dundee.
References
- Rankin EB, Giaccia AJ. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ 2008; 15:678
- Yang QC, Zeng BF, Dong Y, Shi ZM, Jiang ZM, Huang J. Overexpression of hypoxia-inducible factor-1alpha in human osteosarcoma: correlation with clinicopathological parameters and survival outcome. Jpn J Clin Oncol 2007; 37:127 - 134
- Yoshimura H, Dhar DK, Kohno H, Kubota H, Fujii T, Ueda S, et al. Prognostic Impact of Hypoxia-Inducible Factors 1α and 2α in Colorectal Cancer Patients. Clinical Cancer Research 2004; 10:8554 - 8560
- Shibaji T, Nagao M, Ikeda N, Kanehiro H, Hisanaga M, Ko S, et al. Prognostic significance of HIF-1alpha overexpression in human pancreatic cancer. Anticancer Res 2003; 23:4721 - 4727
- Schindl M, Schoppmann SF, Samonigg H, Hausmaninger H, Kwasny W, Gnant M, et al. Overexpression of hypoxia-inducible factor 1alpha is associated with an unfavorable prognosis in lymph node-positive breast cancer. Clin Cancer Res 2002; 8:1831 - 1837
- Osada R, Horiuchi A, Kikuchi N, Yoshida J, Hayashi A, Ota M, et al. Expression of hypoxia-inducible factor 1alpha, hypoxia-inducible factor 2alpha and von Hippel-Lindau protein in epithelial ovarian neoplasms and allelic loss of von Hippel-Lindau gene: nuclear expression of hypoxia-inducible factor 1alpha is an independent prognostic factor in ovarian carcinoma. Hum Pathol 2007; 38:1310 - 1320
- Winter SC, Shah KA, Han C, Campo L, Turley H, Leek R, et al. The relation between hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha expression with anemia and outcome in surgically treated head and neck cancer. Cancer 2006; 107:757 - 766
- Mizokami K, Kakeji Y, Oda S, Maehara Y. Relationship of hypoxia-inducible factor 1alpha and p21WAF1/CIP1 expression to cell apoptosis and clinical outcome in patients with gastric cancer. World J Surg Oncol 2006; 4:94
- Birner P, Schindl M, Obermair A, Plank C, Breitenecker G, Oberhuber G. Overexpression of hypoxia-inducible factor 1alpha is a marker for an unfavorable prognosis in early-stage invasive cervical cancer. Cancer Res 2000; 60:4693 - 4696
- Theodoropoulos VE, Lazaris A, Sofras F, Gerzelis I, Tsoukala V, Ghikonti I, et al. Hypoxia-inducible factor 1alpha expression correlates with angiogenesis and unfavorable prognosis in bladder cancer. Eur Urol 2004; 46:200 - 208
- Aebersold DM, Burri P, Beer KT, Laissue J, Djonov V, Greiner RH, Semenza GL. Expression of hypoxia-inducible factor-1alpha: a novel predictive and prognostic parameter in the radiotherapy of oropharyngeal cancer. Cancer Res 2001; 61:2911 - 2916
- Unruh A, Ressel A, Mohamed HG, Johnson RS, Nadrowitz R, Richter E, et al. The hypoxia-inducible factor-1 alpha is a negative factor for tumor therapy. Oncogene 2003; 22:3213 - 3220
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999; 399:271 - 275
- Iliopoulos O, Levy AP, Jiang C, Kaelin WG Jr, Goldberg MA. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci USA 1996; 93:10595 - 10599
- Jiang BH, Jiang G, Zheng JZ, Lu Z, Hunter T, Vogt PK. Phosphatidylinositol-3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth Differ 2001; 12:363 - 369
- Fandrey J, Gorr TA, Gassmann M. Regulating cellular oxygen sensing by hydroxylation. Cardiovasc Res 2006; 71:642 - 651
- Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 2008; 30:393 - 402
- Gorlach A, Bonello S. The cross-talk between NFkappaB and HIF-1: further evidence for a significant liaison. Biochem J 2008; 412:17 - 19
- van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1alpha by NFkappaB. Biochem J 2008; 412:477 - 484
- Bonello S, Zahringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, et al. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler Thromb Vasc Biol 2007; 27:755 - 761
- Deudero JJ, Caramelo C, Castellanos MC, Neria F, Fernandez-Sanchez R, Calabia O, et al. Induction of hypoxia-inducible factor 1alpha gene expression by vascular endothelial growth factor. J Biol Chem 2008; 283:11435 - 11444
- Zelzer E, Levy Y, Kahana C, Shilo BZ, Rubinstein M, Cohen B. Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1alpha/ARNT. EMBO J 1998; 17:5085 - 5094
- Feldser D, Agani F, Iyer NV, Pak B, Ferreira G, Semenza GL. Reciprocal positive regulation of hypoxia-inducible factor 1alpha and insulin-like growth factor 2. Cancer Res 1999; 59:3915 - 3918
- Hellwig-Burgel T, Rutkowski K, Metzen E, Fandrey J, Jelkmann W. Interleukin-1beta and tumor necrosis factor-alpha stimulate DNA binding of hypoxia-inducible factor-1. Blood 1999; 94:1561 - 1567
- Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol-3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res 2000; 60:1541 - 1545
- Gorlach A, Diebold I, Schini-Kerth VB, Berchner-Pfannschmidt U, Roth U, Brandes RP, et al. Thrombin activates the hypoxia-inducible factor-1 signaling pathway in vascular smooth muscle cells: Role of the p22(phox)-containing NADPH oxidase. Circ Res 2001; 89:47 - 54
- Spinella F, Rosano L, Di Castro V, Natali PG, Bagnato A. Endothelin-1 induces vascular endothelial growth factor by increasing hypoxia-inducible factor-1alpha in ovarian carcinoma cells. J Biol Chem 2002; 277:27850 - 27855
- Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol 2001; 21:3995 - 4004
- Bardos JI, Chau NM, Ashcroft M. Growth factor-mediated induction of HDM2 positively regulates hypoxia-inducible factor 1alpha expression. Mol Cell Biol 2004; 24:2905 - 2914
- Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J 2003; 22:4082 - 4090
- Blundell RA. The Biology of p21—Review Paper. American Journal of Biochemistry and Biotechnology 2006; 2:33 - 40
- Morris MR, Hughes DJ, Tian YM, Ricketts CJ, Lau KW, Gentle D, et al. Mutation Analysis of Hypoxia-inducible Factors HIF1A and HIF2A in Renal Cell Carcinoma. Anticancer Research 2009; 29:4337 - 4343
- Bates S, Ryan KM, Phillips AC, Vousden KH. Cell cycle arrest and DNA endoreduplication following p21Waf1/Cip1 expression. Oncogene 1998; 17:1691 - 1703
- Dotto GP. p21(WAF1/Cip1): more than a break to the cell cycle?. Biochim Biophys Acta 2000; 1471:43 - 56
- Fujiwara K, Daido S, Yamamoto A, Kobayashi R, Yokoyama T, Aoki H, et al. Pivotal role of the cyclin-dependent kinase inhibitor p21WAF1/CIP1 in apoptosis and autophagy. J Biol Chem 2008; 283:388 - 397
- Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther 2002; 1:639 - 649
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993; 75:805 - 816
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev 1995; 9:1149 - 1163
- Costanzi-Strauss E, Strauss BE, Naviaux RK, Haas M. Restoration of growth arrest by p16INK4, p21WAF1, pRB and p53 is dependent on the integrity of the endogenous cell cycle control pathways in human glioblastoma cell lines. Exp Cell Res 1998; 238:51 - 62
- Murakami H, Nurse P. DNA replication and damage checkpoints and meiotic cell cycle controls in the fission and budding yeasts. Biochem J 2000; 349:1 - 12
- Kennett SB, Udvadia AJ, Horowitz JM. Sp3 encodes multiple proteins that differ in their capacity to stimulate or repress transcription. Nucleic Acids Res 1997; 25:3110 - 3117
- Lee SJ, Ha MJ, Lee J, Nguyen P, Choi YH, Pirnia F, et al. Inhibition of the 3-hydroxy-3-methylglutaryl-coenzyme A reductase pathway induces p53-independent transcriptional regulation of p21(WAF1/CIP1) in human prostate carcinoma cells. J Biol Chem 1998; 273:10618 - 10623
- Somasundaram K, Zhang H, Zeng YX, Houvras Y, Peng Y, Zhang H, et al. Arrest of the cell cycle by the tumour-suppressor BRCA1 requires the CDK-inhibitor p21WAF1/CiP1. Nature 1997; 389:187 - 190
- Li JM, Datto MB, Shen X, Hu PP, Yu Y, Wang XF. Sp1, but not Sp3, functions to mediate promoter activation by TGFbeta through canonical Sp1 binding sites. Nucleic Acids Res 1998; 26:2449 - 2456
- Prowse DM, Bolgan L, Molnar A, Dotto GP. Involvement of the Sp3 transcription factor in induction of p21Cip1/WAF1 in keratinocyte differentiation. J Biol Chem 1997; 272:1308 - 1314
- Nakano K, Mizuno T, Sowa Y, Orita T, Yoshino T, Okuyama Y, et al. Butyrate activates the WAF1/Cip1 gene promoter through Sp1 sites in a p53-negative human colon cancer cell line. J Biol Chem 1997; 272:22199 - 22206
- Billon N, van Grunsven LA, Rudkin BB. The CDK inhibitor p21WAF1/Cip1 is induced through a p300-dependent mechanism during NGF-mediated neuronal differentiation of PC12 cells. Oncogene 1996; 13:2047 - 2054
- Deniaud E, Baguet J, Chalard R, Blanquier B, Brinza L, Meunier J, et al. Overexpression of transcription factor Sp1 leads to gene expression perturbations and cell cycle inhibition. PLoS One 2009; 4:7035
- Gervais JL, Seth P, Zhang H. Cleavage of CDK inhibitor p21(Cip1/Waf1) by caspases is an early event during DNA damage-induced apoptosis. J Biol Chem 1998; 273:19207 - 19212
- Bose R, Verheij M, Haimovitz-Friedman A, Scotto K, Fuks Z, Kolesnick R. Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals. Cell 1995; 82:405 - 414
- Lacour S, Hammann A, Grazide S, Lagadic-Gossmann D, Athias A, Sergent O, et al. Cisplatin-induced CD95 redistribution into membrane lipid rafts of HT29 human colon cancer cells. Cancer Res 2004; 64:3593 - 3598
- Liu YY, Yu JY, Yin D, Patwardhan GA, Gupta V, Hirabayashi Y, et al. A role for ceramide in driving cancer cell resistance to doxorubicin. Faseb J 2008; 22:2541 - 2551
- Blagosklonny MV, Robey R, Bates S, Fojo T. Pretreatment with DNA-damaging agents permits selective killing of checkpoint-deficient cells by microtubule-active drugs. J Clin Invest 2000; 105:533 - 539
- Carbonaro M, O'Brate A, Giannakakou P. Microtubule disruption targets HIF-1alpha mRNA to cytoplasmic P-bodies for translational repression. J Cell Biol 2011; 192:83 - 99
- Levine B, Yuan J. Autophagy in cell death: an innocent convict?. J Clin Invest 2005; 115:2679 - 2688
- Tiwari M, Bajpai VK, Sahasrabuddhe AA, Kumar A, Sinha RA, Behari S, Godbole MM. Inhibition of N-(4-hydroxyphenyl)retinamide-induced autophagy at a lower dose enhances cell death in malignant glioma cells. Carcinogenesis 2008; 29:600 - 609
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117:326 - 336
- Schumm K, Rocha S, Caamano J, Perkins ND. Regulation of p53 tumour suppressor target gene expression by the p52 NFkappaB subunit. EMBO J 2006; 25:4820 - 4832
- Rocha S, Campbell KJ, Perkins ND. p53- and Mdm2-independent repression of NFkappaB transactivation by the ARF tumor suppressor. Mol Cell 2003; 12:15 - 25
- Kenneth NS, Mudie S, van Uden P, Rocha S. SWI/SNF regulates the cellular response to hypoxia. J Biol Chem 2009; 284:4123 - 4131
- Culver C, Sundqvist A, Mudie S, Melvin A, Xirodimas D, Rocha S. Mechanism of Hypoxia-Induced NF{kappa}B. Mol Cell Biol 2010; 30:4901 - 4921