Abstract
Chromosome mis-segregation and aneuploidy are greatly induced in Alzheimer’s disease and models thereof by mutant forms of the APP and PS proteins and by their product, the Ab peptide. Here we employ human somatic cells and Xenopus egg extracts to show that Aβ impairs the assembly and maintenance of the mitotic spindle. Mechanistically, these defects result from Aβ’s inhibition of mitotic motor kinesins, including Eg5, KIF4A and MCAK. In vitro studies show that oligomeric Aβ directly inhibits recombinant MCAK by a noncompetitive mechanism. In contrast, inhibition of Eg5 and KIF4A is competitive with respect to both ATP and microtubules, indicating that Aβ interferes with their interactions with the microtubules of the mitotic spindle. Consistently, increased levels of polymerized microtubules or of the microtubule stabilizing protein Tau significantly decrease the inhibitory effect of Aβ on Eg5 and KIF4A. Together, these results indicate that by disrupting the interaction between specific kinesins and microtubules and by exerting a direct inhibitory effect on the motor activity, excess Ab deregulates the mechanical forces that govern the spindle and thereby leads to the generation of defective mitotic structures. The resulting defect in neurogenesis can account for the over 30% aneuploid/hyperploid, degeneration-prone neurons observed in Alzheimer disease brain. The finding of mitotic motors including Eg5 in mature post-mitotic neurons implies that their inhibition by Ab may also disrupt neuronal function and plasticity.
Introduction
Alzheimer disease (AD) is the most common form of dementia, affecting one-third to one-half of the human population above age 85.Citation1,Citation2 Several pathological features characterize the brains of AD patients, most notably, accumulation of extracellular amyloid deposits and soluble oligomeric forms of Aβ peptide. Both biochemical and genetic studies indicate that the Aβ plays a key role in Alzheimer disease pathogenesis.Citation2 Aβ is cleaved from a membrane-traversing amyloid precursor protein (APP) by the β-amyloid cleaving enzyme (BACE) and the presenilin (PS)- containing γ-secretase enzyme complex.Citation3 Our current understanding of the pathogenesis of AD links the overproduction of Aβ, particularly the Aβ 1–42 form, and its subsequent self-aggregation to the progressive loss of neurons and impairment of their function in the AD brain. However, the quantity of extracellular amyloid deposits in the brain does not correlate well with the severity of clinical symptoms, indicating that Aβ may also contribute to the AD neurodegeneration without involving large extracellular amyloid formations.Citation4–Citation6 This underscores the importance of further elucidating the biological roles and molecular targets of Aβ 1–42 species in order to develop a better understanding of AD pathogenesis.Citation7
Recently, it has become clear that AD is also characterized by cell cycle defects.Citation8,Citation9 For example, the findings that Down syndrome/trisomy 21 patients express increased levels of Aβ and develop AD pathology by age 40 and that the APP gene resides on chromosome 21,Citation10 suggested that a genetic or environmentally induced tendency to mitotic chromosomal mis-segregation and subsequent aneuploidy might lead to trisomy 21 and other mosaicism and thus contribute to both sporadic and familial AD.Citation11 Indeed, AD patients, including those with FAD mutant APP or PS and those with sporadic disease develop up to 30% aneuploid cells both in brain and peripheral tissues, indicating the presence of widespread chromosome partitioning defects in AD.Citation12–Citation21 Furthermore, premature centromere division, a mitotic defect associated with chromosome mis-segregation is increased in ADCitation12,Citation22,Citation23 A causative rather than merely correlative link between aneuploidy and AD pathogenesis is supported by the facts that (1) members of families with inherited AD have an increased risk of giving birth to a Down syndrome childCitation10,Citation24,Citation25 and (2) young mothers of Down syndrome children are generally prone to chromosome mis-segregation and have a five-fold increase that they will develop AD later in life.Citation26,Citation27
Recently we demonstrated that AD transgenic mouse models (which produce increased levels of Aβ 1–42) develop up to 25% aneuploid cells in both the brain and peripheral tissues, and that tissue culture cells transiently expressing FAD-mutant PS or APP or exposed to Aβ become similarly highly aneuploid within 48 hours.Citation28,Citation29 Overexpression of normal and particularly mutant PS in cultured cells also leads to abnormal mitotic spindles.Citation28 Interestingly, both PS, its substrate APP and its product Aβ localize to the mitotic spindle, especially the spindle poles and kinetochores.Citation30 These results indicate that chromosomal aneuploidy occurs in an Aβ 1–42 dependent manner prior to and during the development of AD neuropathology. Indeed the >30% of aneuploid/hyperploid neurons that arise in AD are particularly prone to degeneration and may account for 90% of the neuronal loss that characterizes late-stage AD.Citation21
The mitotic spindle is the macromolecular machine that separates duplicated chromosomes equally during mitosis through the controlled polymerization and depolymerization of microtubules (MTs) and the integrated forces of several MT-based motor proteins that properly arrange and segregate the chromosomes.Citation31 Numerous MT-based motor proteins associate with the mitotic spindle, control its shape and enable its function.Citation32 These include the dynein complex and at least eight different kinesin family members, which translocate their cargo (chromosomes, kinetochores, MTs) along the MTs of the mitotic spindle. Interference with MT dynamics or inhibition of mitotic motor proteins generates defective spindles and promotes chromosome mis-segregation and aneuploidy.Citation33–Citation35 To determine whether and how the mitotic spindle is affected in AD, we used primary human cells in culture and cell-free Xenopus egg extracts to examine the structure of the mitotic spindle exposed to Aβ. From the experiments described below we discovered that AD-causing Aβ directly disrupts the assembly and maintenance of the mitotic spindle by inhibiting specific MT-based mitotic motors, such as Eg5, KIF4A and MCAK. These results suggest a mechanism in which Aβ, through its binding to the motor proteins, both directly abrogates their functions and interferes with their association with MTs of the mitotic spindle, thus generating a deficit and imbalance in motor forces. We propose that this imbalance leads to the generation of defective and dysfunctional mitotic spindles and induces the aneuploidy associated with AD pathology.
Results
Aβ directly disrupts the assembly and maintenance of the mitotic spindle.
Over 20% aneuploidy is induced by the AD-causing Aβ peptide in cultured cells, suggesting that the peptide interferes with proper chromosome segregation during mitosis.Citation29 To test this prediction, we added Aβ to primary human mammary epithelial cells immortalized with hTERT and found that Aβ peptides, particularly Aβ 1–42, induced abnormal spindle phenotypes within 48 hrs, including ring-like structures, shortened spindles and spindles with disorganized MTs, all of which displayed misaligned chromosomes ( and B).
A substantial amount of research demonstrates that cells take up extracellular Aβ into the cytoplasm and that intracellular Aβ may be a key player in AD pathogenesis.Citation36 Thus it seemed plausible that in our tissue culture experiments, the added Aβ became internalized and directly impaired the process of cell division. To address this possibility, the effects of Aβ 1–42 on the mitotic spindle were assessed in detail using the cell-free system of cytostatic factor (CSF)-arrested Xenopus egg extracts, which can form large numbers of mitotic spindles in vitro and allows addition of Aβ directly to the cytosol of the extracts.Citation37 Our results showed that addition of Aβ 1–42 (but not Aβ with a scrambled amino acid sequence) to the extracts induced the development of abnormal spindles with distinct phenotypes, including shortened and bent spindles ( and D, Sup. Fig. 1A). The abnormal spindles also displayed disorganized MTs and MT fibers that protruded outside of the spindles, often associated with chromosomes lying away from the metaphase plate (Sup. Fig. 1B).
In addition to compromising the formation of mitotic spindles, Aβ also impaired the maintenance of spindles: addition of Aβ 1–42, but not control scrambled Aβ, to extracts containing normal fully formed spindles led to their shortening, collapsing or bending ( and F). Together, these data demonstrate that intracellular Aβ 1–42 can directly abrogate both the assembly and maintenance of the mitotic spindle. Furthermore, Aβ was observed to be associated with the mitotic spindle, indicating that its effect may be mediated through direct interaction with spindle components (Sup. Fig. 2).
Because the shortened spindle was the predominant phenotype observed in Aβ treated extracts, we asked whether Aβ inhibits MT polymerization by examining its effects on aster formation induced in egg extracts by DMSO.Citation38 Addition of DMSO and Aβ 1–42 or scrambled Aβ to extracts revealed no differences in shapes or sizes of MT asters, nor in the quantity of polymerized MTs precipitated from extracts (Sup. Fig. 3A and B), indicating that Aβ does not affect MT polymerization in the extracts.
Aβ 1–42 impairs functions of motor kinesins.
The shape and length of the mitotic spindle are also governed by microtubule movement/motility, which is mediated by mechanical forces generated by MT-based motor proteins.Citation31 We used time-lapse fluorescence microscopy to investigate the effects of Aβ on MT motility in CSF-arrested Xenopus egg extracts. Under control conditions we observed the anticipated rapid MT movements and the appearance and disappearance of MTs as they moved in and out of the plane of focus. Upon the addition of increasing concentrations of Aβ 1–42, but not scrambled peptide), the movement of MTs was reduced in a dose-dependent manner, suggesting that Aβ inhibits mitotic MT-based motors (Sup. Fig. 4A and B and Sup. Vid.).
Spindle assembly in Xenopus egg extracts requires the action of molecular motor proteins to regulate the assembly and dynamics of MTs in the spindle.Citation31 We therefore evaluated whether the Aβ-induced spindle phenotypes could be explained by the peptide's ability to inhibit individual motors previously shown to be critical for spindle assembly in Xenopus CSF extracts. Shortened and bent spindles (such as in ) result from the inhibition of chromokinesin KIF4A,Citation39,Citation40 and collapsed spindles (such as in and E) are observed following inhibition of Eg5.Citation40,Citation41 Complete blocking of MCAK function causes excessive MT polymerization, while inhibition of centromeric MCAK leads to shortened spindles with misaligned chromosomes.Citation42 To test whether Aβ-induced spindle abnormalities in the extracts might be due to inhibition of Eg5, KIF4A or MCAK, we individually immunopurified each motor protein from extracts supplemented with 0.5, 1.0 and 2.0 µM Aβ 1–42 or 2.0 µM scrambled Aβ and subjected the immunopurified motors to an in vitro MT-dependent ATPase assay. All three motors displayed a dose-dependent Aβ 1–42 specific inhibition within the time-frame of the appearance of the Aβ-induced spindle abnormalities in the extracts ().
Rescue of mitotic spindle abnormalities by addition of recombinant motor proteins.
To directly address whether Aβ-mediated inhibition of the motor proteins underlies its generation of abnormal spindles, we asked whether the spindle defects observed in the presence of Aβ can be rescued by addition of increased amounts of pure motor proteins. Xenopus egg extracts were supplemented with the recombinant motor domains of Eg5, KIF4A or MCAK prior to the addition of Aβ. Each recombinant motor reversed the spindle disruptive effects of Aβ to the levels of controls, probably functioning by decoying Aβ away from the endogenous motor (). The statistical significance of these results was increased when extracts were rescued with a combination of the three motors. All together these results strongly support that the observed spindle defects are caused by Aβ-mediated impairment of kinesins function.
Oligomeric Aβ 1–42 directly inhibits recombinant Eg5, KIF4A and MCAK in vitro.
The direct effect of Aβ on the MT-dependent ATPase activity of the motor domains of recombinant human mitotic kinesins Eg5, KIF4A, MCAK and the KHC (kinesin heavy chain or kinesin-1) as a non-mitotic kinesin control was determined at the endogenous concentrations of the motors. The results showed a pronounced Aβ 1–42 specific inhibition of mitotic kinesins Eg5, KIF4A and MCAK but no inhibition of KHC/kinesin 1 (). Such inhibition required the full length Aβ peptide, including the C-terminus, as truncated fragments of Aβ were increasingly less inhibitory (). Oligomeric forms of Aβ, which numerous studies have shown to be the most toxic species,Citation43 were more effective MT motor inhibitors than fibers, with protofibrils having intermediate activity ( and Sup. Fig. 5). Together these results identify mitotic motor kinesins, including Eg5, KIF4A and MCAK, as novel direct targets of the Alzheimer Aβ 1–42 peptide.
Kinetics of kinesin inhibition by Aβ.
To further characterize the inhibition of the mitotic kinesins by Aβ and to define its mechanism, we studied the effects of Aβ 1–42 on the enzyme kinetics of Eg5, KIF4A, MCAK, CENP-E and KHC. The inhibitory effects of Aβ on Eg5 and KIF4A were reduced in the presence of high ATP concentrations, indicating a competitive mode of inhibition ( and B), quantitatively confirmed by a pronounced increase in the Km values (). Higher concentrations of Aβ also mildly reduced the Vmax of KIF4A, implying the existence of non-competitive component in the Aβ's inhibition of this motor ( and ). In contrast, MCAK revealed a noncompetitive inhibition by Aβ, which clearly reduced Vmax of MCAK's enzymatic reaction in a dose-dependent manner, but did not affect the Km ( and ). Consistent with data shown in , Aβ did not change the parameters of KHC and CENP-E enzymatic kinetics in a dose-dependent manner ( and and E).
ATP hydrolysis by kinesin motors is stimulated many fold upon their association with MTs. Therefore, the effect of Aβ on the motor's ATP cycle may be due to interference with the motor-MT interaction. To explore this possibility, we assayed [MT]-dependent enzyme kinetics of the motors. Interestingly, Aβ significantly increased the Km for MTs of Eg5 and KIF4A (Sup. Table 1), without pronounced effects on Vmax (Sup. Table 1 and and G). This result demonstrates that the competitive nature of Aβ inhibition of Eg5 and KIF4A is most likely caused by Aβ's inhibition of the motors' interactions with MTs. Consistent with our findings with ATP, studies of the [MT]- dependent kinetics of MCAK confirmed a noncompetitive mode of inhibition by Aβ (Sup. Table 1 and ).
Aβ interacts with mitotic kinesins and MTs.
To explore the mechanism by which Aβ affects the activity of mitotic motors, we asked whether Aβ directly associates with each motor. The results showed that Aβ is co-immunoprecipitated with endogenous Eg5, KIF4A and MCAK from Xenopus egg extracts preincubated with 1.0 µM Aβ 1–42 (). Furthermore, Eg5, KIF4A and MCAK in the extract readily bound to added Aβ 1–42 (but not scrambled Aβ) conjugated to Sepharose resin (AnaSpec Inc.) and could be recovered by centrifugaiton (). Consistent with and , we did not detect Aβ in KHC immunoprecipitates () nor any interaction of KHC with Aβ 1–42 conjugated beads ().
The ATPase activity of kinesin motors depends on the binding of the motors to MTs, and indeed, significant levels of Aβ co-pelleted with polymerized MTs from the Xenopus egg extracts supplemented with 1.0 µM Aβ 1–42 (). To exclude the possibility that the observed Aβ in these precipitates could be due to its own independent aggregation, a control MT pull down was conducted in the presence of nocodazole, a potent MT depolymerizing drug. No Aβ was spun down from these extracts (), demonstrating that the Aβ recovered in the MT pellets must be associated with the MT fibers. Furthermore, the fact that addition of nocodazole, failed to prevent the co-immunoprecipitation of Aβ with mitotic kinesins () shows that the interaction between Aβ and the kinesins is direct, and not merely mediated by both these proteins interacting with MTs.
The clear selectivity in Aβ's effects on different motor proteins is consistent with other differences between the motors. For example, Aβ-related inhibition of KHC-dependent vesicle transport has been reported, but is not direct, requiring additional factors.Citation44 The fact that the kinetochore-associated kinesin-like motor protein CENP-E is unaffected by Aβ is consistent with the absence in Aβ-treated Xenopus egg extracts of the profound chromosome misalignment defects that are found in extracts with compromised CENP-E function.Citation45 Finally, unlike Eg5 (and potentially, KIF4A), both kinesin-1 and CENP-E, are highly processive motors,Citation46,Citation47 which reduces the probability of their detachment from MT tracks and may allow them to tolerate the addition of Aβ.
Aβ interferes with kinesin/MT binding.
To explore the possibility that Aβ may impair functions of mitotic motors by affecting their association with MTs, we examined the localization of each motor protein to MTs of the mitotic spindle in the presence of Aβ. Control spindles and spindles formed in extracts supplemented with scrambled peptide displayed the previously described normal distribution of the mitotic kinesins Eg5, KIF4A and MCAK. Specifically, Eg5 was enriched at the spindle poles and the overlap region of the spindle, the chromokinesin KIF4A was concentrated predominantly on the chromosomes arms and the spindle polesCitation39 and MCAK was localized at the spindle poles and MT fibers.Citation48 In contrast, abnormal spindles from Aβ 1–42 treated extracts displayed dramatically reduced quantities of associated Eg5 and KIF4A, and somewhat reduced levels of MCAK (). These data demonstrate that Aβ 1–42 indeed interferes with the normal association of the mitotic kinesins Eg5, KIF4A and, to a lesser degree, MCAK, with MTs.
Motor activities are rescued by added MTs, Tau.
As kinesin motor activity depends on binding to MTs, one unifying explanation for the results is that Aβ hampers the interaction between Eg5 and KIF4A and MTs and thereby inhibits the motors' function. If this interpretation is correct, then an increase in MT concentration should decrease the inhibitory effects of Aβ as did an increase in motor concentration. We conducted the MT-dependent ATPase assays in the presence of higher concentrations of purified and stabilized MTs, and found that the additional MTs virtually reversed Aβ's inhibition of Eg5 and KIF4A ( and B). Interestingly, the inhibition of MCAK, which displayed a noncompetitive inhibition, remained significantly inhibited by Aβ even in the presence of high concentrations of MTs ().
Under native conditions, the stability of MTs is controlled by MT-associated proteins, of which tau is intimately connected to Aβ toxicity in AD (Mandelkow and Mandelkow, 1998; Roberson et al. 2007).Citation49–Citation51 Consistent with the data described above, we observed that increased levels of Tau also reversed the inhibition of Eg5 and KIF4A by Aβ, possibly by stabilizing MTs ( and E). Inhibition of MCAK was not modified by the presence of Tau (), indicating that its inhibition by Aβ might not affect accessibility of MTs.
In addition to reducing the inhibition of mitotic motors by Aβ, increased levels of polymerized MTs also changed the distribution of mitotic kinesin motors between MT-bound and Aβ-bound states: increasing MT polymerization in Xenopus egg extracts through supplementing them with 5% DMSO increased the amounts of Eg5, KIF4A and MCAK associated with polymerized MTs and decreased the amounts of Eg5 and KIF4A associated with Aβ 1–42 conjugated beads (). These results confirm the existence of competitive interactions between Aβ, the mitotic motors, particularly Eg5 and KIF4A, and MTs.
Inhibition of Eg5 leads to the generation of aneuploidy.
Because Aβ causes chromosome mis-segregation and aneuploidy and inhibits Eg5, our results suggest that partial inhibition of Eg5, as by the Alzheimer Aβ peptide, should lead to chromosome mis-segregation. Indeed, treating hTERT cells with low levels of the Eg5 inhibitor, MonastrolCitation41 for only 48 hours induces chromosome mis-segregation and aneuploidy in more than 50% of cells (). Interestingly polymorphisms in the Eg5/KIF11 gene have been reported to increase the risk of AD (Feuk et al. 2005),Citation52 consistent with the conclusion that impairment of certain motor proteins may lead to the development of AD though disruption of mitosis during neurogenesis. The anti-mitotic activity of Aβ is consistent with the finding that FAD mutant APP and/or PS inhibit neurogenesis.Citation53,Citation54
Discussion
On the basis of the experiments reported here, we propose that by attacking mitotic MT motors excess Aβ can impair mitosis in neuronal precursor cells, leading to chromosome mis-segregation and defective, aneuploid neurons, which, in turn, can lead to deficits in learning and memory and have novel implications for AD diagnosis and treatment ().
It is interesting to note that chromosome and spindle abnormalities have been reported recently to be induced also by Frontal Temporal Dementia and Huntington disease related proteins, reinforcing the general idea that mitotic defects may contribute to many neurodegenerative and other aging-associated diseases.Citation30,Citation55,Citation56
MT motors are particularly critical in neurons, which must transport vesicles, organelles and molecules, including microtubules themselves over great distances.Citation57 Indeed, some studies have shown axonal transport to be disrupted in cell and animal models of AD.Citation58–Citation60 Furthermore, regenerating neurons and neurons responding to learning and memory signals must retain an ability to modify their cytoskeleton to accommodate neurite outgrowth and synapse remodeling. Interestingly, many classically known mitotic motors, including Eg5, KIF4A and MCAK, are expressed in neurons.Citation61–Citation63 Thus, it is possible that the Aβ's inhibition of the neuronal counterparts of the mitotic motors may also lead to neuronal malfunctioning and directly contribute to the brain pathology of AD.
In summary, our data provide a mechanism by which Aβ through inhibition of mitotic kinesins disrupts the mitotic spindle and leads to the chromosome mis-segregation and aneuploidy observed in AD patients, transgenic mice and cultured cells overexpressing or exposed to Aβ (). Given the great conservation of Aβ in evolution,Citation64 we consider it is an interesting possibility that the regulation of MT transport function by Aβ that we describe here may reflect a normal role of the peptide, which becomes disrupted by its overproduction in AD.
Materials and Methods
In vitro incubation of hTERT-HME1 cells with Aβ peptides, immunocytochemistry and FISH analysis.
For mitotic spindles analyses, hTERT-HME1 cells (Clontech) in Mammary Epithelium Basal Medium (MEBM, Lonza) were plated on single chamber slides (BD Falcon) pre-coated with 2 µg/mL poly-L-lysine (Sigma) at the density of 1–2 × 105 cells/8.6 cm2 and treated with various peptides for 48 hours. Then the cells were fixed and stained for α-tubulin (Sigma, clone B-5-1-2) and DAPI (Abbott, Vysis) according to reference Citation28. Synthetic peptides Aβ1–40 and Aβ1–42 were from AnaSpec, BioSource International and American Peptide Company. Synthetic Aβ42-1 was from BioSource International and American Peptide Company. Synthetic Aβ1–11, Aβ1–28 and Aβ1–33 were from AnaSpec. Scrambled Aβ 1–42 was from AnaSpec, Sigma Genosys and Bio-Synthesis. FISH analysis of hTERT cells incubated for 48 hrs with 10 µM Monastrol was performed as described previously in reference Citation29.
Preparation of xenopus egg extracts and in vitro spindle assembly.
Spindles were formed in freshly prepared cytostatic factor (CSF) arrested Xenopus egg extracts as previously described in reference Citation37. To study the effects of Aβ on mitotic spindle assembly, extracts were supplemented with Aβ or control scrambled peptides or corresponding volumes of buffer and incubated on ice for 1.0 hr to ensure even distribution of the peptides prior to the initiation of the spindle assembly. To study the effects of Aβ on the spindle stability, extracts were incubated for 75 min at RT to allow for spindle formation and then treated with Aβ or corresponding volumes of buffer and incubated for an additional 1.0 hr at RT. Rescue of Aβ-induced spindle defects was studied in extracts supplemented with recombinant human domains of Eg5, KIF4A and MCAK (Cytoskeleton, Inc.) in the quantities corresponding to their endogenous concentrations (0.4 µM Eg5, 0.4 µM KIF4A and 0.1 µM MCAK).
Pelleting spindles onto coverslips and immunostaining.
Spindles assembled in CSF arrested Xenopus egg extracts were sedimented onto coverslips and subjected to immunostaining as previously described in reference Citation37. The primary antibodies were added 1:1,000 rabbit polyclonal anti-human Eg5 (ab37009, Abcam), 1:500 rabbit polyclonal anti-human KIF4A (PAB0793 Abnova), 1.5 µg/ml sheep polyclonal anti-Xenopus MCAK and incubated for 1.0 hr at RT. Secondary fluorescently labeled Alexa Fluor 488 anti-rabbit and anti-sheep antibodies (Invitrogen) were used at a 1:5,000 dilution. To visualize localization of Eg5, KIF4A and MCAK, coverslips were mounted with ProLong Gold antifade reagent with DAPI (Invitrogen) and subjected to fluorescent microscopy at constant exposure times.
MT aster formation and MT pelleting from xenopus egg extracts.
Polymerized MTs were pelleted from Xenopus egg extracts as previously described in reference Citation28, except the 0.1% Triton-X100 was used. To observe MT asters, 1–2 µl of extracts were mixed 1:2 with a fixative (25% glycerol, 7.4% formaldehyde, 0.1 mM HEPES pH 7.5) and subjected to fluorescent microscopy.
MT motility assay.
MT motility assays were performed utilizing a Kinesin Motility Assay Biochem kit from Cytoskeleton. Prior to the motility reaction, CSF arrested Xenopus egg extracts and the purified Rhodamine-labeled MTs were pre-incubated at RT with Aβ peptides or buffer. Subsequently, 10 µl of extracts and MTs were sequentially injected into perfusion chambers. MT motility was triggered by perfusion of an ATP-containing buffer and observed by collecting fluorescent time-lapse images for 12 sec with 2 sec intervals. To avoid dilution of Aβ during manipulation with the perfusion chambers, all solutions used in the assay were supplemented with appropriate concentrations of Aβ.
MT-dependent kinesin ATPase assay.
MT-dependent ATPase activities of kinesins were measured by using a Kinesin ATPase End-Point Biochem kit from Cytoskeleton Inc.Citation65 To study activities of endogenous motor proteins from Xenopus egg extracts, Eg5, KIF4A and MCAK immunoprecipitated on protein G Sepharose beads from 25 µl of control or Aβ-treated CSF extracts were applied to a single assay reaction. To study effects of Aβ on activities of recombinant kinesin motors, 0.2 µg (0.085 µM) of human recombinant motor domain of KHC, 0.75 µg (0.3 µM) of human recombinant motor domain of Eg5, 1.0 µg (0.4 µM) of human recombinant motor domain of Chromokinesin KIF4A or 0.3 µg (0.1 µM) of human recombinant motor domain of MCAK (in concentrations corresponding to their endogenous levels) were pre-incubated with 1.2 µM (0.067 mg/ml) of purified MTs and Aβ for 1.0 hr at RT. The ATPase reaction was triggered by the addition of ATP, and the levels of produced non-organic phosphate were determined spectrophotometrically according to the manufacturer's protocol.
Oligomeric and polymeric Aβ.
The majority of the experiments were carried out with a preparation of Aβ that included monomeric and oligomeric species. To determine which species of Aβ exhibited the strongest inhibitory activity against MT motors, monomers, oligopmer and fibrils were prepared according to Stine and colleagues.Citation66
Enzyme kinetics assay.
200 nM of each recombinant motor domains of human Eg5, KIF4A, MCAK, CENP-E and 80 nM of KHC were pre-incubated with Aβ 1–42 for 30 min at room temperature and subjected to enzymatic kinetics assay based on Kinesin ELIPA Biochem kit (Cytoskeleton Inc.). Each reaction was measured every 30 sec for 20 min and replicated in at least three independent experiments. The average speed of reaction (V) was calculated by using Excel software. The plots V = f([ATP]) and V = f([MT]) were fitted into Michaelis-Menten equation V = Vmax × [S]/(Km + [S]) by using SigmaPlot software with the goodness of fit >0.97. Vmax and Km for each condition were calculated by using SigmaPlot. Statistical analysis was performed by calculating t-values according to the equation:
t = [x1 − x2]/√s12/n1 + s22.
Immunoprecipitation.
Endogenous Xenopus motor proteins for MT-dependent ATPase activity assay or co-immunoprecipitation analysis were immunoprecipitated as described in reference Citation67, with anti-human KHC (Abcam, ab42492, 0.1 mg/ml), anti-human Eg5 (Abcam, ab37009, 1:5 dilution), anti-human KIF4A (Abnova, PAB0793, 1:5 dilution) or anti-Xenopus MCAK (0.1 mg/ml) or equivalent concentrations of isotype control antibodies.
Aβ-conjugated sepharose bead pull downs.
CSF arrested Xenopus egg extracts were diluted 1:1 with EB buffer (80 mM β-glycerol phosphate, pH 7.3, 15 mM MgCl2, 20 mM EGTA, 25 mM NaF, 1 mM Na3VO4) supplemented with 0.1% Triton ×100 and 10 µg/ml each of pepstatin, leupeptin and chymostatin and incubated for 2.0 hrs with pre-equilibrated Sepharose beads conjugated with Aβ 1–42 or scrambled Aβ (AnaSpec). Bead-associated complexes were isolated by a quick centrifugation, washed with copious amount of EB buffer and analyzed by western blotting.
Western blotting.
Analyses were performed using 1:5,000 diluted monoclonal anti-sea urchin α-Tubulin (Sigma, T5168), 1.0 µg/ml monoclonal anti-human Aβ 1–42 (MABTECH, 3740-5-1000), 1:500 diluted monoclonal anti-human Eg5 (Santa Cruz, sc-53691), 1:500 diluted polyclonal anti-human KIF4A (Santa Cruz, sc-48570), 0.3 µg/ml polyclonal anti-Xenopus MCAK, 1.0 µg/ml polyclonal anti-human KHC (Abcam, ab42492).
Figures and Tables
Figure 1 Aβ disrupts the assembly and maintenance of the mitotic spindle by impairing functions of MT-based mitotic motor proteins. Representative mitotic spindles from hTERT-HME1 cells incubated with Aβ 1–40, Aβ 1–42 or reverse Aβ 42-1 for 48 hrs are shown in (A) and quantification of Aβ-induced abnormalities in (B). Immunofluorescent images (C) of mitotic spindles formed in CSF Xenopus egg extracts in the presence of Aβ 1–42 and scrambled Aβ were assessed for phenotype (D). 1,614 spindle structures were scored in 5 independent experiments. (E and F) Mitotic spindles preformed in CSF Xenopus egg extracts deteriorate after exposure to 0.5, 1.0 and 2.0 µM Aβ 1–42 but not 2.0 µM scrambled Aβ and became shortened, collapsed and bent. 1,045 spindle structures were analyzed in 3 independent experiments. Rhodamine-tubulin (red). Hoechst (blue) is DNA stain. For all figures, *p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.0005.
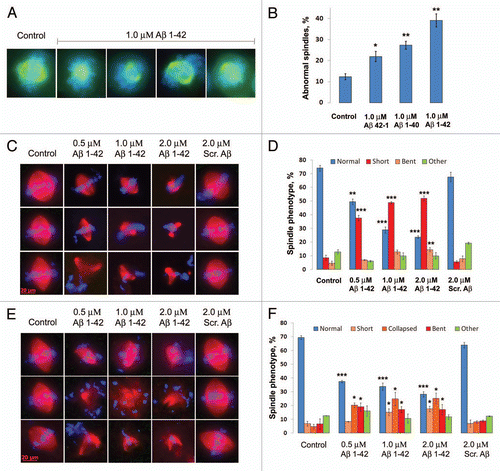
Figure 2 The appearance of defective spindles in Xenopus egg extracts coincides with inhibition of endogenous mitotic motor proteins and is rescued by the addition of recombinant Eg5, KIF4A and MCAK. Endogenous motor proteins Eg5 (A), KIF4A (B) and MCAK (C) were immunopurified from CSF arrested Xenopus egg extracts supplemented with 0.5, 1.0 and 2.0 µM Aβ 1–42 and 2.0 µM scrambled Aβ and subjected to an in vitro MT-dependent ATP ase assay to assess their activities. (D) Spindle abnormalities induced by 1 µM Aβ are rescued by supplementing CSF Xenopus egg extracts with recombinant motor domains of human Eg5, KIF4A and MCAK (1x of endogenous levels). 4,120 spindle structures were scored in three independent experiments.
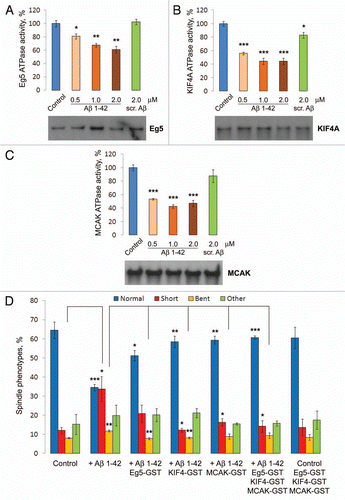
Figure 3 Oligomeric “full-length” Aβ 1–42 directly inhibits motor proteins Eg5, KIF4A and MCAK in vitro. MT-dependent ATP ase activity of human recombinant motor domains of Eg5, KIF4A and MCAK were inhibited after incubation in vitro in the presence of Aβ 1–42 or scrambled Aβ (A). Human recombinant motor domains of Eg5 (B), KIF4A (C) and MCAK (D) were pre-incubated with “full-length” 1–42 and truncated versions of Aβ and subjected to the MT-dependent ATP ase assay to assess their activities, revealing that full length Aβ is the most effective motor inhibitor. All of the experiments reported thus far used a preparation of Aβ that contained a mixture of its monomeric and oligomeric species. To determine whether more polymerized species possess different inhibiting effects on mitotic motor kinesins, freshly-prepared (oligomers), incubated overnight at 4°C (protofibrils) or 37°C (fibers) Aβ 1–42 species were prepared as described. Human recombinant motor domains of Eg5 (E), KIF4A (F) and MCAK (G) were pre-incubated with prepared Aβ 1–42 species and subjected to the in vitro MT-dependent ATP ase activity assay.
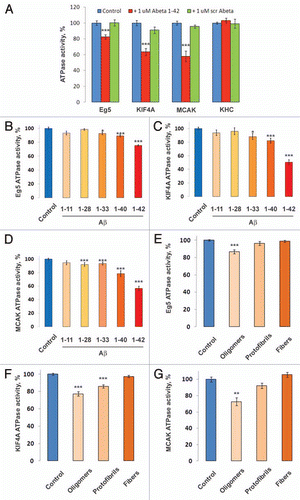
Figure 4 Inhibition of Eg5 and KIF4A enzyme kinetics by Aβ 1–42 displays a competitive mechanism of inhibition, whereas inhibition of MCAK by Aβ 1–42 displays a non-competitive mechanism. Effects of 50 nM, 0.5 and 5.0 µM Aβ 1–42 on [ATP] (A–E) and [MT] (F–H) dependent kinetics of recombinant motor domains of human Eg5 (A and F), KIF4A (B and G), MCAK (C and H), CENP-E (D) and KHC (E) studied by using ELIPA kinetics kit (Cytoskeleton Inc.).
![Figure 4 Inhibition of Eg5 and KIF4A enzyme kinetics by Aβ 1–42 displays a competitive mechanism of inhibition, whereas inhibition of MCAK by Aβ 1–42 displays a non-competitive mechanism. Effects of 50 nM, 0.5 and 5.0 µM Aβ 1–42 on [ATP] (A–E) and [MT] (F–H) dependent kinetics of recombinant motor domains of human Eg5 (A and F), KIF4A (B and G), MCAK (C and H), CENP-E (D) and KHC (E) studied by using ELIPA kinetics kit (Cytoskeleton Inc.).](/cms/asset/257237a6-5018-44b0-bb24-c78e83c3bd0b/kccy_a_10915478_f0004.gif)
Figure 5 Aβ associates with mitotic motors and polymerized MTs. Western blot analysis of the levels of Aβ 1–42 and Eg5 (A), KIF4A (B), MCAK (C) and KHC (D) immunoprecipitated from Xenopus egg extracts supplemented with Aβ 1–42 and nocodazole, a MT destabilizing agent, by antibodies to the respective motor proteins. (E) Western blot analysis of the levels of Eg5, KIF4A, MCAK and KHC affinity-purified from Xenopus egg extracts by the means of Aβ 1–42 or scrambled Aβ conjugated Sepharose beads. (F) Western blot analysis of the levels of polymerized α-Tubulin and MT-associated Aβ 1–42 purified from Xenopus egg extracts supplemented with Aβ 1–42 or scrambled Aβ in the presence and absence of nocodazole.
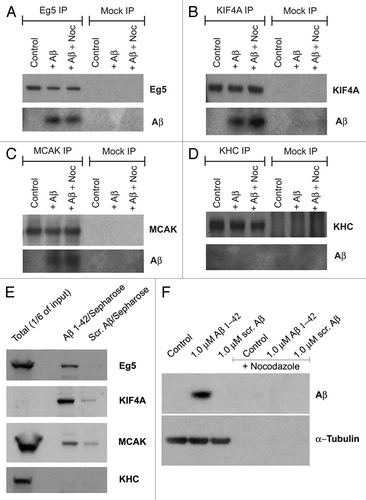
Figure 6 Aβ blocks motor access to MTs of the mitotic spindle. Fluorescent images showing that Aβ reduces endogenous Eg5, KIF4A and MCAK (black and white, green) association with mitotic spindles (red) in Xenopus egg extracts.
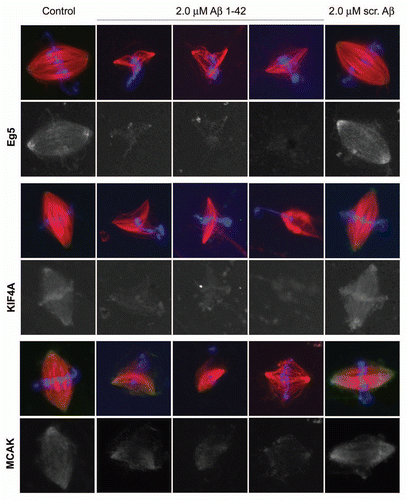
Figure 7 Aβ inhibition of mitotic motors is reversed by added MTs or Tau. Aβ inhibition of Eg5 (A), KIF4A (B) and to a lesser extent MCAK (C) is reversed by increased concentrations of MTs in the in vitro reaction. Increasing concentrations of Tau, a MT stabilizing protein, reverses inhibition of Eg5 (D) and KIF4A (E) by Aβ, but does not affect Aβ's inhibition of MCAK (F). (G) Western blot analysis showing the decrease in the levels of Eg5 and KIF4A affinity-purified with Aβ 1–42 conjugated Sepharose beads and the increase in the levels of these motors co-purified with polymerized MTs from Xenopus egg extracts supplemented with 5% DMSO to induce a robust MT polymerization.
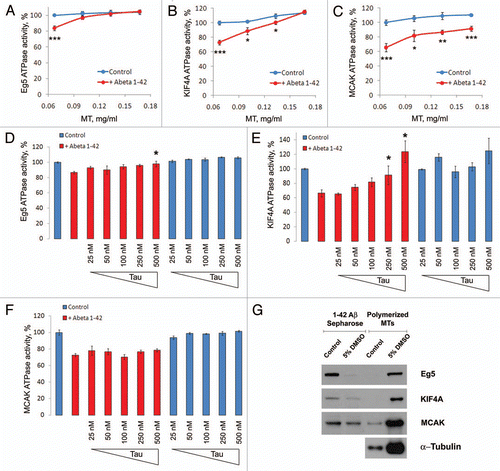
Figure 8 Induction of aneuploidy by monastrol, a small molecule inhibitor of Eg5. hTERT cells were grown for 48 hours in the presence of a low concentration (10 µM) of Monastrol and then subjected to chromosome 12 and 21 FISH analysis. In each culture, at least 500 cells were each scored for chromosome 21 trisomy and tetrasomy (A) and for chromosome 12 trisomy and tetrasomy (B).
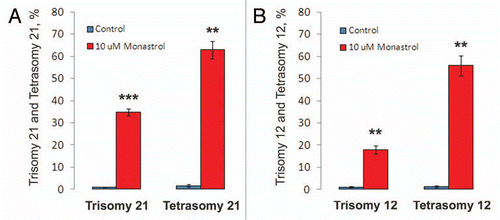
Figure 9 Inhibition of the mitotic kinesins by Aβ 1–42 leads to disruption of proper mitosis and contributes to the development of Alzheimer disease. Alzheimer Aβ through direct inhibition of the mitotic motors Eg5, KIF4A and MCAK, and/or by binding to MTs and abrogating the normal interaction of Eg5 and KIF4A with MTs may reduce the amount of functional motor proteins participating in the assembly and maintenance of the mitotic spindle. This leads to the generation of aberrant and defective mitotic structures incapable of faithful chromosome partitioning during anaphase, which in turn, provokes chromosomal mis-segregation and the generation of aneuploid cells. Aneuploidy within the neuronal progenitor cells might lead to the depletion of normal neurogenesis and contribute to the generation of Trisomy 21 and other aneuploid neurons prone to apoptosis and overproduction of Aβ peptide. Neuronal loss caused by the aberrant chromosomal content may contribute to the brain malfunctions associated with AD.
Table 1 Effects of Aβ 1–42 on Vmax and Km of [ATP]-dependent enzymatic kinetics of mitotic motors
Additional material
Download Zip (1.2 MB)Acknowledgments
We dedicate this paper to the memory of Mark A. Smith, whose open-minded pursuit and support of research on the role of the cell cycle in Alzheimer disease were appreciated by all. We miss him and wish his family well. Supported by Byrd Alzheimer's Institute, Eric Pfeiffer Chair for Research on Alzheimer's Disease, NIA (AG25711), and the Martha and James Porter Equipment grant to USF/Byrd. We thank Dr. Chad Dickey (University of South Florida) for recombinant tau protein, Drs. Gloria Ferreira and Greg Hunter (University of South Florida) for guidance in conducting and analysis of enzyme kinetics experiments and Dr. Ren Chen (Biostatistics Core, University of South Florida) for guidance in statistical analysis.
References
- Hebert LE, Scherr PA, Beckett LA, Albert MS, Pilgrim DM, Chown MJ, et al. Age-specific incidence of Alzheimer's disease in a community population. JAMA 1995; 273:1354 - 1359
- Hardy J. A hundred years of Alzheimer's disease research. Neuron 2006; 52:3 - 13
- Wolfe MS. The secretases of Alzheimer's disease. Curr Top Dev Biol 2003; 54:233 - 261
- Chui DH, Tanahashi H, Ozawa K, Ikeda S, Checler F, Ueda O, et al. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med 1999; 5:560 - 564
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, et al. High-level neuronal expression of Abeta 1–42 in wild-type human amyloid protein pre-cursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 2000; 20:4050 - 4058
- Takeuchi A, Irizarry MC, Duff K, Saido TC, Hsiao Ashe K, Hasegawa M, et al. Age-related amyloid beta deposition in transgenic mice overexpressing both Alzheimer mutant presenilin 1 and amyloid beta pre-cursor protein Swedish mutant is not associated with global neuronal loss. Am J Pathol 2000; 157:331 - 339
- Hardy J. The amyloid hypothesis for Alzheimer's disease: a critical reappraisal. J Neurochem 2009; 110:1129 - 1134
- Snape M, Lee HG, Casadesus G, Smith MA. Cell cycle aberrations in Alzheimer's disease: a novel therapeutic opportunity. Expert Rev Neurother 2009; 9:1579 - 1580
- Potter H. Padmanabhan J. Numbers count: Trisomy 21 and APP duplication as a cause of Alzheimer's disease. Cell regulators in Alzheimer's Disease 2010; Kerala Transworld Research Network
- Epstein CJ. The consequences of chromosome imbalance. Am J Med Genet Suppl 1990; 7:31 - 37
- Potter H. Review and hypothesis: Alzheimer disease and Down syndrome—chromosome 21 nondisjunction may underlie both disorders. Am J Hum Genet 1991; 48:1192 - 1200
- Potter H, Ma J, Das S, Geller LN, Benjamin M, Kayyali US, Dressler D. Iqbal K MJA, Winblad B, Wisniewski HM. Beyond beta-protein: New steps in the pathogenic pathway to Alzheimer's disease. Research Advances in Alzheimer's Disease and Related Disorders 1995; New York John Wiley and Sons Ltd. 643 - 654
- Migliore L, Testa A, Scarpato R, Pavese N, Petrozzi L, Bonuccelli U. Spontaneous and induced aneuploidy in peripheral blood lymphocytes of patients with Alzheimer's disease. Hum Genet 1997; 101:299 - 305
- Geller LN, Potter H. Chromosome missegregation and trisomy 21 mosaicism in Alzheimer's disease. Neurobiol Dis 1999; 6:167 - 179
- Migliore L, Botto N, Scarpato R, Petrozzi L, Cipriani G, Bonuccelli U. 1999 Preferential occurrence of chromosome 21 malsegregation in peripheral blood lymphocytes of Alzheimer disease patients. Cytogenet Cell Genet 1999; 87:41 - 46
- Trippi F, Botto N, Scarpato R, Petrozzi L, Bonuccelli U, Latorraca S, et al. Spontaneous and induced chromosome damage in somatic cells of sporadic and familial Alzheimer's disease patients. Mutagenesis 2001; 16:323 - 327
- Mosch B, Morawski M, Mittag A, Lenz D, Tarnok A, Arendt T. Aneuploidy and DNA replication in the normal human brain and Alzheimer's disease. J Neurosci 2007; 27:6859 - 6867
- Ringman JM, Rao PN, Lu PH, Cederbaum S. Mosaicism for trisomy 21 in a patient with young-onset dementia: a case report and brief literature review. Arch Neurol 2008; 65:412 - 415
- Thomas P, Fenech M. Chromosome 17 and 21 aneuploidy in buccal cells is increased with ageing and in Alzheimer's disease. Mutagenesis 2008; 23:57 - 65
- Iourov IY, Vorsanova SG, Liehr T, Yurov YB. Aneuploidy in the normal, Alzheimer's disease and ataxia-telangiectasia brain: differential expression and pathological meaning. Neurobiol Dis 2009; 34:212 - 220
- Arendt T, Bruckner MK, Mosch B, Losche A. Selective cell death of hyperploid neurons in Alzheimer's disease. Am J Pathol 2010; 177:15 - 20
- Moorhead PS, Heyman A. Chromosome studies of patients with Alzheimer disease. Am J Med Genet 1983; 14:545 - 556
- Zivkovic L, Spremo-Potparevic B, Plecas-Solarovic B, Djelic N, Ocic G, Smiljkovic P, et al. Premature centromere division of metaphase chromosomes in peripheral blood lymphocytes of Alzheimer's disease patients: relation to gender and age. J Gerontol A Biol Sci Med Sci 2010; 65:1269 - 1274
- Lucarelli P, Piciullo A, Palmarino M, Verdecchia M, Saccucci P, Arpino C, et al. Association between presenilin-1-48C/T polymorphism and Down's syndrome. Neurosci Lett 2004; 367:88 - 91
- Petersen MB, Karadima G, Samaritaki M, Avramopoulos D, Vassilopoulos D, Mikkelsen M. Association between presenilin-1 polymorphism and maternal meiosis II errors in Down syndrome. Am J Med Genet 2000; 93:366 - 372
- Schupf N, Kapell D, Lee JH, Ottman R, Mayeux R. Increased risk of Alzheimer's disease in mothers of adults with Down's syndrome. Lancet 1994; 344:353 - 356
- Migliore L, Boni G, Bernardini R, Trippi F, Colognato R, Fontana I, et al. Susceptibility to chromosome malsegregation in lymphocytes of women who had a Down syndrome child in young age. Neurobiol Aging 2006; 27:710 - 716
- Boeras DI, Granic A, Padmanabhan J, Crespo NC, Rojiani AM, Potter H. Alzheimer's presenilin 1 causes chromosome missegregation and aneuploidy. Neurobiol Aging 2008; 29:319 - 328
- Granic A, Padmanabhan J, Norden M, Potter H. Alzheimer Abeta peptide induces chromosome mis-segregation and aneuploidy, including trisomy 21: requirement for tau and APP. Mol Biol Cell 2010; 21:511 - 520
- Li J, Xu M, Zhou H, Ma J, Potter H. Alzheimer presenilins in the nuclear membrane, interphase kinetochores and centrosomes suggest a role in chromosome segregation. Cell 1997; 90:917 - 927
- Walczak CE, Heald R. Mechanisms of mitotic spindle assembly and function. Int Rev Cytol 2008; 265:111 - 158
- Heald R, Walczak CE. Microtubule-based motor function in mitosis. Curr Opin Struct Biol 1999; 9:268 - 274
- Hsu TC, Satya-Prakash KL. Aneuploidy induction by mitotic arrestants in animal cell systems: possible mechanisms. Basic Life Sci 1985; 36:279 - 289
- Mailhes JB, Mastromatteo C, Fuseler JW. Transient exposure to the Eg5 kinesin inhibitor monastrol leads to syntelic orientation of chromosomes and aneuploidy in mouse oocytes. Mutat Res 2004; 559:153 - 167
- Mazumdar M, Sundareshan S, Misteli T. Human chromokinesin KIF4A functions in chromosome condensation and segregation. J Cell Biol 2004; 166:613 - 620
- Bayer TA, Wirths O. Intracellular accumulation of amyloid-Beta—a predictor for synaptic dysfunction and neuron loss in Alzheimer's disease. Front Aging Neurosci 2010; 2:8
- Desai A, Murray A, Mitchison TJ, Walczak CE. The use of Xenopus egg extracts to study mitotic spindle assembly and function in vitro. Methods Cell Biol 1999; 61:385 - 412
- Budde PP, Desai A, Heald R. Analysis of microtubule polymerization in vitro and during the cell cycle in Xenopus egg extracts. Methods 2006; 38:29 - 34
- Vernos I, Raats J, Hirano T, Heasman J, Karsenti E, Wylie C. Xklp1, a chromosomal Xenopus kinesin-like protein essential for spindle organization and chromosome positioning. Cell 1995; 81:117 - 127
- Walczak CE, Vernos I, Mitchison TJ, Karsenti E, Heald R. A model for the proposed roles of different microtubule-based motor proteins in establishing spindle bipolarity. Curr Biol 1998; 8:903 - 913
- Kapoor TM, Mayer TU, Coughlin ML, Mitchison TJ. Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J Cell Biol 2000; 150:975 - 988
- Kline-Smith SL, Khodjakov A, Hergert P, Walczak CE. Depletion of centromeric MCAK leads to chromosome congression and segregation defects due to improper kinetochore attachments. Mol Biol Cell 2004; 15:1146 - 1159
- Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem 283; 29639 - 29643
- Pigino G, Morfini G, Atagi Y, Deshpande A, Yu C, Jungbauer L, et al. Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc Natl Acad Sci USA 2009; 106:5907 - 5912
- Wood KW, Sakowicz R, Goldstein LS, Cleveland DW. CENP-E is a plus end-directed kinetochore motor required for metaphase chromosome alignment. Cell 1997; 91:357 - 366
- Rosenfeld SS, Fordyce PM, Jefferson GM, King PH, Block SM. Stepping and stretching. How kinesin uses internal strain to walk processively. J Biol Chem 2003; 278:18550 - 18556
- Yardimci H, van Duffelen M, Mao Y, Rosenfeld SS, Selvin PR. The mitotic kinesin CENP-E is a processive transport motor. Proc Natl Acad Sci USA 2008; 105:6016 - 6021
- Walczak CE, Mitchison TJ, Desai A. XKCM1: a Xenopus kinesin-related protein that regulates microtubule dynamics during mitotic spindle assembly. Cell 1996; 84:37 - 47
- Mandelkow EM, Mandelkow E. Tau in Alzheimer's disease. Trends Cell Biol 1998; 8:425 - 427
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta-amyloid-induced neurotoxicity. Proc Natl Acad Sci USA 2002; 99:6364 - 6369
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science 2007; 316:750 - 754
- Feuk L, McCarthy S, Andersson B, Prince JA, Brookes AJ. Mutation screening of a haplotype block around the insulin degrading enzyme gene and association with Alzheimer's disease. Am J Med Genet B Neuropsychiatr Genet 2005; 69 - 71
- Wen PH, Hof PR, Chen X, Gluck K, Austin G, Younkin SG, et al. The presenilin-1 familial Alzheimer disease mutant P117L impairs neurogenesis in the hippocampus of adult mice. Exp Neurol 2004; 188:224 - 237
- Zhang C, McNeil E, Dressler L, Siman R. Long-lasting impairment in hippocampal neurogenesis associated with amyloid deposition in a knock-in mouse model of familial Alzheimer's disease. Exp Neurol 2007; 204:77 - 87
- Rossi G, Dalpra L, Crosti F, Lissoni S, Sciacca FL, Catania M, et al. A new function of microtubule-associated protein tau: involvement in chromosome stability. Cell Cycle 2008; 7:1788 - 1794
- Godin JD, Colombo K, Molina-Calavita M, Keryer G, Zala D, Charrin BC, et al. Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron 2010; 67:392 - 406
- Goldstein LS. Molecular motors: from one motor many tails to one motor many tales. Trends Cell Biol 2001; 11:477 - 482
- Tezapsidis N, Merz PA, Merz G, Hong H. Microtubular interactions of presenilin direct kinesis of Abeta peptide and its precursors. FASEB J 2003; 17:1322 - 1324
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science 2005; 307:1282 - 1288
- Lazarov O, Morfini GA, Pigino G, Gadadhar A, Chen X, Robinson J, et al. Impairments in fast axonal transport and motor neuron deficits in transgenic mice expressing familial Alzheimer's disease-linked mutant presenilin 1. J Neurosci 2007; 27:7011 - 7020
- Noda Y, Sato-Yoshitake R, Kondo S, Nangaku M, Hirokawa N. KIF2 is a new microtubule-based antero-grade motor that transports membranous organelles distinct from those carried by kinesin heavy chain or KIF3A/B. J Cell Biol 1995; 129:157 - 167
- Takemura R, Nakata T, Okada Y, Yamazaki H, Zhang Z, Hirokawa N. mRNA expression of KIF1A, KIF1B, KIF2, KIF3A, KIF3B, KIF4, KIF5 and cytoplasmic dynein during axonal regeneration. J Neurosci 1996; 16:31 - 35
- Ferhat L, Cook C, Chauviere M, Harper M, Kress M, Lyons GE, et al. Expression of the mitotic motor protein Eg5 in postmitotic neurons: implications for neuronal development. J Neurosci 1998; 18:7822 - 7835
- Coulson EJ, Paliga K, Beyreuther K, Masters CL. What the evolution of the amyloid protein precursor super-gene family tells us about its function. Neurochem Int 2000; 36:175 - 184
- Funk CJ, Davis AS, Hopkins JA, Middleton KM. Development of high-throughput screens for discovery of kinesin adenosine triphosphatase modulators. Anal Biochem 2004; 329:68 - 76
- Stine WB Jr, Dahlgren KN, Krafft GA, LaDu MJ. In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J Biol Chem 2003; 278:11612 - 11622
- Borysov SI, Guadagno TM. A novel role for Cdk1/cyclin B in regulating B-raf activation at mitosis. Mol Biol Cell 2008; 19:2907 - 2915