Abstract
The activation of nuclear factor (NF)κB pathway and its transducing signaling cascade has been associated with the pathogenesis of many inflammatory diseases. The central role that IκBα and p65 phosphorylation play in regulating NFκB signalling in response to inflammatory stimuli made these proteins attractive targets for therapeutic strategies. Although several chemical classes of NFκB inhibitors have been identified, it is only for a few of those that a safety assessment based on a comprehensive understanding of their pharmacologic mechanism of action has been reported. Here, we describe the specific anti-inflammatory effect of bindarit, an indazolic derivative that has been proven to have anti-inflammatory activity in a variety of models of inflammatory diseases (including lupus nephritis, arthritis and pancreatitis). The therapeutic effects of bindarit have been associated with its ability to selectively interfere with monocyte recruitment and the "early inflammatory response," although its specific molecular mechanisms have remained ill-defined. For this purpose, we investigated the effect of bindarit on the LPS-induced production of inflammatory cytokines (MCP-1 and MCPs, IL-12β/p40, IL-6 and IL-8/KC) in both a mouse leukaemic monocyte-macrophage cell line and bone marrow derived macrophages (BMDM). Bindarit inhibits the LPS-induced MCP-1 and IL-12β/p40 expression without affecting other analyzed cytokines. The effect of bindarit is mediated by the downregulation of the classical NFκB pathway, involving a reduction of IκBα and p65 phosphorylation, a reduced activation of NFκB dimers and a subsequently reduced nuclear translocation and DNA binding. Bindarit showed a specific inhibitory effect on the p65 and p65/p50 induced MCP-1 promoter activation, with no effect on other tested activated promoters. We conclude that bindarit acts on a specific subpopulation of NFκB isoforms and selects its targets wihtin the whole NFκB inflammatory pathway. These findings pave the way for future applications of bindarit as modulator of the inflammatory response.
Introduction
The NFκB family of inducible transcription factors plays a crucial role in regulating inflammatory and immune responses in addition to cell survival, proliferation and differentiation.Citation1 Deregulated NFκB signaling has been associated with the onset of several human pathologies, including cancer and inflammatory diseases.Citation2–Citation4 In mammals the NFκB family of transcription factors is comprised of five different isoforms: p65/Rel-A, c-Rel, Rel-B, p50 and p52.Citation5 These structurally related proteins share extensive sequence and structure similarities within their N-terminal Rel homology domain (RHD) that enables them to dimerize, translocate into the nucleus and bind to specific DNA sequences (κB consensus sites). Only p65/Rel-A, c-Rel and Rel-B contain a C-terminal transcriptional activation domain (TAD) and, therefore, the ability to directly activate the transcription.Citation6,Citation7
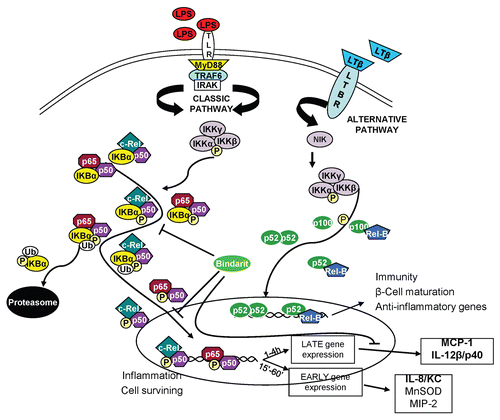
All these proteins form homodimeric and heterodimeric complexes, the activity of which is regulated by two major pathways. The first pathway is the classical NFκB activation pathway (which mainly applies to p65, p50 and c-rel combination of dimers), which, following stimulation with a broad range of stimuli (such as TNFα, LPS, IL-1, viruses and genotoxic agents), activates the transcription of various target genes controlling innate immunity and inflammation.Citation8,Citation9 The so-called alternative or non-canonical NFκB signaling pathway, is induced by a more restricted set of cytokines of the TNF superfamily [lymphotoxin β (LTβ), BAFF and CD40 ligand (CD40L)] that, through RelB/p52 and RelB/p50 activation, controls the development, organization and function, of secondary lymphoid organs.Citation10,Citation11 These functions include B-cell maturation and cell survival as well as the negative loop by which inflammation is downregulated.Citation12,Citation13
We selected bindarit (2-methyl-2-[(1-[phenylmethyl]-1H-indazol-3yl)methoxy]propanoic acid), a small molecule with proved anti-inflammatory activity in various inflammatory diseases in a wide variety of animal models,Citation14–Citation18 to explore its action on the NFκB pathway. Several studies demonstrated that this molecule is able to prevent the chronicity of inflammation, thereby reducing the potentially serious cytotoxic effects of this process at local and systemic levels.Citation19–Citation22 Yet, the molecular mechanism of its action remains to be elucidated.
Bindarit specifically downregulates the stimulus-induced gene expression of monocyte chemoattractant protein-1 (MCP-1) and of the other members of the MCPs cluster (MCP2/CCL8, MCP-3/CCL7),Citation21–Citation24 which collaboratively promote prolonged and sustained inflammation through monocyte-macrophage recruitment, playing a central role in homeostatic positioning and trafficking of immune cells. This process is controlled by multiple pathways, which strictly relate to the structure of the MCP-1 promoter. The murine MCP-1 promoter consists of two main regulatory regions: the distal (−2,650/−2,450) and the proximal (−248/0), both of which are specifically involved in the transcriptional activation of gene expression.Citation25–Citation27 The physiological oscillations in MCP-1 expression are mainly controlled by growth factors (PDGF, TGFβ, IFNγ), mechanic stress signals and other physiologic mediators that collaboratively manage the relative homeostatic balance of MCP-1 and of the whole “chemokinome.” The majority of these mediators control MCP-1 gene expression by specifically acting on the proximal regulatory region of its promoter through the Sp1/AP-1-controlled transcription of the gene.Citation26,Citation28 The situation differs for MCP-1 expression after an insult, however. One of the classic modes of activation of this inflammatory pathway is the one controlled by the Toll-like receptor-ligand system.Citation29–Citation33 The best characterized and known TLR4 ligand is lipopolisaccaride (LPS), a critical component of the Gram-negative bacteria cell wall that induces the activation of a complex signaling cascade system (MyD88-dependent and MyD88-independent pathways), producing a significant activation of the NFκB system.Citation34,Citation35 This involves the quick release of the IκBα-sequestred NFκB inactive dimers into the active conformation through the phosphorylation and subsequent ubiquitin-mediated proteasomic degradation of IκBα, activating the canonical NFκB pathway.Citation5–Citation7 This early-activated pathway, that involves p65/Rel-A, p50 and c-rel homodimeric/heterodimeric combinations of dimers exclusively, induces the timing-specific transcription of various target genes controlling innate immunity and inflammation.Citation8,Citation9 Acetylation and/or phosphorylation of the NFκB dimers may also occur in the cytoplasm and the nucleus, further modulating their nuclear translocation and DNA binding.Citation36,Citation37 This process is accompanied by a parallel increase in histone-acetylation at both the distal and proximal regions as well as within the intervening sequences separating the two regulatory regions of the MCP-1 promoter,Citation38 increasing the NFκB recruitment on the κB consensus sites and then significantly inducing MCP-1 expression.Citation27,Citation28
MCP-1 is the best-characterized target of bindarit, and its “stimulus-induced gene-expression” is mainly controlled by the p65 isoform of the NFκB classical pathway.Citation39 We reasoned that elucidation of the moecular mechanism by which bindarit modulates MCP-1 expression and the NFκB pathway would provide essential information toward the application of this drug. The capacity of bindarit to downregulate the MCP-1/MCPs and IL-12β/p40Citation40 gene expression is mediated by a specific subpopulation of NFκB isoforms, leading to a reduced chemotactic process and cytotoxic persistance of inflammation.
Results
Bindarit specifically inhibits LPS-mediated MCP-1/CCL2 (and MCPs) and IL-12β/p40 induction.
As shown by in vivo and in vitro studies,Citation14,Citation15,Citation41,Citation42 bindarit specifically inhibits MCP-1 and MCPs production in monocytic-macrophagic cells, significantly downregulating chronic inflammation, reducing the chemotactic process by which the inflammation persists at the lesion/infection sites.Citation43,Citation44
All these CC chemokines are strongly conserved in both human and mouse, forming a gene cluster and co-localizing on the same chromosome (chromosome 17 in human and 11 in mouse). The MCPs members of both species share protein sequence/folding and gene and promoter organization/regulation, even if their expression levels are different in various tissues (Fig. S1).Citation45,Citation46
To further investigate the mechanism by which bindarit exerts its anti-inflammatory action, we stimulated Raw 264.7 cells, a mouse leukemic monocyte macrophage cell line, with LPS (1 µg/ml) at different time points (1, 2, 4, 8, 24 h) in presence or absence of bindarit (300 µM, 1 h pre-treatment). The expression levels of different cytokines involved in early inflammation were measured. As shown in , bindarit pre-treatment significantly reduced MCP-1 mRNA levels, with a more potent effect at the highest LPS-induced peak of expression (4 h). We found the same inhibitory trend for p40 expression, the β-subunit of IL-12. Together with p35, these form one of the most important mediators of inflammation that modulates various biological activities on T- and natural killer (NK) cells, including induction of IFNγ production, enhancement of cell-mediated cytotoxicity and comitogenic effects on resting T cells.Citation40,Citation47–Citation49
IL-8/KC and IL-6, two other key players of the early inflammatory response, showed a different timing in their activation, and their levels were not altered by bindarit pre-treatment ().
As shown in , 1 h pre-treatment with bindarit also induces a significant downregulation in mRNA production for the other members of MCPs cluster.Citation23,Citation24 At their respective LPS-induced peaks of expression (4 h), MCP-2 and MCP-3 gene expression was inhibited by 24% and 36%, respectively.
When Raw 264.7 cells were pretreated with bindarit for 1 h, washed with PBS to remove bindarit and, finally, stimulated with LPS, mRNA expression was not significantly modified from the same samples without pretreatment (data not shown). This finding indicates that the persistent presence of bindarit is crucial for its action.
We also tested whether bindarit action would affect the mRNA stability of selected targets. To confirm/exclude this possibility, we examined the effect of bindarit 300 µM 1 h pretreatment on MCP-1, IL-12β/p40 and IL-8/KC expression after the addition of actinomycin D (5 µg/ml).Citation50 The addition of actinomycin D (for 30 or 90 min) strongly decreased the mRNA production of MCP-1 (70%), IL-12β/p40 (80%) and IL-8/KC (60%) at their peaks of expression (4 h, 4 h and 1 h, respectively) induced by LPS treatment. Bindarit pretreatment had no additive effect on the transcriptional block induced by actinomycin D, suggesting that it exerts its action at the transcriptional level ().
These results were confirmed by similar experiments conducted in a primary line of macrophagic cells derived from mice bone marrow (BMDM) pre-treated for 1 h with bindarit (300 µM) and then stimulated with LPS (1 µg/ml). As shown in , bindarit's effects were assessed on MCP-1, IL-12β/p40 and IL-8/KC LPS-induced expression at their respective peaks of expression (4 h, 4 h and 1 h, respectively). Similarly to the results obtained in Raw 264.7 cells, bindarit showed a significative inhibition on MCP-1 and IL-12β/p40 mRNA levels in BMDM primary cultures, with no-effects on IL-8/KC gene expression.
Bindarit effects on NFκB pathway activation.
As described above, the lack of a bindarit effect measured after PBS wash suggested that the inhibitory action was exerted in the cell without interfering with the ligand-receptor interaction.
While the importance of LPS-induced signal through the TLR4 receptors is documented, activation of NFκB pathway plays a crucial role in the activation of the inflammatory system and specifically in the expression of the two identified targets, MCPs and IL-12β/p40. To determine whether the reduced MCP-1 and IL-12β/p40 mRNA production was due to a downregulation of the NFκB pathway, as suggested by studies where p65 proves to have a crucial importance in their activation,Citation39,Citation40 we analyzed by western blot and immunofluorescence the LPS-induced NFκB pathway activation in Raw 264.7 cells in the presence or absence of bindarit.
LPS treatment (0, 15, 30, 45, 60, 120 min) produced a powerful induction of the NFκB pathway through TLR4 activation and subsequent kinases cascade activation. As shown in , IκBα phosphorylation (on Ser32/36) initiates at 15 min and peaks between 30–60 min. This phosphorylation, mediated by IKKs, induces a quick degradation of IκBα that is almost absent at 15 min and re-synthesized starting from 30 min through the ubiquitin-proteasome system. At the same time (15 min since LPS stimulation), p65 reached its phosphorylation-peak on Ser536, which, since this time point, started to decrease, almost expiring at 120 min. Three different sets of samples were used to analyze the effect of 1 h bindarit pre-treatment at the maximum time-point of activation (30 min) of the NFκB pathway by western blot on total extracts (). MG132 (proteasome inhibitor) and Tocris PF184 (potent and selective small-molecule IKK-2 inhibitors)Citation51 were also used as negative and positive controls, respectively, for the ability to induce accumulation or degradation of the proteins of the pathway. In agreement with previously shown qPCR data, western blots (and respective densitometry) confirmed that bindarit exerts a significant inhibitory effect both on IκBα Ser32/36 and p65 Ser536 LPS-induced phosphorylation. Vehicle showed no effect on that pathway, while a pre-treatment for 1 h with MG132 and Tocris PF184 was able to, respectively, enhance and block the accumulation of the activated forms of the NFκB proteins. It should be noted that the effective downstream effect of MG132 on the NFκB pathway is still to be defined. The amount of p65 seems to have remained unchanged in the total extracted proteins. For this reason, we tried to find out if bindarit was able to modify/reduce the p65 translocation into the nucleus. This was performed by making nuclear and cytosolic extracts. shows that 1 h bindarit pre-treatment (300 µM) has a significant effect on p65 nuclear translocation at both tested time points (15, 30 min) without modifying the cytosolic amount of the protein. This confirms its effect on the downregulation of the LPS-activated NFκB pathway. These results were confirmed in Raw 264.7 cells by immunofluorescence experiments and relative densitometric analysis of the p65 nuclear staining. As shown in , pre-treatment with bindarit (300 µM) for 1 h produced a significant reduction on the LPS-induced p65 nuclear translocation after 30 min. Analogous results were obtained with western blot in BMDMs ().
Bindarit inhibits the LPS-induced p65 binding to MCP-1 promoter.
The western blot and qPCR experiments demonstrated that bindarit reduced the NFκB activation and its nuclear translocation following LPS stimulation. These findings indicated that the effects on the NFκB pathway in the cytoplasm could also affect the interaction of NFκB-with DNA target sites and thereby the expression of specific target genes. To assess this possibility, we performed ChIP experiments in Raw 264.7 cells and determined the effect of bindarit on p65 recruitment on MCP-1 promoter. As shown in previous studies,Citation25–Citation27 a bioinformatic analysis of the 3 kb putative promoter region of murine MCP-1 gene revealed two regions: the distal regulatory enhancer region (DDR: −2,650/−2,450), with three κB consensus sites, and the proximal regulatory region (PRR: −250/−25) that includes the TATA-box, one AP-1 site, a Sp1 site and one κB site ( and ).
We used ChIP assaysCitation52 to measure LPS-induced p65 recruitment to the distal and proximal regulatory region of murine MCP-1 promoter. As shown by Saccani et al., the maximum recruitment of p65 at the MCP-1 promoter occurs in Raw 264.7 cells 90 min after LPS stimulation. Thus, we choose this timing of LPS treatment and anti-p65 antibody (SC-372). While LPS treatment induced p65 recruitment to both DRR and PRR, pretreatment with bindarit significantly affected p65 binding only on the PRR (). To confirm the specificity of this action we also tested bindarit's effect on another canonical p65-dependent gene, IκBα. shows that after 20 min of LPS stimulation, when the recruitment of p65 on the IκBα promoter is at its peak,Citation53 bindarit pretreatment had no effect. This result supports the notion that bindarit specifically targets a subpopulation of NFκB isoforms.
The demonstrated reduced activation of the NFκB pathway and the subsequent reduced nuclear translocation is consistent with the decreased recruitment on the MCP-1 promoter.
Bindarit specifically inhibits the p65 and p65/p50 mediated MCP-1 promoter activation.
To study the effect of bindarit on MCP-1 expression, we cloned the mouse promoter region (−2,816/−9 with respect to the ATG translational starting site) in the pGL3b (Promega) reporter system ( and ). To confirm the responsiveness of this promoter region to NFκB, we used the Addgene NFκB coding plasmids and co-transfected them in Raw 264.7 cells. As shown in , only p65 and p65/p50 co-transfection induce a significant MCP-1 transcriptional activation, while a possible involvement of c-Rel cannot be excluded. One-hour bindarit pretreatment (300 µM) shows a significant inhibitory effect, reducing p65 and p65/p50 recruitment and MCP-1 promoter activity by 61 and 35%, respectively. To test for specificity, we also analyzed three other promoter systems: (1) the Dbp-promoter-luc with its transcription activators CLOCK and BMAL1 (a PAR leucine zipper transcription factor that play a crucial role in the circadian oscillations);Citation54,Citation55 (2) the ApolipoproteinB-promoter-luc and its specific activator HNF-4 (the primary system responsible for carrying cholesterol to tissues that plays a crucial role in metabolism);Citation56 (3) the PPARγ-TK-luc and its activator PPARγ (the glitazone receptor and ligand that regulate the fatty acid storage and glucose metabolism).Citation57,Citation58 Bindarit had no inhibitory effect on the activation of these promoters, underscoring its specificity for the p65- and p65/p50-mediated activation of the MCP-1 promoter.
Discussion
The orchestration of the inflammatory responses to both infection and tissue damage is arguably the key physiological function of NFκB pathway, a signaling transduction system that plays a pivotal role in many different physiological responses, conveying information and stimuli from the extracellular environments to cellular response. Interference with the activation of NFκB signaling has been widely and convincingly associated to the onset of many inflammatory diseases.
The cytokines/chemokines network is central in the early development of inflammation and cellular response to a wide range of insults, many of which are coordinated by the NFκB family of transcription factors. In the whole chemokinome a crucial role is played by both the monocyte chemoattractant proteins group (mainly by MCP-1) and IL-12. These molecules act as mediators of inflammation, managing cellular recruitment via autocrine/paracrine chemotactic comunication and the regulation of T- and natural killer (NK) cell activity, respectively. The expression of both MCP-1 and IL-12 is induced by receptor systems activation and downstream activation of the NFκB family of transcriptional factors. Considering these notions and the tight link between the activation of NFκB pathway and specific mediators that manage the inflammatory response, we tested the effect of bindarit, an indazolic-derivative with proven anti-infiammatory effects,Citation41,Citation42 on the stimulus-induced activation of NFκB pathway that leads to inflammation.
We have addressed the effect of bindarit on the NFκB canonical pathway induced by LPS-treatment and the consequent downregulation of MCP-1 (MCPs) and IL-12β/p40 gene expression and protein production. All our results converge toward a scenario in which bindarit downregulates p65 and p65/p50-mediated MCP-1 gene expression through a reduction in IκBα phosphorylation and corresponding NFκB dimers' nuclear translocation and transcriptional activity. The downstream effects of this action, as shown by ChIP experiments, include reduced binding to the proximal regulatory region of the murine MCP-1 promoter, the reduced gene-expression () and, consequently, lower levels of secreted protein.Citation26 Similar effects were observed for MCP-2 and MCP-3, most likely because of the high degree of homology that these members of the CC cluster share both in protein organization and promoter structure.Citation45,Citation46 On the contrary, our assays indicate that bindarit acts specifically on the NFκB system, with no effect on other tested promoters and respective activators.
Further confirmation of the bindarit-specific action on the classical pathway of NFκB is also represented by preexisting evidences about the lack of immunosuppressive effects at the systemic level of this molecule. These notions underscore the non-involvement of the NFκB alternative pathway, which through p52 and Rel-B modulates the homeostasis and development of the immunosuppressive system.Citation9,Citation12
The present study reveals the molecular mechanism by which bindarit acts as NFκB modulator. In addition to the proven anti-inflammatory properties of this molecule in a variety of inflammatory diseases, bindarit showed a potent effect in the modulation of NFκB activated pathway, reducing IκBα/p65 phosphorylation and the nuclear localization of NFκB dimers. The ultimate, therapeutically valuable effect of bindarit is to prevent an excessive production of chemokines and cytokines at the lesion site, avoiding the cytotoxic chronicity of this process that has been directly linked with the onset of many pathologies.
Materials and Methods
Chemicals.
Bindarit was synthesized by and obtained from Angelini (Angelini Research Center-ACRAF) and prepared as a 100 mM stock solution in 1 N NaOH, which was diluted to test concentrations in phosphate-buffered saline (PBS) at pH 7.4; Ultrapure LPS was purchased from Invitrogen; PF184 (IKK-2 and NFκB inhibitor) from Tocris Bioscences (4238); MG132 and actinomycin D from Sigma-Aldrich.
Cell culture.
Raw 264.7, a mouse macrophage-like cell line, was obtained from the American Type Culture Collection (Cryosite). Dulbecco's modified Eagle medium (DMEM) was purchased from Thermo Scientific, and fetal bovine serum and penicillin/streptomycin were purchased from Invitrogen Technologies. Raw 264.7 cells were grown in DMEM with 10% fetal bovine serum, 100 U/ml penicillin and 100 µg/ml streptomycin at 37°C in 5% CO2 humidified air. For all the experiments described here, cells were used between the 5th and 10th passage.
BMDM primary cell culture.
All animal experimental protocols were approved by an institutional animal care committee. WT C57BL/6 mice were euthanized by cervical dislocation, and femurs were isolated by surgical resection. Bone marrow-derived macrophages (BMDMs) were collected from femurs as described by Srivastava and De Larrea.Citation59 The ends of femurs were trimmed, and the effluent bone marrow stem cells were collected injecting cold RPMI. The cells were then spun and cultured in 5 Petri dishes with 20 ml of bone marrow macrophages media [BMMM: RPMI, 10% heat-inactivated FBS, 1% L-Glu, 1% Antibiotics/Antimiotics, 30% L929 (murine fibroblast)-cell conditioned medium].Citation57,Citation60 At day 3, the macrophages were fed with 10 ml of BMMM. At day 7, the media was removed, and the cells were resuspended in 25 ml of RPMI + 1% L-Glu + 10% heat-inactivated FBS (no Ab/Am) for cell counting. Appropriate amount of BMDMs were plated for qPCR, immunohistochemistry or western analysis.
Preparation of cytoplasmic and nuclear protein extracts.
Extracts were prepared for analysis from whole cells or nuclei by a rapid method modified from Osborn et al. Briefly, for cellular extracts, 107 cells were washed once with phosphate-buffered saline and twice with buffer A (10 mM HEPES-KOH pH 7.9; 1.5 mM MgCl2; 10 mM KCl; 0.5 mM DTT; protease inhibitors). The cell pellet was suspended in buffer A/0.1% Nonidet P-40. After incubating for 10 min on ice, the lysed cellular suspension was briefly mixed on a vortex and micro-centrifuged for 10 min at 4°C. The supernatant is the cytosolic material (removed and stored at −70°C). The nuclear pellet was re-suspended in 15 µl of buffer C (20 mM HEPES-KOH pH 7.9; 25% v/v glycerol; 0.42 M NaCl; 1.5 mM MgCl2; 0.2 mM EDTA; 0.5 mM PMSF; 0.5 mM DTT; protease inhibitors), incubated 15 min at 4°C, mixed briefly and micro-centrifuged for 10 min at 4°C. The supernatant contained the nuclear extract and was stored at −70°C.
Western blot.
Samples of cytoplasmic, nuclear or total protein were loaded into each lane and electrophoresed on a 10% sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE) under reducing conditions. The proteins were transferred onto a 0.45 µm pure nitrocellulose membrane (Bio-Rad Laboratories, Inc.). To block nonspecific binding sites, the membrane was incubated sequentially with 5% milk-TBS-T solution and then primary antibodies for IκBα (1:2,000), phospho-IκBα α (1:1,000), p65/Rel-A (1:1,000), phospho-p65/Rel-A (Ser536) (1:1,000), c-Rel (1:1,000), IKKβ (1:1,000), phosphor-IKKα/β (Ser176/180) (1:1,000) (Cell Signaling Technology), α-Tubulin (Sigma-Aldrich), TBP-α (Euromedex)Citation62 overnight at 4°C. Membranes were washed three times and incubated with appropriate peroxidase-conjugated secondary rabbit (Cell Signaling Technology) and mouse (Millipore Corporation) antibodies (1:10,000). Blots were developed using the Chemiluminescent HRP substrate (Millipore Corporation).
ChIP assay.
Dual crosslinking ChIP assayCitation52 was used with slight modifications. After LPS stimulation in presence/absence of bindarit (300 µM, 1 h pre-treatment), cells were washed three times with room temperature PBS then PBS/1 mM MgCl2 was added. Disuccinimidyl Glutalate (DSG) was added to a final concentration of 2 mM for crosslinking. After 45 min at room temperature, formaldehyde was added to a final concentration of 1% (v/v) and cells were incubated for 15 min. After dual cross-linking, glycine was added to a final concentration of 0.1 M and incubated for 10 min to quench formaldehyde out. After harvesting, cells were lysed in 500 ml ice-cold cell lysis buffer [50 mM Tris/HCl pH 8.0, 85 mM KCl, 0.5% NP-40, 1 mM PMSF, 13 protease inhibitor cocktail (Roche)] for 10 min on ice. Nuclei were precipitated by centrifugation (3,000 g for 5 min), resuspended in 600 µl ice-cold RIPA buffer [50 mM Tris/HCl pH 8.0, 150 mM NaCl, 1 mM EDTA pH 8.0, 1% Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate, 1 mM PMSF, 13 protease inhibitor cocktail (Roche)] and incubated on ice for 30 min. Sonication was done to obtain 100–600 bp DNA fragments.
Extracts were precleared for 4 h with 80 µl of a 50% suspension of salmon sperm-saturated proteinG (ss-proteinG). Immunoprecipitations were performed at 4°C overnight with 2 ug of anti-p65 antibody (Santa Cruz, SC-372). Immune complexes were collected with 25 µl of ss-proteinG for 3 h at 4°C and washed three times (5 min each) with high-salt buffer (washing buffer: 20 mM Tris, pH 8.0, 0.1% SDS, 1% NP-40, 2 mM EDTA, 500 mM NaCl), once with LiCl ChIP wash buffer (0.25 mM LiCl, 1% IGEPAL630, 1% deoxycholate, 1 mM EDTA, 10 mM TRIS-HCl pH 8.0) and twice with TE buffer (10 mM Tris-KCl pH 8.0, 1 mM EDTA). Immune complexes were extracted, and protein-DNA cross-links were reverted by heating at 65°C overnight. After proteinase K digestion (55°C for 1 h), DNA was extracted by phenol-chloroform and ethanol was precipitated. Three µl of 30 µl total used for resuspension of DNA were used in each PCR. To assess the effects of bindarit on LPS-induced p65/Rel-A recruitment on distal (DDR) and proximal (PRR) regulatory regions of MCP-1 promoter, the following primers were used: mDRR-MCP-1 sense 5′-AGTCCTCACCCCATTACATCTC-3′, antisense 5′-CAGAGTAGGATAAGAGCGTGGA-3′; mPRR-MCP-1 sense 5′-AGCTTCATTTGCTCCCAGGAG-3′, antisense 5′-GAGGCAGCCTTTTATTGTAAGC-3′.
Immunocytochemistry.
Cells were fixed using 4% paraformaldehyde in PBS. Blocking was performed using 3% BSA diluted in PBS for 30 min at room temperature. Primary antibody used: anti-p65 (SantaCruz SC-372, 1:400 dilution) was incubated over night. Secondary antibody was conjugated with Alexa-488 goat anti-rabbit (Invitrogen). Nuclei were stained with DRAQ-5 [-(dimethylamino) ethylamino-4,8-dihydroxyanthracene-9,10-dione] (Biostatus Limited). Immunolabelled sections were examined with a Leica confocal microscope SP5 (DMRE, Leica). Controls were always performed by omitting primary antibodies. Intensity of fluorescence is evaluated as pixels/µm2. The Leica SP5 software LAS AF was used for quantification.
Plasmid construction.
Murine MCP-1 promoter (−2,816/−9 respect to the ATG translational starting site) was cloned into the pGL3-basic vector plasmid (Promega) from murine WT C57BL/6 mice genomic DNA (the PCR templates were amplified using sense primer 5′-TGG CTA GCT GAA CTG TAC AAG TGG CCA G-3′ and antisense primer 3′-TCC TCG AGT GCT CTG AGG CAG CCT TTT A-5′).
The five full-length cFlag-pcDNA3-NFκB isoforms codifying plasmids (p50, p52, p65/Rel-A, c-Rel, Rel-B) were purchased form Addgene Inc.
The pGL4.11-DBP promoter plasmid and the respective activator plasmids expressing myc-tagged mClock (pSG5-Myc-mCLOCK) and FLAG-tagged mBmal1 (pCS2-Flag-Myc-mBMAL1) were described previously in reference Citation55.
The pSG5-Flag-PPARγ and PPRE3-TK-luc containing three PPAR responsive elements (PPREs) upstream of the TK minimal promoter controlling the luciferase expression was kindly provided by Bruce Spiegelman.Citation58,Citation63
The ApoB-E4-luc and Flag-HNF-4 were kindly provided by Frances M. Sladek.
Plasmid expressing β-galactosidase (pGL3-lacZ) was used as transfection efficiency control.
Cell transfection and luciferase assay.
Raw264.7 cells were plated at 1 × 105 cells per well in a 24-well plate for 24 h. Using the BioT transfectant agent (Bioland Scientific LLC) and following the manufacturer instructions, 60–80% confluence cells were transiently transfected with 500 ng of total DNA: MCP-1prom-luc (100 ng), p65/Rel-A (100 ng), c-Rel (100 ng), Rel-B (100 ng), p50 (25 ng), p52 (25 ng); DBPprom-luc (100 ng), Clock (100 ng), Bmal1 (50 ng); ApoB-E4-luc (100 ng), HNF-4 (100 ng); PPARγ-RE-Lc (100 ng), PPARγ; and Luc-Z reporter (125 ng, as control for transfection efficiency). Bindarit (300 µM) was added 6 h after the transfection. 24 h following transfection, cells were lysed and luciferase activity determined with the luminometer and β-gal/lac-Z assay (Abs 495 nm) for transfection efficiency normalization. A minimum of three independent experiments was performed in triplicate for each assay.
RNA extraction and RT-PCR and mRNA stability studies.
Total cellular RNA was isolated from unstimulated or LPS-stimulated Raw 264.7 cells following, or not, bindarit and or actinomycin D (5 µg/ml; from Sigma-Aldrich) Citation43 treatment using TRIZOL reagent (Invitrogen). Superscript II Reverse Transcriptase (Invitrogen) was then used with oligo-dT primers (Thermo Scientific) to prepare cDNA from 1 µg of total RNA (50 min at 42°C; 15 min at 70°C). QPCR was performed using 2 µl of cDNA with specific primers for qPCR.
Quantitative RT-PCR.
Each quantitative real-time RT-PCR was performed in triplicate using the Chromo4 real-time detection system (Bio-Rad) with Sybr-green master mix (SABiosciences). The reactions were performed under the amplification condition of 40 cycles of 95°C for 30 sec and 60°C for 60 sec with gene-specific primers based on sequences from NCBI-genebank, which were as follows: mMCP-1 sense 5′-CTT CTG GGC CTG CTG TTC A-3′, antisense 5′-CCA GCC TAC TCA TTG GGA TCA-3′; mMCP-2 sense 5′-TCT TTG CCT GCT GCT CAT AGC TG-3′, antisense 5′-GCG CAG ACT TCC ATG CCC TTC TT-3′; mMCP-3 sense 5′-TCA GCA TCC AAG TGT GGG CCC A-3′, antisense 5′-GCG CAG ACT TCC ATG CCC TTC TT-3′; mIL-12β/p40 sense 5′-TGG TTT GCC ATC GTT TTG CTG-3′, antisense 5′-ACA GGT GAG GTT CAC TGT TTC T-3′; mIL-8/KC sense 5′-TGC ACC CAA ACC GAA GTC AT-3′, antisense 5′-TTG TCA GAA GCC AGC GTT CAC-3′; mIL-6 sense 5′-TTC CAT CCA GTT GCC TTC TT-3′, antisense 5′-CAG AAT TGC CAT TGC ACA AC-3′. mPPIA sense 5′-TCT CGG AGC GCA ATA TGA AGG TG-3, antisense 5′-TCC AAA GAG TCC AAA GAC GAC TCG-3′;Citation64 mβ-actin sense 5′-CCA GTT GGT AAC AAT GCC ATG T-3′, antisense 5′-GGC TGT ATT CCC CTC CAT CG-3′ were used for double normalization.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Figures and Tables
Figure 1 Bindarit transcriptional effects on LPS-induced inflammatory chemokine. (A) Raw 264.7 cells were stimulated with LPS (1 µg/ml) in presence or absence of bindarit (300 µM 1 h pre-treatment) for the indicated time points. After RNA extraction and reverse transcription MCP-1, IL-12β/P40, IL-8/KC and IL-6 gene expression was measured. At the same time points, bindarit (300 µM 1 h pre-treatment) showed a specific inhibitory effect only for MCP-1 and p40 subunit of IL-12 without effects on IL-8/KC and IL-6. (B) Bindarit (300 µM, 1 h pre-treatment) effect was assessed in Raw 264.7 on the expression of the other members of the murine CC chemochine gene cluster (MCP-2/CCL8, MCP-3/CCL7). After LPS induction (1 µg/ml), gene expression was measured with qPCR 1 and 4 h after LPS induction in presence or absence of bindarit. (C) Bindarit's effects on MCP-1, IL-12β/P40 and IL-8/KC mRNA stability after LPS induction. Effect of bindarit (300 µM), Actinomycin D (5 µg/ml) and co-treatment on indicated chemokines gene expression was assessed in Raw 264.7 cells after 1 µg/ml LPS induction for 4 and 1 h respectively. (D) Bone marrow-derived macrophages (BMDMs) were stimulated with LPS (1 µg/ml) in presence or absence of bindarit (300 µM, 1 h pre-treatment) for 1 and 4 h. After RNA extraction and reverse transcription MCP-1, IL-12β/P40 and IL-8/KC gene expression was measured. At the relative maximum peak of expression, bindarit (300 µM 1 h pre-treatment) showed a specific inhibitory effect only for MCP-1 and p40 subunit of IL-12 without effects on IL-8/KC gene expression. A p-value of less than 0.05 was considered statistically significant (*p < 0.01, **p < 0.001). For all the data shown, the average of three different experiments is presented, normalized for both β-Actin and PPIA expression.
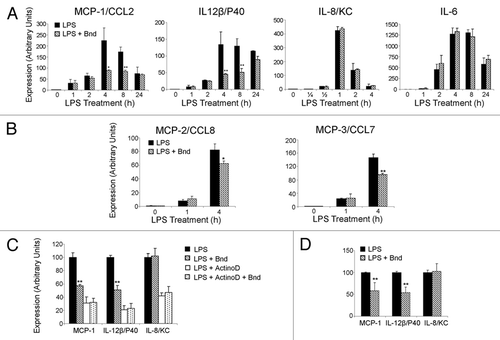
Figure 2 Bindarit's effect on the LPS-induced NFκB pathway activation. (A) Raw 264.7 cells were treated at different timing (0, 15, 30, 45, 60, 120 min) with 1 µg/ml LPS. For all the different time points, western analysis for IκBα, p65 and respective phosphorylated isoforms on total extracts were performed as described in Materials and Methods. α-Tubulin was used as load control. (B) Raw 264.7 cells were treated for 30 min with 1 µg/ml LPS after 1 h pre-treatment with vehicle (V), bindarit (300 and 600 µM) (B), MG132 (20 µM) (MG), and Tocris PF184 (1 µM) (T). Western analysis and respective densitometries for p65 and IκBα phosphorylation are presented. (C) Raw 264.7 cells were treated for 15 and 30 min with 1 µg/ml LPS after 1 h bindarit pre-treatment (300 µM). Western analysis and respective densitometries on cytosolic (Cy) and nuclear (Nu) extracts were performed to assess bindarit effects on p65 cytosolic-nuclear traslocation at both the indicated time points. α-Tubulin and TBP-α were used as load control for cytosolic and nuclear proteins, respectively. (D) Raw 264.7 cells were treated with LPS (1 µg/ml) for 30 min, and then the intracellular location of p65 was determined by immunofluorescence confocal scanning microscope. Double staining (Merge) with p65 (green) and DAPI (blue) staining demonstrates the p65 nuclear localization after LPS treatment. One-hour bindarit pre-treatment effect was assessed on p65 nuclear translocation. Densitometry chart of p65 nuclear staining for the three different conditions of treatment is also presented. (E) Bone marrow-derived macrophages (BMDMs) were treated with LPS (1 µg/ml) for 30 min. Western blot analysis and respective densitometries on cytosolic (Cy) and nuclear (Nu) extracts were performed for p65 nuclear translocation and IκBα cytosolic phosphorylation in presence/absence of bindarit (300 µM, 1 h pre-treatment). α-Tubulin and TBPa were used as load control for cytosolic and nuclear proteins, respectively. All the presented data are representative of three different experiments. A p-value of less than 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001).
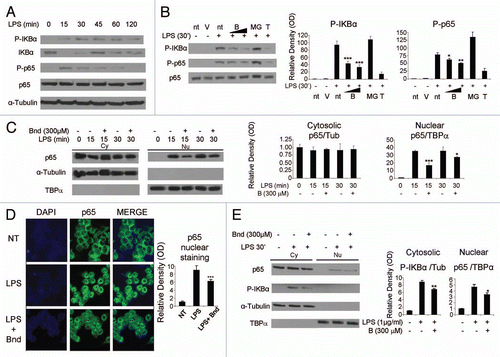
Figure 3 Sequence of the enhancer and promoter region of the murine monocyte chemoattractant protein-1 (mMCP-1) gene. (A) The 3 KB long sequence of the murine MCP-1 putative promoter is presented. Two key regions have been identified within this sequence: the distal regulatory region (−2,650/−2,450) and the proximal regulatory region (−2,48/0). The NFκB binding sites are highlighted in both enhancer (κB2a: −2,460/−2,450; κB2b: −2,486/−2,476; κB3: −2,635/−2,625) and promoter (κB1: −152/−142) regulatory regions. The specific transcriptor factor (Sp1) binding site (−252/−242), the activated protein-1 (AP-1) binding site (−130/−121) and the TATAbox (−32/−24) are underlined in promoter region. The translational starting site (ATG) is located in position +85 with respect to the transcription starting site (+1). (B) Recruitment of p65/Rel-A to the different regulatory region of MCP-1 promoter was assessed in LPS-stimulated Raw 264.7 cells. After 90 min of LPS stimulation (1 µg/ml), ChIP assay using p65 (SC-372) antibody was performed in presence/absence of bindarit (300 µM 1 h pre-treatment) as described in Materials and Methods. Real-time PCR was then performed on immunoprecipitated DNA using primers amplifying the proximal regulatory region (PRR) and the distal regulatory region (DRR) of the murine MCP-1 promoter. (C) Recruitment of p65/Rel-A to the two NFκB sites of IκBα promoter was assessed in LPS-stimulated Raw 264.7 cells. After 20 min of LPS stimulation (1 µg/ml), ChIP assay using p65 (SC-372) antibody was performed in presence/absence of bindarit (300 µM 1 h pre-treatment) as described in Materials and Methods. Real-time PCR was then performed on immunoprecipitated DNA using primers amplifying the NFκB-containing region of the IκBα promoter. A p-value of less than 0.05 was considered statistically significant (*p < 0.01). Data are representative of three indipendent experiments.
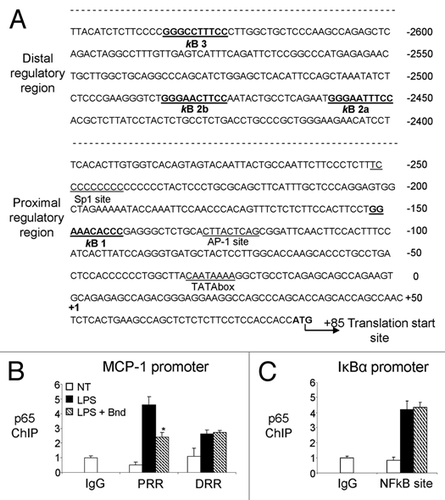
Figure 4 Specific bindarit effect on NFκB-induced MCP-1prom-luc expression. (A) The map of the transcription factors' consensus sites within the murine MCP-1 putative promoter is presented. The 2,807 bp region (−2,816/−9) of murine MCP-1 promoter, including both the enhancer region (distal regulatory region) and the promoter region (proximal regulatory region) has been cloned in pGL3basic vector using C57BL/6 mice genomic DNA as described in Materials and Methods. (B) Raw 264.7 cells were transiently transfected with MCP-1prom-luc construct (100 ng) together with an expression vector for full-length c-Rel (100 ng), p65 (100 ng), p50 (25 ng), rel-B (100 ng) and p52 (25 ng) as described in Materials and Methods. Bindarit's (300 µM, added 6 h after transfection) effect on MCP-1prom-luc Luciferase expression was measured 24 h after transfection. (C) The specificity of bindarit action was tested on four different responsive element/activator systems. Raw 264.7 cells were transiently transfected with 100 ng of four different promoter-luc constructs (MCP-1prom-luc, Dbp-promoter-luc; ApolipoproteinB-promoter-luc; PPARγ-promoter-luc) and 50–100 ng of their respective co-activator expressing plasmids (p65; Clock/Bmal; HNF-4; PPARγ) as described in Materials and Methods. Bindarit's (300 µM, added 6 h after transfection) effects was measured 24 h after transfection. For (B and C), the promoter activity (mean ± SD) is presented as fold increase over cells transfected with empty vector. To normalize the transfection efficency across samples, the β-galactosidase activity (pGL3-LacZ expression) was used as internal control. A p-value of less than 0.05 was considered statistically significant (*p < 0.001). Data are representative of five independent experiments.
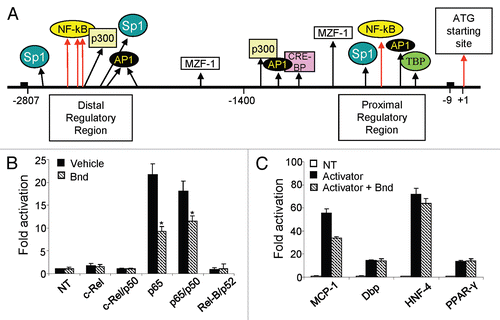
Figure 5 Proposed model for bindarit action. The potential mechanism of action of bindarit is presented. The stimulus-induced activation of NFκB pathway is modulated by bindarit pre-treatment. This anti-inflammatory molecule produces a reduction of IκBα and p65 phosphorylation, a subsequent significant reduction of the nuclear translocation of p65-constituted dimers and, consequently, a reduced recruitment of these transcription factor to the κB sites of specific promoters of inflammatory genes. The overall effect of this combined action is the significant inhibition of MCP-1 (/MCPs) and IL-12β/p40 expression that ultimately led to a significant downregulation of the inflammation, especially at the lesion site.
Additional material
Download Zip (1.2 MB)Acknowledgments
We wish to thank all the members of the Sassone-Corsi lab and the Dr. Francesca Mancini of the of the Angelini ACRAF Spa for help, discussions and reagents. E.M. was supported by a fellowship of the AIRC (Associazione Italiana Ricerca Cancro) Prot n 338/2010/MEP/ac. Our research is supported by grants from the Natonal Institute of Health, the INSERM (Institut National de la Sante et de la Recherche Medicale-France) and Angelini R&D-ACRAF SpA.
References
- Baeuerle PA, Henkel T. Function and activation of NFkappaB in the immune system. Annu Rev Immunol 1994; 12:141 - 179; PMID: 8011280; http://dx.doi.org/10.1146/annurev.iy.12.040194.001041
- Sun XF, Zhang H. NFκB and NFκBI polymorphisms in relation to susceptibility of tumor and other diseases. Histol Histopathol 2007; 22:1387 - 1398; PMID: 17701919
- Mantovani A. Molecular pathways linking inflammation and cancer. Curr Mol Med 2010; 10:369 - 373; PMID: 20455855; http://dx.doi.org/10.2174/156652410791316968
- Chefetz I, Holmberg JC, Alvero AB, Visintin I, Mor G. Inhibition of Aurora-A kinase induces cell cycle arrest in epithelial ovarian cancer stem cells by affecting NFκB pathway. Cell Cycle 2011; 10:2206 - 2214; PMID: 21623171; http://dx.doi.org/10.4161/cc.10.13.16348
- Hayden MS, Ghosh S. Signaling to NFkappaB. Genes Dev 2004; 18:2195 - 2224; PMID: 15371334; http://dx.doi.org/10.1101/gad.1228704
- Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NFkappaB. Annu Rev Cell Biol 1994; 10:405 - 455; PMID: 7888182; http://dx.doi.org/10.1146/annurev.cb.10.110194.002201
- Pahl HL. Activators and target genes of Rel/NFkappaB transcription factors. Oncogene 1999; 18:6853 - 6866; PMID: 10602461; http://dx.doi.org/10.1038/sj.onc.1203239
- Ghosh S, Karin M. Missing pieces in the NFκB puzzle. Cell 2002; 109:81 - 96; PMID: 11983155; http://dx.doi.org/10.1016/S0092-8674(02)00703-1
- Bonizzi G, Karin M. The two NFκB activation pathways and their role in innate and adaptive immunity. Trends Immunol 2004; 25:280 - 288; PMID: 15145317; http://dx.doi.org/10.1016/j.it.2004.03.008
- Derudder E, Dejardin E, Pritchard LL, Green DR, Korner M, Baud V. RelB/p50 dimers are differentially regulated by tumor necrosis factora and lymphotoxinß receptor activation. J Biol Chem 2003; 278:23278 - 23284; PMID: 12709443; http://dx.doi.org/10.1074/jbc.M300106200
- Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NFκB2 in maturing B cells. Nat Immunol 2002; 3:958 - 965; PMID: 12352969; http://dx.doi.org/10.1038/ni842
- Dejardin E. The alternative NFκB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem Pharmacol 2006; 72:1161 - 1179; PMID: 16970925; http://dx.doi.org/10.1016/j.bcp.2006.08.007
- Mihalcik SA, Tschumper RC, Jelinek DF. Transcriptional and post-transcriptional mechanisms of BAFF-receptor dysregulation in human B lineage malignancies. Cell Cycle 2010; 9:4884 - 4892; PMID: 21099364; http://dx.doi.org/10.4161/cc.9.24.14156
- Guglielmotti A, Silvestrini B, Saso L, Zwain I, Cheng CY. Chronic inflammatory response in the rat can be blocked by bindarit. Biochem Mol Biol Int 1993; 29:747 - 756; PMID: 7683947
- Guglielmotti A, D'Onofrio E, Coletta I, Aquilini L, Milanese C, Pinza M. Amelioration of rat adjuvant arthritis by therapeutic treatment with bindarit, an inhibitor of MCP-1 and TNFalpha production. Inflamm Res 2002; 51:252 - 258; PMID: 12056513; http://dx.doi.org/10.1007/PL00000301
- Ialenti A, Grassia G, Gordon P, Maddaluno M, Di Lauro MV, Baker AH, et al. Inhibition of In-Stent Stenosis by Oral Administration of Bindarit in Porcine Coronary Arteries. Arterioscler Thromb Vasc Biol 2011; 31:2448 - 2454; PMID: 21852559; http://dx.doi.org/10.1161/ATVBAHA.111.230078
- Zoja C, Corna D, Benedetti G, Morigi M, Donadelli R, Guglielmotti A, et al. Bindarit retards renal disease and prolongs survival in murine lupus autoimmune disease. Kidney Int 1998; 53:726 - 734; PMID: 9507220; http://dx.doi.org/10.1046/j.1523-1755.1998.00804.x
- Ramnath RD, Ng SW, Guglielmotti A, Bhatia M. Role of MCP-1 in endotoxemia and sepsis. Int Immunopharmacol 2008; 8:810 - 818; PMID: 18442784; http://dx.doi.org/10.1016/j.intimp.2008.01.033
- Conductier G, Blondeau N, Guyon A, Nahon JL, Rovíre C. The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J Neuroimmunol 2010; 224:93 - 100; PMID: 20681057; http://dx.doi.org/10.1016/j.jneuroim.2010.05.010
- Lloyd CM, Dorf ME, Proydfoot A, Salant DJ, Gutierrez-Ramos JC. Role of MCP-1 and RANTES in inflammation and progression to fibrosis during murine crescentic nephritis. J Leukoc Biol 1997; 62:676; PMID: 9365123
- Luster AD, Rothenberg ME. Role of the monocyte chemoattractant protein and eotaxin subfamily of chemokines in allergic inflammation. J Leukoc Biol 1997; 62:620; PMID: 9365117
- Baggiolini M, Dahinden CA. CC chemokines in allergic inflammation. Immunol Today 1994; 15:127; PMID: 8172645; http://dx.doi.org/10.1016/0167-5699(94)90156-2
- Jia T, Serbina NV, Brandl K, Zhong MX, Leiner IM, Charo IF, et al. Additive roles for MCP-1 and MCP-3 in CCR2-mediated recruitment of inflammatory monocytes during Listeria monocytogenes infection. J Immunol 2008; 180:6846 - 6853; PMID: 18453605
- Van Damme J, Proost P, Lenaerts JP, Opdenakker G. Structural and functional identification of two human, tumor-derived monocyte chemotactic proteins (MCP-2 and MCP-3) belonging to the chemokine family. J Exp Med 1992; 176:59 - 65; PMID: 1613466; http://dx.doi.org/10.1084/jem.176.1.59
- Ping D, Boekhoudt GH, Rogers EM, Boss JM. Nuclear factor-kappaB p65 mediates the assembly and activation of the TNF-responsive element of the murine monocyte chemoattractant-1 gene. J Immunol 1999; 162:727 - 734; PMID: 9916692
- Ping D, Jones PL, Boss JM. TNF regulates the in vivo occupancy of both distal and proximal regulatory regions of the MCP-1/JE gene. Immunity 1996; 4:455 - 469; PMID: 8630731; http://dx.doi.org/10.1016/S1074-7613(00)80412-4
- Boekhoudt GH, Guo, Beresford GW, Boss JM. Communication between NFkappaB and Sp1 controls histone acetylation within the proximal promoter of the monocyte chemoattractant protein 1 gene. J Immunol 2003; 170:4139 - 4147; PMID: 12682245
- Ping D, Boekhoudt G, Zhang F, Morris A, Philipsen S, Warren ST, et al. Sp1 binding is critical for promoter assembly and activation of the MCP-1 gene by tumor necrosis factor. J Biol Chem 2000; 275:1708 - 1714; PMID: 10636866; http://dx.doi.org/10.1074/jbc.275.3.1708
- Akira S, Takeda K, Kaisho T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat Immunol 2001; 2:675 - 680; PMID: 11477402; http://dx.doi.org/10.1038/90609
- Takeuchi O, Hemmi H, Akira S. Interferon response induced by Toll-like receptor signaling. J Endotoxin Res 2004; 10:252 - 256; PMID: 15373970
- Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol 2002; 20:197 - 216; PMID: 11861602; http://dx.doi.org/10.1146/annurev.immunol.20.083001.084359
- O'Neill LA. How Toll-like receptors signal: what we know and what we don't know. Curr Opin Immunol 2006; 18:3 - 9; PMID: 16343886; http://dx.doi.org/10.1016/j.coi.2005.11.012
- Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine 2008; 42:145 - 151; PMID: 18304834; http://dx.doi.org/10.1016/j.cyto.2008.01.006
- Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science 2003; 300:1524 - 1525; PMID: 12791976; http://dx.doi.org/10.1126/science.1085536
- Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2001; 2:675 - 680; PMID: 11477402; http://dx.doi.org/10.1038/90609
- Schmitz ML, Bacher S, Kracht M. Ikappa B-independent control of NFkappaB activity by modulatory phosphorylations. Trends Biochem Sci 2001; 26:186 - 190; PMID: 11246025; http://dx.doi.org/10.1016/S0968-0004(00)01753-9
- Chen LF, Williams SA, Mu Y, Nakano H, Duerr JM, BuckBinder L, et al. NFkappaB RelA phosphorylation regulates RelA acetylation. Mol Cell Biol 2005; 25:7966 - 7975; PMID: 16135789; http://dx.doi.org/10.1128/MCB.25.18.7966-7975.2005
- Teferedegne B, Green MR, Guo Z, Boss JM. Mechanism of action of a distal NFkappaB-dependent enhancer. Mol Cell Biol 2006; 26:5759 - 5770; PMID: 16847329; http://dx.doi.org/10.1128/MCB.00271-06
- Ueda A, Okuda K, Ohno S, Shirai A, Igarashi T, Matsunaga K, et al. NFkappaB and Sp1 regulate transcription of the human monocyte chemoattractant protein-1 gene. J Immunol 1994; 153:2052 - 2063; PMID: 8051410
- Murphy TL, Cleveland MG, Kulesza P, Magram J, Murphy KM. Regulation of interleukin 12 p40 expression through an NFkappaB half-site. Mol Cell Biol 1995; 15:5258 - 5267; PMID: 7565674
- Sironi M, Guglielmotti A, Polentarutti N, Fioretti F, Milanese C, Romano M, et al. A small synthetic molecule capable of preferentially inhibiting the production of the CC chemokine monocyte chemotactic protein-1. Eur Cytokine Netw 1999; 10:437 - 442; PMID: 10477401
- Mirolo M, Fabbri M, Sironi M, Vecchi A, Guglielmotti A, Mangano G, et al. Impact of the anti-inflammatory agent bindarit on the chemokinome: selective inhibition of the monocyte chemotactic proteins. Eur Cytokine Netw 2008; 19:119 - 122; PMID: 18775807
- Akira S, Uematsu O, Takeuchi. Pathogen recognition and innate immunity. Cell 2006; 124:783 - 801; PMID: 16497588; http://dx.doi.org/10.1016/j.cell.2006.02.015
- Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol 2004; 22:891 - 928; PMID: 15032599; http://dx.doi.org/10.1146/annurev.immunol.22.012703.104543
- DeVries ME, Kelvin AA, Xu L, Ran L, Robinson J, Kelvin DJ. Defining the origins and evolution of the chemokine/chemokine receptor system. J Immunol 2006; 176:401 - 415; PMID: 16365434
- Zlotnik A, Yoshie O, Nomiyama H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol 2006; 7:243; PMID: 17201934; http://dx.doi.org/10.1186/gb-2006-7-12-243
- Lee SM, Suen Y, Qian J, Knoppel E, Cairo MS. The regulation and biological activity of interleukin 12. Leuk Lymphoma 1998; 29:427 - 438; PMID: 9643557; http://dx.doi.org/10.3109/10428199809050903
- Del Vecchio M, Bajetta E, Canova S, Lotze MT, Wesa A, Parmiani G, et al. Interleukin-12: biological properties and clinical application. Clin Cancer Res 2007; 13:4677 - 4685; PMID: 17699845; http://dx.doi.org/10.1158/1078-0432.CCR-07-0776
- Kim S, Chung EY, Ma X. Immunological consequences of macrophage-mediated clearance of apoptotic cells. Cell Cycle 2005; 4:231 - 234; PMID: 15655362; http://dx.doi.org/10.4161/cc.4.2.1428
- Sobell HM. Actinomycin and DNA transcription. Proc Natl Acad Sci USA 1985; 82:5328 - 5331; PMID: 2410919; http://dx.doi.org/10.1073/pnas.82.16.5328
- Sommers CD, Thompson JM, Guzova JA, Bonar SL, Rader RK, Mathialagan S, et al. Novel tight-binding inhibitory factor-kappaB kinase (IKK-2) inhibitors demonstrate target-specific anti-inflammatory activities in cellular assays and following oral and local delivery in an in vivo model of airway inflammation. J Pharmacol Exp Ther 2009; 330:377 - 388; PMID: 19478133; http://dx.doi.org/10.1124/jpet.108.147538
- Nowak DE, Tian B, Brasier AR. Two-step cross-linking method for identification of NFkappaB gene network by chromatin immunoprecipitation. Biotechniques 2005; 39:715 - 725; PMID: 16315372; http://dx.doi.org/10.2144/000112014
- Saccani S, Pantano S, Natoli G. Two waves of nuclear factor kappaB recruitment to target promoters. J Exp Med 2001; 193:1351 - 1359; PMID: 11413190; http://dx.doi.org/10.1084/jem.193.12.1351
- Grimaldi B, Nakahata Y, Sahar S, Kaluzova M, Gauthier D, Pham K, et al. Chromatin remodeling and circadian control: master regulator CLOCK is an enzyme. Cold Spring Harb Symp Quant Biol 2007; 72:105 - 112; PMID: 18419267; http://dx.doi.org/10.1101/sqb.2007.72.049
- Cardone L, Hirayama J, Giordano F, Tamaru T, Palvimo JJ, Sassone-Corsi P. Circadian clock control by SUMOylation of BMAL1. Science 2005; 309:1390 - 1394; PMID: 16109848; http://dx.doi.org/10.1126/science.1110689
- Nóvak EM, Dantas KC, Charbel CE, Bydlowski SP. Association of hepatic nuclear factor-4 in the apolipoprotein B promoter: a preliminary report. Braz J Med Biol Res 1998; 31:1405 - 1408; PMID: 9921275
- Hosoe S, Ogura T, Hayashi S, Komuta K, Ikeda T, Shirasaka T, et al. Induction of tumoricidal macrophages from bone marrow cells of normal mice or mice bearing a colony-stimulating-factor-producing tumor. Cancer Immunol Immunother 1989; 28:116 - 122; PMID: 2645051; http://dx.doi.org/10.1007/BF00199111
- Kim JB, Wright HM, Wright M, Spiegelman BM. ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc Natl Acad Sci USA 1998; 95:4333 - 4337; PMID: 9539737; http://dx.doi.org/10.1073/pnas.95.8.4333
- Srivastava GK, de Larrea GZ. UVB-induced murine bone marrow derived macrophages and apoptosis. Immunol Invest 2008; 37:293 - 313; PMID: 18569072; http://dx.doi.org/10.1080/08820130802083689
- Waheed A, Shadduck RK. Purification and properties of L cell-derived colony-stimulating factor. J Lab Clin Med 1979; 94:180 - 193; PMID: 313966
- Osborn L, Kunkel S, Nabel GJ. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappaB. Proc Natl Acad Sci USA 1989; 86:2336 - 2340; PMID: 2494664; http://dx.doi.org/10.1073/pnas.86.7.2336
- Brou C, Chaudhary S, Davidson I, Lutz Y, Wu J, Egly JM, et al. Distinct TFIID complexes mediate the effect of different transcriptional activators. EMBO J 1993; 12:489 - 499; PMID: 8440239
- Grimaldi B, Bellet MM, Katada S, Astarita G, Hirayama J, Amin RH, et al. PER2 controls lipid metabolism by direct regulation of PPARγ. Cell Metab 2010; 12:509 - 520; PMID: 21035761; http://dx.doi.org/10.1016/j.cmet.2010.10.005
- Piehler AP, Grimholt RM, Ovstebø R, Berg JP. Gene expression results in lipopolysaccharide-stimulated monocytes depend significantly on the choice of reference genes. BMC Immunol 2010; 11:21; PMID: 20441576; http://dx.doi.org/10.1186/1471-2172-11-21