Abstract
Aurora kinases are mitotic serine/threonine protein kinases and are attractive novel targets for anticancer therapy. Many small-molecule inhibitors of Aurora kinases are currently undergoing clinical trials. Aurora A kinase is essential for successful mitotic transition. MK8745 is a novel and selective small-molecule inhibitor of Aurora A kinase. MK8745 induced apoptotic cell death in a p53-dependent manner when tested in vitro in cell lines of multiple lineages. Cells expressing wild-type p53 showed a short delay in mitosis followed by cytokinesis, resulting in 2N cells along with apoptosis. However, cells lacking or with mutant p53 resulted in a prolonged arrest in mitosis followed by endoreduplication and polyploidy. Cytokinesis was completely inhibited in p53-deficient cells, as observed by the absence of 2N cell population. The induction of apoptosis in p53-proficient cells was associated with activation of caspase 3 and release of cytochrome c but was independent of p21. Exposure of p53 wild-type cells to MK8745 resulted in the induction of p53 phosphorylation (ser15) and an increase in p53 protein expression. p53-dependent apoptosis by MK8745 was further confirmed in HCT 116 p53-/- cells transfected with wild-type p53. Transient knockdown of Aurora A by specific siRNA recapitulated these p53- dependent effects, with greater percent induction of apoptosis in p53 wild-type cells. In conclusion, our studies show p53 as a determining factor for induction of apoptosis vs. polyploidy upon inhibition of Aurora A.
Introduction
Non-diploid DNA, a phenotypic hallmark of almost all human cancers often arises from inappropriate completion of mitosis. This finding led to the emergence of taxanes and the vinca alkaloids to block tumor progression by targeting the mitotic machinery. Later on, upon the discovery of mitotic signaling kinases, such as Aurora kinases, polo-like kinase (PLK) and kinesins, attempts to develop novel anticancer agents targeting these kinases began. Overexpression of Aurora A and B proteins are observed in many cancers.Citation1 Aurora A kinase is one of the many genes that can generate polyploid cells when it is overexpressed (80% of aneuploid breast cancers) due to abortive mitosis.Citation2 Other genes overexpressed, underexpressed or mutated in cancers have roles in cell cycle progression.Citation3,Citation4 Depletion of Aurora B activity causes premature exit from mitosis without cell division. This leads to endoreduplication, resulting in large polyploid nuclei containing multiple copies of the DNA.Citation5 On the other hand, depletion of Aurora A activity results in mitotic defect followed by apoptosis.Citation6 However, preclinical studies using small-molecule inhibitors targeting mitotic proteins/kinases suppress tumor growth by inducing polyploidy.Citation7–Citation10 In spite of the selectivity, the Aurora kinase inhibitors developed, including ZM447439, Hesperadin, AZD 1152HQPA (selective for Aurora B), MK0457 (pan Aurora inhibitor) and MLN 8054 (selective for Aurora A), are potent in suppressing tumor cell growth, mainly by inducing polyploidy.Citation11–Citation15 p53-dependent polyploidy check point activation was observed upon exposure to ZM447439 and MK0457.Citation11,Citation16 MLN 8054 has been shown to induce cellular senescence in a p53/p21 manner.Citation17 However, we have shown previously that AZD 1152HQPA, a specific inhibitor of Aurora B kinase, induces polyploidy independent of p53 status of the cell line.Citation15,Citation18 Even though many small-molecule inhibitors are undergoing clinical trials, it is still unclear whether polyploidy can be taken as a mechanism of cell death. It is also not certain whether polyploid cells eventually die or become aneuploid.
In a study using squamous cell cancer of the head and neck, overexpression of Aurora kinase A is associated with an additive antiproliferative effect upon combining pan Aurora inhibitor with EGFRCitation19 by inducing more polyploidy. Another study used Actinomycin D in normal fibroblasts with wild-type p53 to rescue cells from VX-680-induced polyploidy and cellular abnormalities.Citation20 The mechanism by which cells result in polyploidy is also not clear. p53-dependent post-mitotic check point activation is assumed to prevent cells from undergoing endoreduplication and polyploidy. However, effects of different Aurora kinase inhibitors are inconclusive on the p53 dependency.
In the current study, we utilized MK 8745, a selective inhibitor for Aurora A kinase with more than 100-fold selectivity for Aurora A over other kinases, including Aurora B (IC50 Aurora A, 0.6 nM and IC50 Aurora B, 280 nMa) (acorrespondence with Merck), as a tool to study the polyploid effect in the p53 background. Unlike Aurora B inhibition, MK8745 induced phospho H3 (ser10) and Aurora A protein level indicative of mitotic accumulation, making it difficult to assess the target specificity by checking the auto-phosphorylation (T288) of Aurora A. Therefore, we checked the specificity of MK8745 by in vitro kinase assay using a PLK1 peptide and Aurora A as substrates and either purified Aurora A or immunoprecipitated Aurora A from cells treated with or without MK8745 as kinase. Even though the IC50 for Aurora B has been reported to be in the nM range, we observed no inhibition of phospho histone H3 (ser10), an immediate target of Aurora B, with MK8745 concentrations up to 10 µM. We then explored the mechanism by which MK8745 induces proliferation inhibition in cell lines of multiple lineages, such as colon, melanoma, sarcoma and pancreatic. In general, the cell lines expressing wild-type p53 showed a short delay in mitosis followed by cytokinesis with significant apoptosis upon exposure to MK8745 in a dose-dependent manner. However, cell lines with null or mutant p53 underwent endoreduplication, resulting in polyploidy after a prolonged delay (about 40 h) in mitosis with no apoptosis. This was further confirmed by using isogenic knockout variants of colon carcinoma cell line, HCT.116. These results suggest p53 status could have a biologic impact on patients receiving Aurora A-targeted therapy.
Results
MK8745 is a specific inhibitor of Aurora A kinase.
In order to test the target specificity of MK8745 (, chemical structure) for Aurora A, an in vitro kinase assay was performed using purified Aurora A kinase and biotinylated PLK1 peptide (Ser137), known substrates of Aurora A, in the presence or absence of MK8745 (500 nM). As shown in , phosphorylation of PLK1 peptide was inhibited when the kinase assay was done in the presence of MK8745 but not with an Aurora B-specific inhibitor (ABI), AZD1152. A similar effect was observed when histone H3 was used as the substrate. T-288, an autophosphorylation site of Aurora A, was also inhibited () upon incubation with MK8745 when compared with vehicle control. The experiment was repeated using Aurora B kinase and histone H3 as substrate. No inhibition of Aurora B was observed in phosphorylating histone H3 upon incubation with MK8745 (), indicating the specificity of MK8745 for Aurora A and not Aurora B. shows induction of phospho H3 (ser10) and Aurora A protein due to mitotic accumulation. Mitotic delay and accumulation upon MK8745 treatment is depicted in by measuring cells positive for phospho MPM2, a mitotic marker. It has been shown that Aurora A phosphorylates p53 on ser315,Citation21 and therefore, we tested the effect of MK8745 on inhibiting phosphorylation of p53 on Ser 315 in HCT 116 cells (p53−/−) overexpressing p53. As expected, a decrease in phosphorylation was observed with MK8745 (), further confirming the specificity of MK8745 for Aurora A kinase. An immunoprecipitation assay using an antibody for p53 followed by western blot analysis for Aurora A shows an association of Aurora A with p53 in HCT 116 wild-type cells but no interaction with Aurora B ().
Aurora A inhibition induces apoptosis or polyploidy according to the p53 status of the cell.
We have previously shown that Aurora B inhibition induces polyploidy in a series of cell lines of multiple lineages independent of their p53 status without any cell cycle delay or arrest.Citation18 Therefore, in this study, we elected to examine the effects of MK8745 on cell cycle as well as on mitosis in HCT 116 isogenic clones (parental, p53−/− and p21−/−) by flow cytometry analysis after MPM2 and propidium iodide (PI) staining. As shown in , HCT 116 parental cell populations exposed to 5 µM MK8745 result in an accumulation of 4N DNA cells by 6 h. By 17 h, cells exit from mitosis, undergo cytokinesis, and apoptosis, as indicated by an increase in cells with 2N DNA along with an increased S phase, indicating DNA synthesis along with the appearance of apoptotic sub-G1 peak. Upon prolonged exposure, more cells undergo cytokinesis (increased 2N cells at 22, 30 and 40 h) along with an increase in apoptosis (sub-G1 cells with less than 2N DNA). The same experiment was repeated in HCT 116 p53−/−, and, as shown in (lower part), by 6 h, about 80% of the cells had 4N DNA with a disappearance of 2N cells. There was almost complete inhibition of cytokinesis and induction of endoreduplication as evidenced by the disappearance of 2N peak and a continuous increase of the 8N peak from 16% at 17 h to 60% at 40 h. The effects in HCT116 p21−/− cells (wild-type for p53, middle part of ) were similar to that of the parental cells (mitotic as well as apoptotic). As shown in (last column), the Aurora B inhibitor (ABI) induces over 70% polyploidy in all isogenic clones of HCT 116, showing p53 independence in induction of polyploidy upon Aurora B inhibition.
summarizes the end result of MK8745 on HCT cells at 24 h: parental cells undergo apoptosis; cells lacking p53 undergo polyploidy, and cells lacking p21 undergo apoptosis. Similar effects were observed at MK8745 concentrations ranging from 1 to 10 µM (data not shown).
In order to examine the changes in mitosis relative to the onset of cytokinesis (2N), polypolidy (> 4N) or apoptosis (< 2N), HCT 116 parental and p53−/− cells were released into 1 µM MK8745 after first being synchronized in G1 with thymidine. As shown in , HCT116 parental cells delay in mitosis (12–18 h, green) exit and undergo cytokinesis with increased apoptotic (red) and 2N (blue) population. On the other hand, p53−/− cells undergo a 40% mitotic arrest (green) for about 30–40 h, followed by exit and endoreduplication of DNA (increased polyploidy, gray and unchanged 2N, blue) as shown in . In Mad2 haploinsufficient cells, MK8745, as expected, lacked any mitotic effect but did induce a partial cytokinesis and endoreduplication effect, with the accumulation of both 2N and 8N cells (32%) (data not shown), though less 8N cells than observed in drug-treated p53-null cells (60%). This data would suggest that the manner of growth inhibition by MK8745 is dependent on the p53 status of the cells. Cells with wild-type p53 undergo apoptosis following Aurora A inhibition, whereas cells with mutant p53 that undergo endoreduplication result in polyploidy.
Induction of apoptosis upon Aurora A inhibition is dependent on p53 in all cell lineages tested.
This p53-dependent effect was further tested in cell lines of other lineages, including pancreatic, sarcoma and melanoma. shows the requirement of p53 in the induction of apoptosis upon Aurora A inhibition by MK8745. Pancreatic cells CAPAN2 (p53-positive) treated with 5 µM MK8745 for 48 h result in 15% apoptosis, and PANC1 (p53 mutant) results in 56% polyploidy. Similarly, the desmoplastic small round cell sarcoma cell line, DSCRT (p53-positive) treated with MK8745 results in 8% apoptosis, and the malignant peripheral nerve sheath cell line ST88 (p53 mutant) results in 18% polyploidy. For melanoma cells, SK-Mel32 (p53-positive) treated with MK8745 results in 9.6% apoptosis, and SK-Mel28 (p53 mutant) results in 40% polyploidy. These differences in the induction of apoptosis in these cell lines according to p53 status were confirmed by detecting cleaved PARP in cell lines expressing wild-type p53 (). These results confirm the requirement of p53 in inducing apoptosis when Aurora A kinase is inhibited across the cell lines.
This was further confirmed by time-lapse video microscopy using HCT 116 cells (wild-type for p53 and p53−/−) transiently transfected with GFP H2B followed by treatment with MK8745 (Movies 1A and 1B). depicts clips 4 h post-treatment with MK8745 in HCT116 parental cells clearly showing cytokinesis after a very short delay in mitosis (circled in orange) and apoptotic cell death (circled in red). shows clips from the movie taken after 18 h of exposure to MK8745 showing delay in mitosis (circled in orange), inhibition of cytokinesis and polyploidy (enlarged nuclei, circled in blue) in p53−/− cells. This further confirms the finding that cells with wild-type p53 undergo apoptosis following Aurora A inhibition, whereas cells with mutant p53 undergo endoreduplication and polyploidy. The p53-dependent mitotic exit seems to be unique upon Aurora A inhibition, because taxotere showed similar mitotic delay in both p53-null and wild-type cells (not shown).
Induction of apoptosis and its p53 dependence is due to specific inhibition of Aurora A.
In order to determine whether the effect of MK8745 on apoptosis and polyploidy was simply an off-target effect of the drug or was due to a specific effect of Aurora A inhibition, Aurora A kinase was inhibited by using specific siRNA sequences. is a comparison of flow cytometric analysis of HCT 116 and HCT 116 p53−/− cells following treatment with MK8745 or specific siRNA for Aurora A or Aurora B or Aurora A followed by MK8745. The effect of apoptosis (11.3%) upon exposure to MK8745 for 24 h could be recapitulated by using Aurora A siRNA (19.5%) in HCT parental cells. There was no additional effect upon treatment with MK8745 after downregulating Aurora A, probably due to nearly complete suppression of the target. Similarly, p53-null cells resulted in polyploidy (16.1%) upon Aurora A siRNA treatment, comparable to MK8745 (23.5%) treatment for 24 h. Here also there was no significant increase in polyploidy when Aurora A downregulated cells were treated with MK8745. In contrast, the suppression of Aurora B by siRNA in both isogeneic HCT116 cell lines (wild-type and null for p53) resulted in 50% polyploidy (last column of ). DAPI staining confirmed that inhibition of Aurora A by either MK8745 or siRNA both resulted in de-condensed nuclei, consistent with apoptosis in HCT parental cells, whereas HCT p53−/− cells underwent endoreduplication resulting in large multilobed nuclei (). Induction of apoptosis, as determined by DAPI staining, annexin staining or examination or the sub-G1 peaks, was comparable in HCT parental cells following treatment with control siRNA, MK8745 or Aurora A siRNA for 72 h ().
To further confirm that p53-dependent effects of MK8745 were due to Aurora A inhibition, we compared the effects of MK8745 and siRNA for Aurora A and Aurora B on p53 expression and the induction of apoptosis by western blot. shows an increased phosphorylation of p53 (ser15) with an induction of p53 and p21 proteins upon treatment with MK8745. There was also an increase in cleaved PARP, cleaved caspase 3 and an increase in cytochrome C release to cytosol. As shown, Aurora A downregulation by siRNA could recapitulate the effects of MK8745, supporting the fact that the pro-apoptotic effects of MK8745 were due to its target-specific inhibition of Aurora A. As shown in , the addition of MK8745 to Aurora A downregulated cells did not appreciably increase PARP cleavage, as it was maximally induced with Aurora A siRNA alone. In contrast, the induction of apoptosis in siRNA downregulated Aurora B cells was only induced with MK8745 but not with ABI, supporting the fact that Aurora A targeting is crucial for the induction of apoptosis. Furthermore, apoptosis in HCT 116 p53−/− cells was negligible (no cleaved PARP or caspase 3) upon inhibition of Aurora A by both siRNA or with drug (), indicating the p53 dependency for this process.
Overexpression of p53 in p53-null HCT116 cells induces apoptosis rather than polyploidy.
To further evaluate the role of p53 in inducing apoptosis upon Aurora A inhibition, p53 was overexpressed in the p53−/− cells, and the effect of MK8745 was tested. As shown in , overexpressing p53 in the p53-null background induced a degree of PARP cleavage that was comparable to parental HCT116 cells upon treatment with MK8745. This was confirmed by QFM with DAPI staining (). In the p53-overexpressing cells, MK8745 treatment increased apoptosis from 6% in the vector alone controls to 21% in the p53-overexpressing cells.
We then examined the time course for induction of polyploidy and apoptosis by 5 µM MK8745 in the HCT p53−/− cells overexpressing p53 (p53−/− + p53) and compared this to the effects of the drug in the HCT116 parental and HCT116 p53−/− cells. After 10 h of exposure, parental cells start to undergo apoptosis (indicated by the increased < 2N DNA, , blue line bottom). p53−/−, on the other hand, resulted in little apoptosis (5%, up to 52 h of drug exposure, , red line bottom). p53-null cells overexpressing wild-type p53, however, induced the same amount of apoptosis (20% with 52 h of exposure) as HCT parental cells. But the onset of apoptosis was delayed (20 h) as compared with parental cells (10 h), possibly due to a delay in mitotic exit. Polyploidy was also measured by DNA content, and as shown in (top), parental cells did not result in polyploidy; p53−/− cells started to undergo endoreduplication at 20 h and increased to 60% at 52 h (red). However, p53-null cells overexpressing p53 did not exactly mimic parental cells, but the percentage of polyploid cells was still decreased to 30%.
In order to explain the reduced polyploidy, an immunofluorescence assay was performed for p53 in p53−/− cells overexpressing p53 to check the transfection efficiency. As shown in , DAPI-stained decondensed nuclei (circled, arrowheads) represent apoptosis. Enlarged polyploid nucle were also detected. When the immunodetected p53 expression level was merged with DAPI, p53 was present in most of the decondensed cells. Therefore, it would appear that the inability to completely suppress polyploidy was due to the continued endoreduplication of cells that were not successfully transfected with p53.
Discussion
Aurora kinases were identified as potential targets for cancer therapy based on their overexpression in various tumors as well as the roles they play during mitosis. Many small-molecule inhibitors have been developed and are being tested in clinic. We and others have reported that inhibiting Aurora B results in endoreduplication and polyploidy (> 4N cells) independent of the p53 status of the cell. Many of the small-molecule Aurora kinase inhibitors target both Aurora A and B. However, these inhibitors similarly induce polyploidy. Thus, non-selective Aurora A/B inhibitors essentially act as inhibitors of Aurora B, masking the effects of inhibiting Aurora A. In contrast, specific inhibitors of Aurora A induce an accumulation of cells in mitosis. Two other small-molecule inhibitors shown that selectively targeted Aurora A were MLN8054 and MK-5108.Citation22,Citation23 MK-5108 was shown to inhibit tumor growth in xenograft models. Using the Aurora A-specific inhibitor MK8745, we elected to determine the fate of these cells (polyploidy vs. apoptosis) and to determine whether this was affected by the p53 status of the cell.
MK8745 induced a very significant inhibition of proliferation in almost all the cell lines tested. Upon closer examination, we found a differential effect of MK8745 on the cells depending upon the p53 status of the cell line. In general, cell lines expressing wild-type p53 resulted in significant apoptosis, whereas cells lacking or defective in p53 underwent endoreduplication and polyploidy. Both processes resulted in an inhibition of cell growth. This p53 dependency for Aurora A inhibition is very distinct from that of Aurora B inhibition, in which all cell types resulted in polyploidy independent of the p53 status.Citation18 However, in order to rule out any off-target effect on Aurora B, we used Aurora B kinase with histone H3 in an in vitro kinase assay, and no inhibition was observed, with MK8745 further confirming MK8745 as an inhibitor of Aurora A and not Aurora B. p53 (S315), PLK (S137), Aurora A (T288), known substrates of Aurora ACitation21 also used to further validate target inhibition in the in vitro assay.
A closer examination of Aurora A inhibition on cell cycle showed a delay (up to 18 h) in mitosis in cells expressing wild-type p53, unlike other mitotic spindle poisons. Following the short delay, cells exit mitosis and undergo cytokinesis with an induction in apoptosis (time-lapse, Movie 1A). However, p53-deficient cells manage to arrest cells in mitosis for over 40 h, followed by mitotic exit and endoreduplication (time-lapse, Movie 1B). As we have shown earlier, upon inhibition of Aurora B, cells went on cycling without cytokinesis or any cell cycle checkpoint activation resulting in polyploidy despite the p53 status. Therefore, the inhibition in cell growth occurs either by apoptosis or polyploidy, depending on the p53 status. A p53 dependency on induction of polyploidy was shown earlier using MLN8054; however, unlike their finding,Citation22 polyploidy formation is shown to be independent of p21 in our results. These effects were a direct result of the target-specific inhibition of Aurora A, as confirmed by mimicking the drug effect by specifically downregulating Aurora A using siRNA. The importance of p53 in inducing apoptosis in the context of Aurora A inhibition was further confirmed in p53-null cells transiently overexpressing p53. Though p38α has been shown to be involved in mitotic progression of p53−/− tetraploids, we could not find the effect in our experimental conditions.Citation24
Our results clearly indicate a differential effect of Aurora A kinase inhibition on inhibiting cell growth. In contrast to Aurora B inhibition, which induces polyploidy in all cells, Aurora A inhibition will induce polyploidy or apoptosis according to the cells p53 status. Both processes will induce a decrease in cell number. However, though apoptosis would seem to be the preferred outcome for drug therapy, it is unclear which of these two processes will result in the greatest clinical benefit. Therefore, with the entry of Aurora A inhibitors into the clinic, it will be important to evaluate the p53 status of the cell in order to determine whether there may an identifiable patient population who would most benefit from the targeting of Aurora A.
Materials and Methods
Cell culture.
HCT 116 human colon carcinoma cell line and its isogenic variants for p53-null (HCT p53−/−) and p21-null (HCT p21−/−) were a generous gift from Dr. Bert Vogelstein (Johns Hopkins University). Cultures were maintained at 37°C in the presence of 5% CO2 in McCoy's Media supplemented with 10% heat-inactivated fetal bovine serum, 100 units/mL penicillin and 100 µg/mL streptomycin. PANC1, CAPAN2, DSCRT, HSSY and SYO cells were purchased from American Type Culture Collection (ATCC) and maintained in DME media supplemented with 10% heat inactivated fetal bovine serum and 100 µg/mL streptomycin and 100 units/mL penicillin. Cutaneous melanoma cell lines (SK-mel 11, 19, 23, 28, 29, 30, 32, 103, 130, 133 and 173) were a generous gift from Alan Houghton, MD (Memorial Sloan Kettering Cancer Center) and ocular melanoma cell lines (OMM1.3, Mel202, Mel270) were kindly provided by Dr. Bruce Ksander (Harvard Medical School); OCM1A and 92.1 were from Dr. William Harbour (Washington University), and OCM3 was from Robert Folberg (University of Illinois); all were maintained in RPMI supplemented with 10% heat inactivated fetal bovine serum, 100 µg/ mL streptomycin and 100 units/mL penicillin. Sarcoma cell lines MPNST, ST88, LS141 and DDLS were kindly provided by Sam Singer (Memorial Sloan Kettering Cancer Center) and were maintained in RPMI supplemented with 10% heat inactivated fetal bovine serum and 100 µg/mL streptomycin and 100 units/mL penicillin. MKN-74 (gastric cell line) was maintained in MEM supplemented with 10% heat inactivated fetal bovine serum and 100 µg/mL streptomycin and 100 units/mL penicillin. MK 8745 was obtained from Merck and Co., Inc. AZD 1152 was purchased from Selleckchem.
Colorimetric cell proliferation assay.
The assay was done as per the manufacturer's protocol (Dojindo Molecular Technologies, Inc.). Briefly, 2,000 cells were plated in 100 µL volume per well of a 96-well plate, and treatments were done 24 h after plating. After the desired incubation period with the drug, 10 µL of CCK-8 solution was added to each well, which were further incubated at 37°C for 1 to 4 h. This assay quantifies the amount of formazan dye generated by the activity of dehydrogenases in the cells that is directly proportional to the number of living cells. Then the optical density at 450 nm to determine the cell viability was measured using Spectra Max 340 PC (Molecular Devices Corp.).
Apoptosis assay.
Cells were treated with MK8745 as indicated in each experiment, and FITC Annexin V (BD PharMingen) was used to determine the percentage of cells within a population that were apoptotic as per the manufacturer's protocol.
Quantitative fluorescent microscopy (QFM).
Cells were collected as above after drug treatment and fixed in 3% paraformaldehyde. The nuclear morphology of cells was examined using fluorescence microscopy after staining with 4′,6-diamidino-2-phenylindole (DAPI) to a final concentration of 25 µg/mL. Cells were scored as apoptotic based on the presence of decondensed fragmented chromatin. A minimum of 400 cells were counted for each sample and taken as a percentage of untreated cells.
Flow cytometry.
For flow cytometry, the cells were trypsinized, washed and fixed in 75% ice-cold ethanol. Cells were either stained with propidium iodide (50 µg/mL) containing RNase (5 µg/mL) for the measurement of DNA content or labeled with the phospho MPM-2 monoclonal antibody (Millipore), which recognizes specific phosphorylated epitopes present only during mitosis and subsequently with FITC-conjugated antimouse secondary antibody (ICN/Jackson Immuno Research) for the mitotic index measurement. Cells were then treated with RNase and propidium iodide. Samples were analyzed on a FACScan (Becton Dickinson) for cell cycle distribution and mitotic index (percentage of MPM-2-positive cells) using the Cell Quest software. For this analysis, 10,000 events were examined per sample.
Immunofluorescence assay. HCT-116 p53−/− cells were plated in 2-well chamber slides and transfected with control plasmid or plasmid with wild-type p53. Each group was treated with vehicle or MK8745, and cells were fixed in 4% paraformaldehyde for 10 min at room temperature. The cells were then blocked in 2% goat serum followed by incubation in rabbit polyclonal anti-p53 (Santa Cruz Biotechnology; 1:250) and subsequently with Alexa Fluor-conjugated secondary antibody (Molecular Probes). DNA was labeled with DAPI at a final concentration of 25 µg/ mL for 10 min.
siRNA transfection.
Cells were plated on 60-mm plates, and transfections using lipofectamine RNAiMAX (Invitrogen) were performed according to the manufacturer's protocol. The siRNA sequences for Aurora A (Aurora-A, 725AUG CCC UGU CUU ACU GUC A743)Citation25 and Aurora B (5′-AAC GCG GCA CUU CAC AAU UGA-3′)Citation26 were purchased from Dharmacon Research, and control siRNA was purchased from Santa Cruz Biotechnology.
Plasmid transfections.
For plasmid transfections, cells were plated in 70% confluence, and the transfections were performed on the same day in 60-mm tissue culture plates using Fugene 6 transfection reagent (Roche) in OPTI MEM 1 media (Invitrogen) and 2 µg of pcDNA p53 (Invitrogen), GFP H2B (Addgene) or empty vector were used per plate. Treatments were performed 16 h after transfection.
Cell extraction, immunoprecipitation and immunoblotting.
Cell lysates were prepared by lysis of both floating and adherent cells in whole-cell lysis buffer (50 mmol/L Tris, pH 8, 250 mmol/L NaCl, 0.5% NP-40, 0.2 mmol/L EDTA, 2 mmol/L EGTA, 10% Glycerol, 0.1 mmol/L phenylmethylsulfonylfluoride, 0.1 mmol/L Na3VO4, 1 mmol/L DTT, 1 µg/mL leupeptin, 2 µg/mL aprotinin, 1 µg/mL pepstatin), allowed to lyse on ice for 10 min, syringed and cleared by centrifugation in a microcentrifuge at 13,000 rpm for 10 min at 4°C. Fifty micrograms of protein were fractionated by SDS-PAGE and transferred onto Immobilon membranes (Millipore). After blocking with 5% nonfat milk, membranes were probed with primary antibodies. The following antibodies were used in this study: mouse monoclonal to cleaved poly (ADP-ribose) polymerase (PARP), rabbit polyclonal to phospho p53 (15 and 315), rabbit polyclonal to cleaved caspase 3 mouse monoclonal anti-p21, rabbit polyclonal to Aurora A and rabbit polyclonal to phospho Aurora A were purchased from Cell Signaling, mouse monoclonal to cytochrome C was from BD Biosciences; mouse monoclonal to p53 was from Santa Cruz Biotechnology; α-tubulin was from Millipore. Bound primary antibodies were detected with horseradish peroxidase-conjugated secondary antibodies (ICN/Jackson ImmunoResearch) and visualized by enhanced chemiluminescence reagent (Amersham Pharmacia).
Time-lapse microscopy.
HCT116 cells wild type for p53 or null for p53 were transiently transfected with green fluorescent protein (GFP)-histone 2B (Addgene, 1 Kendall Sq. Ste. B7102, Cambridge, MA 02139) on 4-well chambered coverglass for 24 h and treated with either DMSO vehicle or 1 µM MK8745. The chamber was mounted onto the stage of a Zeiss Axiovert 200 M inverted microscope (Carl Zeiss Microimaging) maintaining normal growth condition with Solent Scientific microscope live imaging incubation system. Confocal images of the cells were acquired by a spinning disk confocal system (UltraView) with a 20x objective lens (0.8 NA) using a cooled EM-CCD camera (iXon+, Andor Technology) every 5 min after 4 h of treatment in parental HCT116 and 18 h after treatment in p53-null cells, with exposure time limited to 4–5 sec/image. For each time point, images were taken with 14 different focal planes along the z-axis 6 µm apart. Imaging data were analyzed using MetaMorph (Molecular Devices).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Figures and Tables
Figure 1 MK8745 is a selective inhibitor of Aurora A kinase. (A) Chemical structure of MK8745. (B) In vitro kinase assay. (i) Using plk peptide or Histone H3 substrate and purified Aurora A Kinase with (MK), without (ND) 500 nM MK8745 or with 500 nM AZD1152 (ABI). Phospho plk1 (S137), phospho H3 (S10) and Aurora A were detected by western blot analysis. (ii) Using purified Aurora A kinase as substrate with or without MK followed by western blot analysis for phospho (T288) Aurora A. (iii) Using Histone H3 as substrate and Aurora B kinase with or without MK followed by western blot analysis for phospho histone (Ser10) H3. (iv) Western blot analysis for phospho H3 (S10), Aurora A and tubulin in lysates of HCT 116 cells treated with MK for 24 h. (v) Flow cytometry analysis of HCT cells treated with (MK) or without (ND) MK for 24 h probed for MPM2, a mitotic marker. (C) Western blot analysis of HCT p53−/− cells overexpressing p53 treated with (MK) or without (ND) probed for phospho (Ser315) p53 and p53. (D) Coimmunoprecipitation of p53 with Aurora A and not with Aurora B in HCT 116 cells. All the results shown are representative of 3–4 independent experiments.
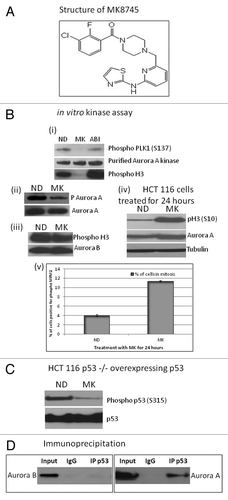
Figure 2 Cell cycle effect and induction of apoptosis by MK8745 (5 µM) in isogenic variants of HCT-116 cells (parental, p53−/−, p21−/−). (A) Flow cytometry analysis of the HCT 116 (top) and its isogenic variants; p21−/− (middle) and p53−/− (bottom) upon exposure to MK for different time points (6, 17, 22, 30 and 40 h) and exposure to ABI (100 nM AZD 1152) for 40 h after Propidium Iodide staining. (B) HCT116, p53−/− and p21−/− cells treated with 5 µM MK for 24 h and analyzed for its DNA content after staining with Propidium Iodide by flow cytometry analysis. Polyploidy (8N, blue bars) and apoptosis (<2N, red bars). (C) Mitotic population determined by flow cytometry analysis after probing for phospho MPM2, a mitotic marker, in all the three cell lines. HCT116 (i) and p53−/− (ii) cells were double thymidine blocked and released to 1 µM MK, harvested at 6, 9, 12, 18 and 30 h and analyzed for its DNA content after staining with Propidium Iodide and mitotic population after phospho MPM2 staining by flow cytometry analysis. All the results are representative of 3–4 independent experiments.
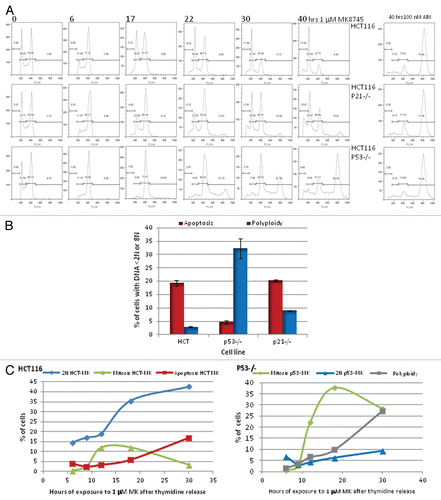
Figure 3 MK 8745 induces apoptosis in a p53 dependent manner. (A) Cell lines different lineages; (i) colon (HCT116 and its p53 null variant), (ii) pancreatic (PANC1, p53 mutant and CAPAN2, p53 wild type), (iii) sarcoma (ST88, p53 mutant and DSCRT, p53 wild type) and (iv) melanoma (SKMel 28, p53 mutant and SKMel 32, p53 wilt type) were treated with MK8745 (5 µM) for 48 h and harvested for flow cytometry for the determination of cell cycle distribution. (B) All the cell lines in (A) were treated with MK8745 and harvested for western blot analysis for induction of PARP cleavage. Results shown are representative of at least three independent experiments.
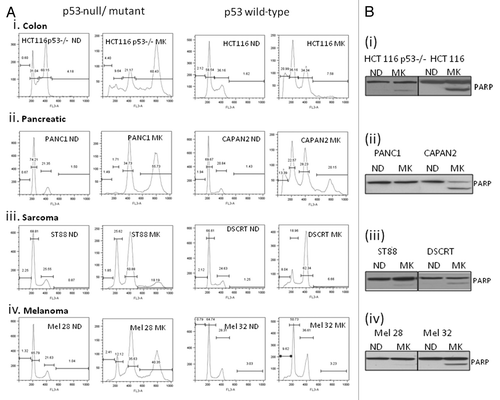
Figure 4 Treatment of MK8745 induces differential effect based on the p53 status. HCT116 and HCT 116 (p53−/−) cells transfected with GFP-histone 2B (A and B) for 24 h were treated with 1 µM MK8745 and live cell imaging was done in parental cells after 4 h and in p53−/− cells 18 h of exposure to MK8745 as described in Materials and Methods. Shown are selected still images from time-lapse movies; the fate of individual cells was tracked over time (in circles). (A) Parental HCT cells enter mitosis (in orange circle) most undergo cytokinesis and eventually undergo apoptosis (in red circle) (Sup. Video 1A data). (B) HCT p53−/− cells enter mitosis (in orange circle), and after a prolonged stay undergo endoreduplication results in polypolidy (in white circle) (Sup. Video 1B data).
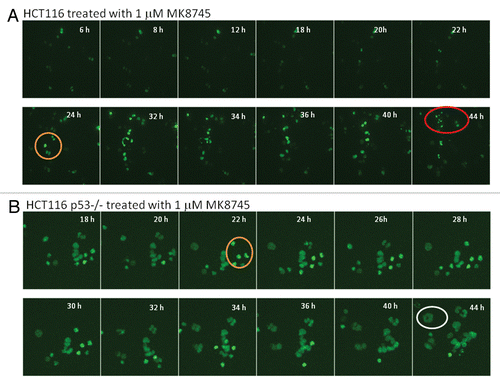
Figure 5 Downregulation of Aurora A recapitulates the effects of MK8745. (A) Aurora A was inhibited in HCT116 and its p53−/− cell lines with 5 µM MK (MK) or by downregulating with specific siRNA sequence (ASi) and treatment with with 5 µM MK (ASi+MK), or by downregulating Aurora B with specific siRNA sequence (BSi) along with control siRNA (CSi) and flow cytometry analysis was performed after staining with propidium iodide. (B) HCT 116 and p53−/− cells were treated with (MK, 5 µM) for 24 h was compared with 48 h of control (CSi) or Aurora A siRNA (ASi) and nuclei were visualized after staining with DAPI. (C) HCT116 cells were treated with 5 µM MK or siRNA specific for Aurora A for 48 h and apoptosis was measured by FACScan analysis after probing for Annexin (gray bar), after staining with Propidium iodide for <2N DNA (white bar) or quantitated using microscopy after staining with DAPI (black bar). (D) (i and iii) HCT 116 and p53−/− cells were treated with (MK, 5 µM) for 24 h was compared with 48 h of control (CSi) or Aurora A siRNA (ASi) and western blot analysis was performed for phospho p53 (ser15), p53, p21, cleaved PARP and cleaved caspase3. Aurora A downregulation is shown and cytochrome C release to cytosol. HCT 116 cells were either transfected with CSi, ASi or BSi were treated with vehicle, MK or ABI (AZD 100 nM) and western blot analyses were performed for Aurora A, phospho Aurora A, Aurora B, cleaved PARP, phospho Histone H3 (ser10) and tubulin. All results are representative of 3–4 independent experiments.
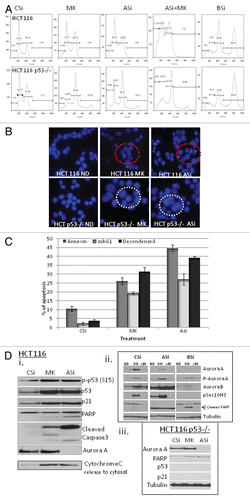
Figure 6 p53 is critical in determining the fate of the cell when Aurora A is inhibited. (A) (i) HCT116 cells and p53−/− cells either transfected with vector or with p53 were treated (MK, 5 µM for 24 h) western blot analysis was performed for PARP cleavage and expression of p53. (ii) Apoptosis was quantitated by microscopy after staining with DAPI (gray, DMSO and black, MK). (B) Polyploidy and apoptosis were quantitated by FACScan analysis after staining with PI in all conditions mentioned in the previous experiment at 0, 6, 18, 24, 30, 44 and 52 h. (C) p53−/− cells overexpressing p53 were treated with MK (5 µM) for 24 h and immunofluorescence assay was performed for p53 (green) followed by DAPI stain (blue). All the results shown are representative of 3–4 independent experiments.
Additional material
Download Zip (113 MB)Acknowledgements
IC50 data was provided by Toshiyasu Shimomura and Mitsuru Ohkubo of Merck and Co., Inc. Merck provided MK8745 for this project.
References
- Naruganahalli KS, Lakshmanan M, Dastidar SG, Ray A. Therapeutic potential of Aurora kinase inhibitors in cancer. Curr Opin Investig Drugs 2006; 7:1044 - 1051; PMID: 17209521
- Meraldi P, Honda R, Nigg EA. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53−/− cells. EMBO J 2002; 21:483 - 492; PMID: 11847097; http://dx.doi.org/10.1093/emboj/21.4.483
- Nigg EA. Centrosome aberrations: cause or consequence of cancer progression?. Nat Rev Cancer 2002; 2:815 - 825; PMID: 12415252; http://dx.doi.org/10.1038/nrc924
- Ho CC, Hau PM, Marxer M, Poon RY. The requirement of p53 for maintaining chromosomal stability during tetraploidization. Oncotarget 2010; 1:583 - 595; PMID: 21317454
- Kawasaki A, Matsumura I, Miyagawa Ji, Ezoe S, Tanaka H, Terada Y, et al. Downregulation of an AIM-1 kinase couples with megakaryocytic polyploidization of human hematopoietic cells. J Cell Biol 2001; 152:275 - 287; PMID: 11266445; http://dx.doi.org/10.1083/jcb.152.2.275
- Rojanala S, Han H, Muñoz RM, Browne W, Nagle R, Von Hoff DD, et al. The mitotic serine threonine kinase, Aurora-2, is a potential target for drug development in human pancreatic cancer. Mol Cancer Ther 2004; 3:451 - 457; PMID: 15078988
- Elhajouji A, Cunha M, Kirsch-Volders M. Spindle poisons can induce polyploidy by mitotic slippage and micronucleate mononucleates in the cytokinesis-block assay. Mutagenesis 1998; 13:193 - 198; PMID: 9568594; http://dx.doi.org/10.1093/mutage/13.2.193
- Cross SM, Sanchez CA, Morgan CA, Schimke MK, Ramel S, Idzerda RL, et al. A p53-dependent mouse spindle checkpoint. Science 1995; 267:1353 - 1356; PMID: 7871434; http://dx.doi.org/10.1126/science.7871434
- Khan SH, Wahl GM. p53 and pRb prevent rereplication in response to microtubule inhibitors by mediating a reversible G1 arrest. Cancer Res 1998; 58:396 - 401; PMID: 9458079
- Lanni JS, Jacks T. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol Cell Biol 1998; 18:1055 - 1064; PMID: 9448003
- Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, et al. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2 and Cenp-E to kinetochores. J Cell Biol 2003; 161:267 - 280; PMID: 12719470; http://dx.doi.org/10.1083/jcb.200208091
- Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol 2003; 161:281 - 294; PMID: 12707311; http://dx.doi.org/10.1083/jcb.200208092
- Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med 2004; 10:262 - 267; PMID: 14981513; http://dx.doi.org/10.1038/nm1003
- Manfredi MG, Ecsedy JA, Meetze KA, Balani SK, Burenkova O, Chen W, et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci USA 2007; 104:4106 - 4111; PMID: 17360485; http://dx.doi.org/10.1073/pnas.0608798104
- Nair JS, de Stanchina E, Schwartz GK. The topoisomerase I poison CPT-11 enhances the effect of the Aurora B kinase inhibitor AZD1152 both in vitro and in vivo. Clin Cancer Res 2009; 15:2022 - 2030; PMID: 19276280; http://dx.doi.org/10.1158/1078-0432.CCR-08-1826
- Gizatullin F, Yao Y, Kung V, Harding MW, Loda M, Shapiro GI. The Aurora kinase inhibitor VX-680 induces endoreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint function. Cancer Res 2006; 66:7668 - 7677; PMID: 16885368; http://dx.doi.org/10.1158/0008-5472.CAN-05-3353
- Schmidt S, Essmann F, Cirtea IC, Kuck F, Thakur HC, Singh M, et al. The centrosome and mitotic spindle apparatus in cancer and senescence. Cell Cycle 2010; 9:4469 - 4473; PMID: 21088502; http://dx.doi.org/10.4161/cc.9.22.13684
- Nair JS, Ho AL, Tse AN, Coward J, Cheema H, Ambrosini G, et al. Aurora B kinase regulates the post-mitotic endoreduplication checkpoint via phosphorylation of the retinoblastoma protein at serine 780. Mol Biol Cell 2009; 20:2218 - 2228; PMID: 19225156; http://dx.doi.org/10.1091/mbc.E08-08-0885
- Hoellein A, Pickhard A, von Keitz F, Schoeffmann S, Piontek G, Rudelius M, et al. Aurora kinase inhibition overcomes cetuximab resistance in squamous cell cancer of the head and neck. Oncotarget 2011; 2:599 - 609; PMID: 21865609
- Rao B, van Leeuwen IM, Higgins M, Campbel J, Thompson AM, Lane DP, et al. Evaluation of an Actinomycin D/VX-680 aurora kinase inhibitor combination in p53-based cyclotherapy. Oncotarget 2010; 1:639 - 650; PMID: 21317459
- Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet 2004; 36:55 - 62; PMID: 14702041; http://dx.doi.org/10.1038/ng1279
- Kaestner P, Stolz A, Bastians H. Determinants for the efficiency of anticancer drugs targeting either Aurora-A or Aurora-B kinases in human colon carcinoma cells. Mol Cancer Ther 2009; 8:2046 - 2056; PMID: 19584233; http://dx.doi.org/10.1158/1535-7163.MCT-09-0323
- Shimomura T, Hasako S, Nakatsuru Y, Mita T, Ichikawa K, Kodera T, et al. MK-5108, a highly selective Aurora-A kinase inhibitor, shows antitumor activity alone and in combination with docetaxel. Mol Cancer Ther 2010; 9:157 - 166; PMID: 20053775; http://dx.doi.org/10.1158/1535-7163.MCT-09-0609
- Vitale I, Jemaà M, Senovilla L, Galluzzi L, Rello-Varona S, Metivier D, et al. Involvement of p38alpha in the mitotic progression of p53(−/−) tetraploid cells. Cell Cycle 2010; 9:2823 - 2829; PMID: 20686359; http://dx.doi.org/10.4161/cc.9.14.12254
- Kufer TA, Silljé HH, Körner R, Gruss OJ, Meraldi P, Nigg EA. Human TPX2 is required for targeting Aurora-A kinase to the spindle. J Cell Biol 2002; 158:617 - 623; PMID: 12177045; http://dx.doi.org/10.1083/jcb.200204155
- Lampson MA, Kapoor TM. The human mitotic checkpoint protein BubR1 regulates chromosome-spindle attachments. Nat Cell Biol 2005; 7:93 - 98; PMID: 15592459; http://dx.doi.org/10.1038/ncb1208