Abstract
The atypical protein kinase C (PKC) isoform zeta (PKCζ) has been implicated in the intracellular transduction of mitogenic and apoptotic signals by acting on different signaling pathways. The key role of these processes in tumorigenesis suggests a possible involvement of PKCζ in this event. PKCζ is activated by cytotoxic treatments, inhibits apoptotic cell death and reduces the sensitivity of cancer cells to chemotherapeutic agents. Here, using pharmacological and DNA recombinant approaches, we show that oxidative stress triggers nuclear translocation of PKCζ and induces resistance to apoptotic agents. Accordingly, chemoresistant cells show accumulation of PKCζ within the nucleus, and a nuclear-targeted PKCζ transfected in tumor cells decreases sensitivity to apoptosis. We thus developed a novel recombinant protein capable of selectively inhibiting the nuclear fraction of PKCζ that restored the susceptibility to apoptosis in cells in which PKCζ was enriched in the nuclear fraction, including chemoresistant cells. These findings establish the importance of PKCζ as a possible target to increase the effectiveness of anticancer therapies and highlight potential sites of intervention.
Introduction
The protein kinase C (PKC) is a ubiquitous family of enzymes that consists of at least 10 structurally related serine/threonine kinases subdivided into three classes: classical (cPKC: α, βI, βII and γ), novel (nPKC: δ, ε, η and θ) and atypical (aPKC: λ, ι and ζ); in particular, the last are insensitive to both Ca2+ and diacylglycerol, whereas they are activated by lipid components, such as phosphatidylinositol, phosphatidic acid, arachidonic acid and ceramide.Citation1
PKCs isoforms are highly conserved in evolutionary terms, and when activated, PKCs can be translocated from one intracellular compartment to another, affecting a wide variety of cellular processes. The high degree of conservation of the individual PKC isoforms across various mammalian species indicates that each could have a specific relevance for the organism.Citation2 In the last years, the PKCs have been the subject of attention in a wide range of pathological processes, ranging from cancer to degenerative disorders.Citation3–Citation5
Several unique structural aspects of PKCs make them highly susceptible targets of oxidative stress. In particular, both the regulatory and the catalytic domains of PKC contain cysteine-rich regions that are targets for redox regulation.Citation6 Modification of these redox-sensitive regions interferes with cellular PKC activity.Citation7 Given that redox stress has been proposed to be involved in the intracellular pathogenesis and maintenance of several neoplastic modifications,Citation8 this class of kinases has become a logical candidate to mediate the pathological transduction of redox stress in cancer.
The atypical PKC isoform zeta (PKCζ) has been implicated in the intracellular transduction of mitogenic and apoptotic signals by acting on different signaling pathways. The key role of these processes in tumorigenesis suggests a possible involvement of PKCζ in this event.Citation9–Citation13 PKCζ has been reported to be antiapoptoticCitation9 and reduces the sensitivity of cancer cells to chemotherapeutic agents.Citation14,Citation15
Here, we demonstrate that atypical PKC isoform ζ, in oxidative stress condition, translocates into the nucleus, inducing resistance to apoptotic agents. Moreover, a recombinant nuclear-PKCζ inhibitor restores apoptotic susceptibility in chemoresistant cells, which present an enrichment of nuclear PKCζ expression. These findings establish the importance of the nuclear PKCζ fraction in sustaining intracellular tumor pathways that allow cancer cells to become drug-resistant.
Results
Oxidative agents induce selective modifications of the intracellular distribution of classical and novel PKC isoforms.Citation16 We therefore decided to investigate whether the distribution of the atypical PKCζ, recently proposed to be involved in the control of apoptosis, is also dependent on oxidative stress. For this purpose, we generated a PKCζ-GFP and investigated its distribution by fluorescence microscopy. In resting conditions, PKCζ-GFP uniformly localizes throughout the cytoplasm with exclusion of the nucleus. Upon oxidative stress challenge (H2O2 1 mM) PKCζ-GFP translocates to the nucleus in different cell type ( and S1A). To exclude that PKCζ translocation was due to its overexpression or to the presence of the GFP tag, we confirmed this observation by monitoring the localization of endogenous PKCζ by immunocytochemistry. We observed the same behavior of the recombinant PKCζ-GFP (). These results were also confirmed using a different oxidative stress stimulus, such as the UVc treatment that also caused PKCζ translocation to the nucleus (Fig. S1B).
We then correlated the nuclear translocation of PKCζ mediated by oxidizing agent with its effect on cell viability. Two approaches were followed: microscopic assessment of cell survival, as previously described in reference Citation17, and the caspase 3 activity assay.
In the first type of experiment, PKCζ-GFP-overexpressing cells displayed an enhanced survival after oxidative stress challenge (H2O2). The effect of H2O2 was dose-dependent: in the experiment of , the increase in the percentage of PKCζ-expressing cells correlated with the H2O2 concentration. Indeed, due to the higher mortality of the cells that do not overexpress PKCζ-GFP, the percentage of living PKCζ-GFP overexpressing cells gradually increased with H2O2 concentration. No change in the percentage of living fluorescent cells was observed when cells were transfected with GFP alone, as all cells (GFP expressing and untransfected) are equally sensitive to H2O2 (Fig. S1C).
These results well matched those in which caspase 3 activity was monitored (). Upon H2O2 challenge, caspase 3 activity in PKCζ-overexpressing cells was lower than in control cells. Overall, these data suggest that oxidative stress induces nuclear translocation of PKCζ, which, in turn, reduces the apoptotic efficacy of the challenge. Moreover, PKCζ did not share this property with all the other isoforms of PKC ().
We then verified whether PKCζ can also protect from other apoptotic stimuli. To this end, we used different apoptotic stimuli: a calcium-dependent agent (ceramide 30 µM), a strong serine-threonine phosphatase inhibitor (okadaic acid 0.5 µM) and a monoclonal anti-FAS antibody (4 µg/ml). None of the apoptotic agents induces, per se, PKCζ-GFP translocation, and interestingly, in contrast to H2O2, PKCζ overexpression did not inhibit apoptosis (, left parts).
Then, we investigated whether nuclear translocation of PKCζ inhibits only oxidative stress-mediated cell death. To induce nuclear translocation of PKCζ-GFP, cells were pre-treated with a mild oxidative stress (100 µM H2O2 for 30 min), a condition inducing the translocation of the kinase in ∼40% of cells and only 4.9 ± 1.1% of cell death. In this case, PKCζ-GFP-overexpressing cells were protected from the apoptotic effect of ceramide and of okadaic acid but not from the apoptotic effect of anti-FAS. Interestingly, the latter observation also confirms the importance of nuclear localization of PKCζ-GFP for cell death inhibition. Indeed, during anti-FAS treatment PKCζ-GFP (as well as other nuclear targeted probe, ) translocates from the nucleus to the cytosol (, right part) due to nuclear envelope permeabilization.
In order to verify whether the pro-survival effect was ensured only by nuclear localization of PKCζ, independently from redox stress conditions and/or by its overexpression, cells were treated with leptomycin B (LMB, 4 µg/ml 2 h), which inhibits the NES-dependent nuclear export of many proteins (and among them also PKCζ by interfering with the binding of NES domain to CRM1/exportinCitation18). Under those conditions, a significant amount of endogenous PKCζ translocates to the nucleus (). Then, we evaluated the effect of nuclear accumulation of endogenous PKCζ accumulation on cell viability in ceramide-treated cells. As shown in , the pre-treated LMB cells were protected from ceramide-induced apoptosis.
The data collected so far clearly suggest a pro-survival pathway mediated by nuclear-localized PKCζ. To better investigate this pathway, we generated a nuclear-targeted PKCζ chimera (nuPKCζ, ) to exclude potential side effects of the pharmacological manipulation of nuclear export. The analysis of the intracellular localization of the nuPKCζ chimera showed both nuclear and cytosolic distribution ( and B). To force nuclear localization of the kinase and enhance its activity, a constitutively active nuclear-targeted PKCζ was obtained from nuPKCζ by removing the regulatory domain containing both the autoinhibitory domain and the nuclear import and export sequences (nuPKCζ-act, and B). In this case, a specific nuclear staining was detected.
In order to confirm the pro-survival role of nuclear PKCζ, we then performed the microscopic assessment of cell survival for PKCζ-GFP, nuPKCζ and nuPKCζ-act-expressing cells exposed to ceramide. As expected from the previous experiments, forced nuclear localization of PKCζ inhibited apoptotic cell death induced by the apoptotic agent (). Interestingly, the nuclear localization of the kinase, if not accompanied by its constitutive activation, only partially affected the apoptotic sensitivity to ceramide (). Altogether, these results suggest that PKCζ reduces the sensitivity of cells to apoptotic agents by acting within the nucleus and, thus, that its nuclear fraction may be a useful target for tumor cell chemosensitization.
In order to obtain a selective nuclear PKCζ inhibitor, we applied the same strategy previously used to generate the nuclear PKCζ-chimeras. The peptide sequence corresponding to the pseudosubstrate domain of PKCζ (13 amino acids in length at positions 113–125), and thereby inhibiting the kinase activity in resting conditions,Citation19,Citation20 (inhPKCζ) was used for generating a chimeric protein including from the N terminus inhPKCζ, a nuclear targeting sequence and GFP. shows a schematic representation of the chimera (named nu-inhPKCζ) and a representative image of its evident nuclear localization. The expression of nu-inhPKCζ did not affect cell viability (data not shown) but contrasted the pro-survival effect of nuclear PKCζ induced by LMB (). The inhibitor, indeed, restored the sensitivity to ceramide-induced apoptosis in LMB pre-treated cells (compare with ).
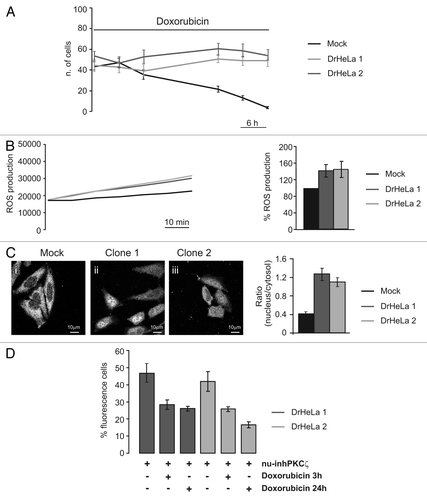
To validate the efficacy of the selectively nuclear PKCζ-inhibitor, we tested it on HeLa cells resistant to the commonly used chemotherapeutic agent doxorubicin. We generated doxorubicin-resistant HeLa clones (DrHeLa) by culturing HeLa cells in the presence of low doxorubicin concentration (2 µM), as described in Methods section. Indeed, as shown in DrHeLa clones showed resistance to high concentration of the antineoplastic drug (20 µM), in contrast to control HeLa clones.
Malignant cells show higher levels of endogenous oxidative stress, mainly due to an increase of ROS production as a consequence of an impaired activity of the respiratory chain due to an accumulation of mtDNA mutations.Citation21 Cancer cells are able to survive intrinsic oxidative stress through an adaptation process involving the activation of redox-sensitive survival machineries, and current evidence suggests the involvement of oxidative stress in anticancer drug resistance.Citation22 In accordance with these observations, we observed a significant increase of ROS production in DrHeLa clones compared with mock cells (). Interestingly, in doxorubicin-resistant HeLa cells, a clear translocation of endogenous PKCζ into the nucleus was observed (), likely to be due to the increased ROS levels of DrHeLa clones that could mimic the treatment with low H2O2 concentration ().
Finally, to verify whether the nuclear activity of PKCζ is a crucial component of the chemoresistance of DrHeLa clones, we analyzed the effect of the nuclear PKCζ inhibitor on cell survival of DrHeLa cells. As previously described, the expression of nu-inhPKCζ did not affect, per se, the viability of DrHeLa clones (). On the contrary, in DrHeLa doxorubicin-treated cells (20 µM for 3 h or 24 h), nu-inhPKCζ expression restores the sensitivity to the anticancer drug in DrHeLa clones ().
Discussion
Resistant tumor cells, with time, develop resistance to anti-neoplastic drugs, because molecular escape routes intervene to promote cancer integrity. Many treatments after a brilliant debut tend to lose their potency, as shown by Gleevec used to treat chronic myelogenous leukemia, where about 17% of patients develop resistance within five years, or Iressa and Tarceva, to which, after about a year, the lung cancer patients suffer drug resistance.Citation23 An increased ability to survive under unfavorable conditions and defects in programmed cell death promote cancer growth and resistance to apoptotic events. Testing combinations of drugs to contrast the tumors are not new in cancer research; a newer approach to cancer therapy is “anticancer targeted therapy,” which combines classical chemotherapy drugs or anticancer approaches with specific signaling transduction inhibitors to block tumor escape routes and render more effective the citotoxicity of therapy. Positive results are emerging in these years on different cancer types, e.g., where the anticancer effect of chemotherapic doxorubicin has been enhanced with specific mTOR inhibitor on breast cancer and in premalignat leukemia cells,Citation24,Citation25 or where the radiotherapyc efficacy has been potentiated with Hsp 90 inhibitor on bladder cancer.Citation18
In this study, we shed light on escape signaling routes that contribute to cancer cell survival, allowing the cells to adapt in presence of drug. The cumulative production of reactive oxygen species through either endogenous or exogenous insults is common for many types of cancer cell that are linked with altered redox regulation of cellular signaling pathways. The “oncogenic-character” of ROS is substantiated by growing body of evidence that ROS within cells act as secondary messengers in intracellular signaling cascades, which induce and maintain the oncogenic phenotype of cancer cells, facilitating mutagenesis, tumor promotion, progression and chemoresistance. As confirmed by Martinez-Outschoorn et al., where the drug combination of Tamoxifen-Dasatinib acts as generalized antioxidant in breast cancer. This association retards tumor-stroma co-evolution and promotes cell death through Warburg effect induction. In this case, the combination therapy interrupt a vicious metabolic cycle between epithelial cancer and associated fibroblasts, reconferring apoptotic sensitivity.Citation26
Pathological activation of certain oncogenes leads to an increase in intracellular ROS through a host of mechanisms involving alterations in scavenging, metabolic rate and potentially activation of intracellular oxidases.Citation14 The effects of ROS, thus, are specifically related to redox-responsive signaling cascades. The redox-sensitive target molecules of these signaling cascades and their chemical modification by oxidative agents are, in most cases, unknown. Thus, understanding the role of signaling proteins and their involvement represents a new exciting challenge in biomedical sciences and, together, allow the design of new drugs that can be used to manipulate cellular signaling networks for antitumorigenic therapeutic purposes.
Oxidative stress induces PKC translocation that is specific for different isoforms and different cell types. For example, in mouse embryonic fibroblasts (MEFs), oxidative stress triggers translocation of PKCα, β, δ and ε isoforms from the cytosol to the plasma membrane. Under the same conditions, PKCζ translocates to the nucleus.Citation27 The intracellular redox state controls the selectivity of PKC isoform activation, changing the sensitivity of the different isoforms to cell stimulation, as observed in the case of PKCα during agonist stimulation after H2O2 pre-treatment.Citation16 Increased PKCs levels have been associated with development of malignant transformation in several cell lines, including breast,Citation28 prostate,Citation29 lung and gastric cancer.Citation30
These aspects suggest a clear role of PKCs in tumorigenesis and, at the same time, spawn questions about the pivotal contribution of individual isozymes. In an elegant set of experiments, Benzombes and colleagues, demonstrated how PKCζ overexpression results in a reduction of drug-induced ROS production, Lyn activation, neutral sphingomyelinase stimulation, ceramide production as well as in apoptosis inhibition and drug resistance.Citation14 Moreover, PKCζ has been identified as ceramide-activated protein kinase that is a critical element in ceramide-induced Jun N-terminal kinase activation and nuclear factor κB translocation.Citation31,Citation32 In this study, we showed a functional role of nuclear PKCζ in regulating cell viability and, in particular, the sensitivity to apoptotic cell death, thus drawing the attention from cytosolic processes, such as sphingomyelinase inactivationCitation14 or caspase 9 activation,Citation33 to the control of nuclear events.
Our data provide the first evidence that nuclear PKCζ inhibition can overcome chemoresistance and thus potentially improve the outcome of therapeutic treatment. A plausible explanation for anti-apoptotic effect of nuclear PKCζ is mostly at the transcriptional level, but little information is still available on the topic. PKCζ was reported to control the expression levels of Bcl-2 family members and other apoptotic regulators.Citation15 The PKCζ inhibition promotes apoptosis in leukemia cells exposed to etoposide and TNFα as well as sensitized tumor cells grown in nude mice to exposed to etoposide.Citation15
Altogether, these results suggest that PKCζ reduces the sensitivity of cancer cells to chemotherapeutic agents and confirms this PKC isoform as a useful target for tumor cell chemosensitization. Indeed, we provide evidence that drug-resistant cells present nuclear PKCζ accumulation as a consequence of ROS production, and that through a specific recombinant nuclear-PKCζ inhibitor, we were able to restore apoptotic sensibility to anti-neoplastic drug. Moreover, the use of a targeted therapy for inhibiting only the nuclear fraction of PKCζ translates into a lower burden of side effects than those caused by traditional inhibitors acting in all cellular compartments. By this approach, it will be possible to preserve the physiological activities of PKCζ and switch-off only the anti-apoptotic effects exerted by the nuclear PKCζ. Although much work remains to be done to support a possible clinical use, the demonstration that nuclear PKCζ plays a key role in the resistance of cancer cells to the effect of chemoterapic drugs opens, in principle, new combined strategies for treating the most aggressive and resistant forms of malignant tumors.
Materials and Methods
Cell culture and transfection.
HeLa cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, in 75-cm2 Falcon flasks. For transient expression experiments, the cells were seeded onto 24-mm glass coverslips and allowed to grow to 75% confluence. At this stage, transfection with 8 µg of the appropriate PKC plasmid DNA or inhibitor of PKCζ were performed as previously described,Citation34 and cell viability measurements, translocation or immunofluorescence assay were performed 36 h after transfection.
Generation of doxorubicin drug-resistant HeLa clone.
The doxorubicin drug-resistant HeLa clone (Dr-HeLa) was established, employing a single-step selection with 20 µM doxorubicin treatment. HeLa cells were cultured in tissue dish to 75% confluence. The cultured cells were selected by the addition of 20 µM doxorubicin to the culture medium, feeding cells every 2–3 days with selection medium and checking cell death after 3–12 days. Resistant cells are observed normally after about 2 weeks of selection. Individual cells were selected randomly and placed in separate wells within a 24-well plate. Clones were grown to confluency and expanded in a maintenance doxorubicin media (2 µM). Expanded clones were retested for drug resistance before any further studies.
Microscopic analysis of PKC translocation.
Images were recorded using a digital imaging system based on a Zeiss Axiovert 200 fluorescence microscope equipped with a back-illuminated CCD camera. The data were acquired and processed using the MetaMorph analyzing program (Universal Imaging Corporation). A high-resolution, 3-D reconstruction of the distribution of a GFP chimera are obtained with the technique of digital image restoration. The recruitment of the kinases are represented as nuclear translocation of different PKCζGFP chimeras, expressed as the increase in fluorescence ratio with respect to time 0 (calculated as the ratio of nucleus and cytosol average intracellular fluorescence, obtained from multiple regions inside the cytosol and on the cell nucleus, measured on single cell). The graphs indicate the levels of PKC translocation, expressed as the fluorescence ratio, to the nucleus, on the average of cytosolic fluorescence intensity. All the stimuli (H2O2, Ceramide, Okadaic acid, anti-FAS and leptomycin B) were added to the Krebs-Ringer buffer (KRB).
Cell viability assays.
After inducing apoptosis, cells were harvested, washed in phosphate-buffered saline (PBS). Caspase 3-like activity was evaluated by using the EnzChek Caspase 3 Assay Kit (Molecular Probes). Enzymatic activity was determined spectrofluorimetrically (LS 50B Perkin Elmer spectrometer) by measuring the kinetics of fluorescence increase at excitation/emission wavelengths of 496/520 nm.
In a cell count assay, the cell viability was evaluated through fluorescent microscopy by counting viable and positive cells, as derived from the analysis of >10 similar experiments. The percentage of living cells are indicated as “Δ% of fluorescent cells,” calculated as the ratio of positive untreated and positive treated cells, obtained from multiple regions inside the coverslip.
Immunofluorescence experiments.
The cells were washed with PBS, warmed at 37°C, and fixed with 4% paraformaldehyde for 20 min at room temperature. Cells were washed twice with PBS and permeabilized with 0.2% Triton X-100 in PBS for 5 min. Cells were incubated overnight with a mouse monoclonal anti-PKCz antibody (Sigma Aldrich). Antibodies binding to PKCz were detected by incubating the cells for 2 h with a FITC-conjugated sheep anti-mouse IgG (Santa Cruz Biotechnology). Fluorescence was visualized using a digital imaging system based on a Zeiss Axiovert 200 fluorescence microscope. Some cells were treated only with secondary antibodies and used to calculate values of background (due to unspecific binding) in the samples.
PKCζ-based constructs.
Human PKCζ cDNA (1,779 bp), fused with the GFP cDNA, was cloned into the expression vector pcDNA3. To obtain the nuclear localization of the kinase, the coding sequence of nuclear localization regions of Glucocorticoid receptors (GR 354 bp) was fused with the chimeric protein PKCζGFP. The constitutively active PKCζ-mutant was obtained by substituting the entire PKCζ cDNA with the catalytic domain (1,014 bp), while the nuclear PKCζ inhibitor was obtained by substituting the entire PKCζ cDNA with the autoinhibitory domain.
Intracellular ROS measurements.
Intracellular ROS generation was measured with 5-(and-6)-chloromethyl-2,7-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) (Invitrogen). After ester hydrolysis and after its oxidation by ROS, 5-(and-6)-chloromethyl-2,7-dichlorofluorescein (CM-DCF) green emission was recorded at 520 nm. Cells were loaded with CM-H2DCFDA at 37°C in KRB. After 30 min, laser scanning confocal microscopy images were obtained. Acquisitions were made every 1 s for 500 s.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
| CM-H2DCFDA | = | 5-(and-6)-chloromethyl-2,7-dichlorodihydrofluorescein diacetate, acetyl ester |
| DrHeLa | = | doxorubicin-resistant HeLa clone |
| GFP | = | green fluorescence protein |
| KRB | = | Krebs-Ringer buffer |
| LMB | = | leptomycin B |
| MEF | = | mouse embryonic fibroblast |
| NES | = | nuclear export system |
| nu-inhPKCζ | = | nuclear-targeted PKCζ inhibitor |
| nuPKCζ | = | nuclear-targeted PKCζ chimera |
| nuPKCζ-act | = | constitutively active nuclear-targeted PKCζ |
| PKC | = | protein kinase C |
| PKCζ | = | protein kinase isoform zeta |
Figures and Tables
Figure 1 Redox stress induces PKCζ nuclear translocation protecting PKCζ-overexpressing HeLa cells by its own apoptotic effects. (A) PKCζGFP-overexpressing HeLa cells in resting condition (left) and after a 30 min treatment with H2O2 1 mM (right); histograms represent the ratio between the nuclear fluorescence and the cytosolic fluorescence, (184 ± 12.1). (B) Endogenous PKCζ in HeLa cells in resting conditions and after 30 min of treatment with H2O2. (C)(i) Increase dose-dependent in cell viability in PKCζGFP overexpressing HeLa cells: the histograms represents apoptotic counts expressed as difference of % fluorescent cells treated (30 min with different H2O2 concentrations) compared with resting condition (50 µM: 1.7 ± 1.3, 100 µM: 4.9 ± 1.1, 1 mM: 15.7 ± 0.8, 2 mM: 18.5 ± 1.3). (C)(ii) Caspase 3 activity assay: gray line, untreated cells; black line, PKCζGFP-overexpressing cells; dotted-gray line, mock cells treated with H2O2; dotted-black line, PKCζGFP-overexpressing cells treated with H2O2. (D) Apoptotic counts in HeLa cells overexpressing different PKC isoforms (PKCα, PKCε, PKCζ) treated with oxidant agent (H2O2 1 mM) and compared with mock cells (Mock: 1.0 ± 2.1; PKCα −4.7 ± 0.3; PKCε 7.3 ± 0.8; PKCζ 15.7 ± 0.8).
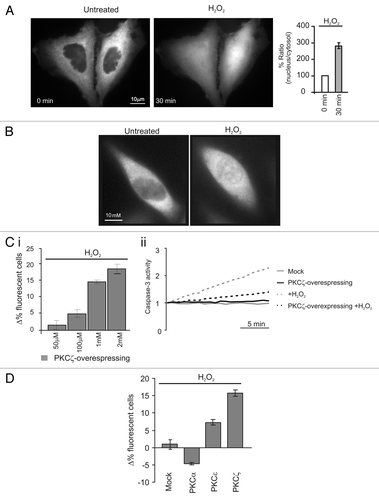
Figure 2 Nuclear PKCζ protects HeLa cells by different apoptotic stimuli. (A) H2O2 (100 µM 30 min) pretreatment induces PKCζ nuclear translocation in PKCζGFP-overexpressing cells, protecting from apoptotic effects of (i) ceramide (30 µM 2 h) and (ii) okadaic acid (0.5 µM 2 h) and (iii) but not from of anti-Fas (4 µg/ml). The upper parts show PKCζGFP-overexpressing HeLa cells following the apoptotic stimulus (left) and following the apoptotic stimulus but previously accompanied by H2O2 100 µM pretreatment (right). The lower parts show the antiapoptotic effects promoted by nuclear translocation of PKCζGFP, mediating H2O2 pre-treatment (cell viability in ceramide-treated cells: Δ% −5.3 ± 1.11 vs. 12.1 ± 0.95 in H2O2 pre-treated HeLa cells; Okadaic acid-treated cells Δ% 2.0 ± 1.21 vs. 14.5 ± 1.23 in H2O2 pre-treated cells; antiFAS-treated cells: Δ% −1.4 ± 1.73 vs. 1.6 ± 1.57 in H2O2 pre-treated HeLa cells). (B) Anti-FAS induces nuclear perturbation as confirmed by nuclear-GFP distribution, where it appears cytosolic-redistributed after 2 h of anti-FAS treatment. (C)(i) LMB (4 µg/ml 2 h) pretreatment induces PKCζ nuclear accumulation in PKCζGFP overexpressing cells, protecting from the apoptotic effects of ceramide (after 2 h of ceramide 30 µM; Δ% cells viability in pre-treated LMB cells: −13.4 ± 7.03 vs. −47.2 ± 8.33 in mock cells. Δ% cells viability in pre-treated LMB cells: −11.0 ± 3.38). (iii) Immunofluorescence of endogenous PKCζ: effect of LMB 4 µg/ml 2 h.
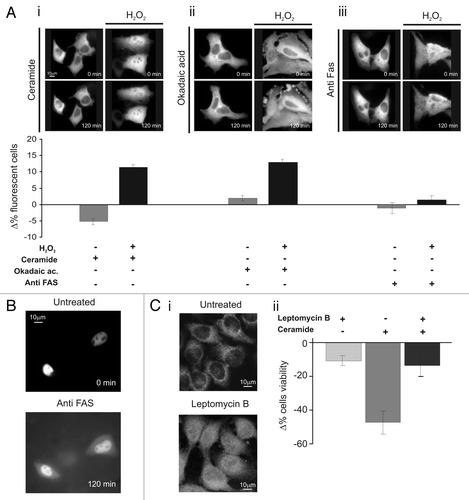
Figure 3 Effects of constitutively nuclear PKCζ (nuPKCζ), constitutively active and constitutively nuclear PKCζ (nuPKCζ-act) and selectively nuclear PKCζ inhibitor (nu-inhPKCζ) on cell viability. (A) Cellular localization and schematic structure of the chimera PKCζ: (i) nuPKCζ and (ii) nuPKCζ-act. (B) Nuclear localization of the different PKCζ chimeras: the histograms represent the ratio between the nuclear and cytosolic average fluorescence intensity, expressed as a percentage, for the three chimera proteins in resting conditions (nuclear distribution of: nuPKCζ-act 4.71 ± 0.80 or nuPKCζ 2.76 ± 0.41 vs. 0.41 ± 0.03 for PKCζ-GFP). (C) Apoptotic counts of HeLa cells expressing the different PKCζ mutants following ceramide treatment (cell viability in nuPKCζ-act and nuPKCζ cells: Δ% 12.6 ± 0.40 or 6.3 ± 1.08 vs. −5.3 ± 1.11 in PKCζGFP expressing HeLa cells, respectively). (D) Cellular localization and schematic structure of the selectively nuclear PKCζ inhibitor (nu-inhPKCζ). (E) Effects of nu-inhPKCζ overexpression on cell viability following LMB pretreatment and/or C2-ceramide treatment (Δ% cells viability in pre-treated LMB cells ceramide untreated: −4.6 ± 2.31 vs. −60.0 ± 8.60 after ceramide treatment or in not LMB pre-treated −48.6 ± 8.71).
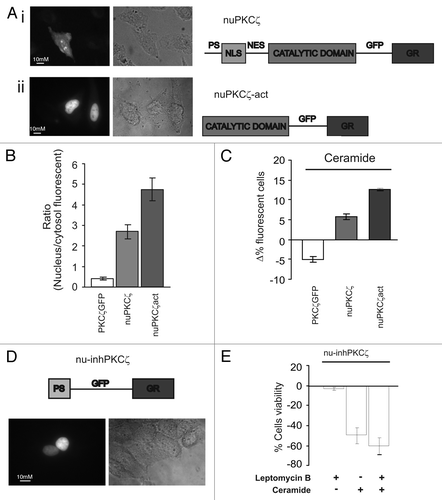
Figure 4 Role of PKCζ nuclear translocation in chemoresistance cells. (A) Doxorubicin resistance of two different clones selected by culturing HeLa cells in presence of Doxorubicin 20 µM. The graph represents a time course of cell viability of three different clones of HeLa cells: a mock clone (black line) and two selected doxorubicin-resistant clones of HeLa cells (gray and dark gray) (Mock: 43.3 ± 4.63, 48.56 ± 4.24, 35.33 ± 4.57, 21.56 ± 3.06, 13.11 ± 2.26, 3.67 ± 0.97; DrHeLa1: 44.25 ± 5.82, 42.75 ± 5.45, 37.50 ± 7.09, 50.75 ± 3.53, 48.88 ± 4.53, 48.88 ± 5.54; DrHeLa2: 56.50 ± 4.42, 48.25 ± 5.58, 55.50 ± 6.61, 63.63 ± 5.48, 61.50 ± 6.31, 56.88 ± 5.03). (B) Time course of ROS production, expressed as normalized values of fluorescence intensity (mock and two doxorubicin-resistant HeLa cells). The histograms represent the increase in ROS production expressed as a percentage respect to mock cells (DrHeLa1 45.1% ± 20.3; DrHeLa2 49.2% ± 25.5). (C) Immunofluorescence of endogenous PKCζ in mock HeLa cells and in two different clones of doxorubicin-resistant HeLa cells. The histograms on the right represent the nuclear distribution of endogenous PKCζ, expressed as ratio between nuclear and cytosolic average fluorescence intensity (mock cells: 0.43 ± 0.04 vs. 1.31 ± 0.16 or 1.11 ± 0.10 in clone 1 or 2, respectively). (D) Apoptotic counts of doxorubicin-resistant HeLa cells transiently expressing nu-inhPKCζ. The histograms represents the difference of percentage of fluorescence following different doxorubicin treatment (20 µM 3 h or 24 h) compared with resting conditions (nu-inhPKCζ expressing DrHeLa1: 46.8 ± 5.23 vs. 28.3 ± 2.75 or 26.1 ± 1.33 after 3 h or 24 h of doxorubicin exposure, respectively; clone2: 41.9 ± 5.66 vs. 25.7 ± 1.45 or 16.6 ± 1.74).
Additional material
Download Zip (264.5 KB)Acknowledgements
This research was supported by: the Italian Ministry of Education (MIUR), FP7 “MyoAGE,” no. 223576, NIH Grant #1P01AG025532-01A1, Cariparo Foundation, Tetethon (GPP1005) and Italian Association for Cancer Research (AIRC) to R.R., the Italian Ministry of Health to A.R. and the Italian Association for Cancer Research (AIRC), Telethon (GGP09128), local funds from the University of Ferrara, the Italian Ministry of Education, University and Research (COFIN, FIRB and Futuro in Ricerca), the Italian Cystic Fibrosis Research Foundation and Italian Ministry of Health to P.P.
References
- Mellor H, Parker PJ. The extended protein kinase C superfamily. Biochem J 1998; 332:281 - 292; PMID: 9601053
- Hug H, Sarre TF. Protein kinase C isoenzymes: divergence in signal transduction?. Biochem J 1993; 291:329 - 343; PMID: 8484714
- Pascale A, Govoni S, Battaini F. Age-related alteration of PKC, a key enzyme in memory processes: physiological and pathological examples. Mol Neurobiol 1998; 16:49 - 62; PMID: 9554701; http://dx.doi.org/10.1007/BF02740602
- O'Brian CA, Ward NE. Biology of the protein kinase C family. Cancer Metastasis Rev 1989; 8:199 - 214; PMID: 2697470; http://dx.doi.org/10.1007/BF00047337
- Nishino N, Kitamura N, Nakai T, Hashimoto T, Tanaka C. Phorbol ester binding sites in human brain: characterization, regional distribution, age-correlation and alterations in Parkinson's disease. J Mol Neurosci 1989; 1:19 - 26; PMID: 2642064; http://dx.doi.org/10.1007/BF02896852
- Giorgi C, Agnoletto C, Baldini C, Bononi A, Bonora M, Marchi S, et al. Redox control of protein kinase C: cell- and disease-specific aspects. Antioxid Redox Signal 2010; 13:1051 - 1085; PMID: 20136499; http://dx.doi.org/10.1089/ars.2009.2825
- Gopalakrishna R, Jaken S. Protein kinase C signaling and oxidative stress. Free Radic Biol Med 2000; 28:1349 - 1361; PMID: 10924854; http://dx.doi.org/10.1016/S0891-5849(00)00221-5
- Cross CE, Halliwell B, Borish ET, Pryor WA, Ames BN, Saul RL, et al. Oxygen radicals and human disease. Ann Intern Med 1987; 107:526 - 545; PMID: 3307585
- Berra E, Diaz-Meco MT, Dominguez I, Municio MM, Sanz L, Lozano J, et al. Protein kinase Czeta isoform is critical for mitogenic signal transduction. Cell 1993; 74:555 - 563; PMID: 7688666; http://dx.doi.org/10.1016/0092-8674(93)80056-K
- Cohen EE, Lingen MW, Zhu B, Zhu H, Straza MW, Pierce C, et al. Protein kinase Czeta mediates epidermal growth factor-induced growth of head and neck tumor cells by regulating mitogen-activated protein kinase. Cancer Res 2006; 66:6296 - 6303; PMID: 16778206; http://dx.doi.org/10.1158/0008-5472.CAN-05-3139
- Kampfer S, Windegger M, Hochholdinger F, Schwaiger W, Pestell RG, Baier G, et al. Protein kinase C isoforms involved in the transcriptional activation of cyclin D1 by transforming Ha-Ras. J Biol Chem 2001; 276:42834 - 42842; PMID: 11551901; http://dx.doi.org/10.1074/jbc.M102047200
- Leroy I, de Thonel A, Laurent G, Quillet-Mary A. Protein kinase Czeta associates with death inducing signaling complex and regulates Fas ligand-induced apoptosis. Cell Signal 2005; 17:1149 - 1157; PMID: 15993755; http://dx.doi.org/10.1016/j.cellsig.2004.12.013
- Moscat J, Rennert P, Diaz-Meco MT. PKCzeta at the crossroad of NFkappaB and Jak1/Stat6 signaling pathways. Cell Death Differ 2006; 13:702 - 711; PMID: 16322752; http://dx.doi.org/10.1038/sj.cdd.4401823
- Bezombes C, de Thonel A, Apostolou A, Louat T, Jaffrézou JP, Laurent G, et al. Overexpression of protein kinase Czeta confers protection against antileukemic drugs by inhibiting the redox-dependent sphingomyelinase activation. Mol Pharmacol 2002; 62:1446 - 1455; PMID: 12435813; http://dx.doi.org/10.1124/mol.62.6.1446
- Filomenko R, Poirson-Bichat F, Billerey C, Belon JP, Garrido C, Solary E, et al. Atypical protein kinase Czeta as a target for chemosensitization of tumor cells. Cancer Res 2002; 62:1815 - 1821; PMID: 11912160
- Rimessi A, Rizzuto R, Pinton P. Differential recruitment of PKC isoforms in HeLa cells during redox stress. Cell Stress Chaperones 2007; 12:291 - 298; PMID: 18229448; http://dx.doi.org/10.1379/CSC-211.1
- Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramideinduced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J 2001; 20:2690 - 2701; PMID: 11387204; http://dx.doi.org/10.1093/emboj/20.11.2690
- Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, et al. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci USA 1999; 96:9112 - 9117; PMID: 10430904; http://dx.doi.org/10.1073/pnas.96.16.9112
- Laudanna C, Mochly-Rosen D, Liron T, Constantin G, Butcher EC. Evidence of zeta protein kinase C involvement in polymorphonuclear neutrophil integrin-dependent adhesion and chemotaxis. J Biol Chem 1998; 273:30306 - 30315; PMID: 9804792; http://dx.doi.org/10.1074/jbc.273.46.30306
- Dang PM, Fontayne A, Hakim J, El Benna J, Périanin A. Protein kinase Czeta phosphorylates a subset of selective sites of the NADPH oxidase component p47phox and participates in formyl peptide-mediated neutrophil respiratory burst. J Immunol 2001; 166:1206 - 1213; PMID: 11145703
- Czarnecka AM, Kukwa W, Krawczyk T, Scinska A, Kukwa A, Cappello F. Mitochondrial DNA mutations in cancer—from bench to bedside. Front Biosci 2010; 15:437 - 460; PMID: 20036829; http://dx.doi.org/10.2741/3629
- Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P. Redox regulation of cell survival. Antioxid Redox Signal 2008; 10:1343 - 1374; PMID: 18522489; http://dx.doi.org/10.1089/ars.2007.1957
- Kaiser J. Combining targeted drugs to stop resistant tumors. Science 2011; 331:1542 - 1545; PMID: 21436437; http://dx.doi.org/10.1126/science.331.6024.1542
- Sokolosky ML, Stadelman KM, Chappell WH, Abrams SL, Martelli AM, Stivala F, et al. Involvement of Akt-1 and mTOR in sensitivity of breast cancer to targeted therapy. Oncotarget 2011; 2:538 - 550
- Abrams SL, Steelman LS, Shelton JG, Chappell W, Bäsecke J, Stivala F, et al. Enhancing therapeutic efficacy by targeting non-oncogene addicted cells with combinations of signal transduction inhibitors and chemotherapy. Cell Cycle 2010; 9:1839 - 1846; PMID: 20436269; http://dx.doi.org/10.4161/cc.9.9.11544
- Martinez-Outschoorn UE, Lin Z, Ko YH, Goldberg AF, Flomenberg N, Wang C, et al. Understanding the metabolic basis of drug resistance: therapeutic induction of the Warburg effect kills cancer cells. Cell Cycle 2011; 10:2521 - 2528; PMID: 21768775; http://dx.doi.org/10.4161/cc.10.15.16584
- Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, Giorgio M, et al. Protein kinase Cbeta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 2007; 315:659 - 663; PMID: 17272725; http://dx.doi.org/10.1126/science.1135380
- O'Brian CA, Liskamp RM, Solomon DH, Weinstein IB. Inhibition of protein kinase C by tamoxifen. Cancer Res 1985; 45:2462 - 2465; PMID: 3157445
- Benavides F, Blando J, Perez CJ, Garg R, Conti CJ, DiGiovanni J, et al. Transgenic overexpression of PKCε in the mouse prostate induces preneoplastic lesions. Cell Cycle 2011; 10:268 - 277; PMID: 21224724; http://dx.doi.org/10.4161/cc.10.2.14469
- Schwartz GK, Jiang J, Kelsen D, Albino AP. Protein kinase C: a novel target for inhibiting gastric cancer cell invasion. J Natl Cancer Inst 1993; 85:402 - 407; PMID: 8433394; http://dx.doi.org/10.1093/jnci/85.5.402
- Malisan F, Testi R. Lipid signaling in CD95-mediated apoptosis. FEBS Lett 1999; 452:100 - 103; PMID: 10376687; http://dx.doi.org/10.1016/S0014-5793(99)00543-8
- Bourbon NA, Yun J, Kester M. Ceramide directly activates protein kinase Czeta to regulate a stress-activated protein kinase signaling complex. J Biol Chem 2000; 275:35617 - 35623; PMID: 10962008; http://dx.doi.org/10.1074/jbc.M007346200
- Brady SC, Allan LA, Clarke PR. Regulation of caspase 9 through phosphorylation by protein kinase Czeta in response to hyperosmotic stress. Mol Cell Biol 2005; 25:10543 - 10555; PMID: 16287866; http://dx.doi.org/10.1128/MCB.25.23.10543-55.2005
- Pinton P, Rimessi A, Romagnoli A, Prandini A, Rizzuto R. Biosensors for the detection of calcium and pH. Methods Cell Biol 2007; 80:297 - 325; PMID: 17445701; http://dx.doi.org/10.1016/S0091-679X(06)80015-4