Abstract
The functional annotation of the cancer genome can reveal new opportunities for cancer therapies. The wealth of genomic data on various cancers has not yet been mined for clinically and therapeutically useful information. We use cross-comparisons of genomic data with the results of unbiased genetic screens to prioritize genomic changes for further study. In this manner, we have identified a soluble variant of the ephrin receptor A7 (EPHA7TR) as a tumor suppressor that is lost in lymphoma. We also developed antibody-based delivery to restore this tumor suppressor to the cancer cells in situ. We will discuss our strategy of screening genomic data, specific findings concerning EPHA7 and the potential for future discoveries.
Key words:
Cancer is a heterogeneous disease characterized by countless genomic alterations. While it has become relatively easy to describe changes in the cancer genomes in great detail, the mining of genomic data for clinically relevant information is only just beginning. We can expect to gain new insight into the molecular origins of cancer and to identify new avenues of cancer therapy. To this end, functional analyses are needed, but—given the wealth of genomic data—where should we begin?
Our laboratory is especially interested in lymphoid cancers, Non-Hodgkin lymphoma and acute lymphatic leukemias (ALL). Great progress has been made in the treatment of childhood ALL, and, similarly, many aggressive forms of Non-Hodgkin lymphoma are largely curable today. This is in contrast to ALL arising in adults (> 35 y of age) and also to so-called indolent lymphomas. These cancers remain incurable except for aggressive transplantation regimens that only a subset of patients are eligible for. Hence, within the category of lymphoid cancers, we focus on these problematic diseases.
Follicular lymphoma (FL) is a common and deadly type of lymphoma. FL is diagnosed in ∼18,000 Americans per year. Clinically, it is characterized by an indolent growth pattern of slow and persistent growth. FLs respond to conventional chemotherapy, but incessant relapses progressively limit marrow and organ function, and frequently, transformation toward a more aggressive cancer leads to patients' demise. Genetically, FLs are characterized by a translocation t(14:18) that activates Bcl2 expression.Citation3,Citation4 Bcl2 blocks cell death and delays cell cycle entry; clearly, additional genetic changes are required for lymphoma development. Unfortunately, a lack of cell lines and murine models of this cancer have hampered research into FL. On the other hand, many excellent cytogenetic studies are available,Citation5–Citation7 and recent sequencing studies have revealed recurrent mutations in several epigenetic regulators, such as MLL2, EZH2, EP300 and CREBBP.Citation8,Citation9 However, the functional consequences of these changes remain to be explored.
Progress in cancer genomics has been immense, and an avalanche of genomic information is published and available. In large part, this reflects advances in sequencing technology but also the ready availability of array comparative genomic hybridization (array CGH), genome-wide methylation profiling and coding and expression data concerning coding and non-coding RNAs. Somatic mutations may be the most readily understood way gene function is affected in cancers. However, alterations in genomic integrity, promoter methylation and microRNA expression affect gene expression and cancer phenotypes. In fact, determining the actual gene dosage in a given cancer is not an easy task and likely requires combining data from different technologies.
Genomic lesions in cancer are increasingly cataloged. As indicated, many different platforms are available to analyze alterations in cancer genomes at high resolution and compare with normal counterparts or similar cancers at different stages. However, these descriptive data do not immediately reveal function, contribution and requirement in cancer cells. Hence, the next challenge is to provide functional annotation of genomic alterations in cancer.
Changes in the cancer genome are often complex and reflect complicated process of malignant transformation. For example, array CGH analyses typically reveal large regions of gains and losses and significant variation between individual cases. Extracting the common patterns is one way to narrow down the list of potential gene targets. Regions of bi-allelic loss provide even more focal information. However, in most cases, even the minimal overlapping regions of change are large, and areas of loss are frequently hemizygous. Hence, often the genomic data alone do not unequivocally identify genetic targets of large-scale changes.
In parallel with advances in genomic technologies, we now have genetic tools to perform large-scale gain- and loss-of-function screens. While long available in simple model organisms like yeast or bacteria, advances in RNAi technology now allows similar genetic approaches in mammalian cells and even in vivo.Citation10–Citation16 Libraries of RNAi constructs, microRNAs or cDNAs are now readily available for gene-by-gene or pooled screening approaches. Clearly, these screening approaches have limitations. For example, RNAi constructs can produce off-target effects, and cDNA expression may cause unphysiological protein levels. Moreover, a screen result—a “hit”—implies biological potential; it does not demonstrate actual relevance to the cancer problem. Hence, results of genetic screens need to be carefully validated and interpreted in the disease context to determine their relevance.
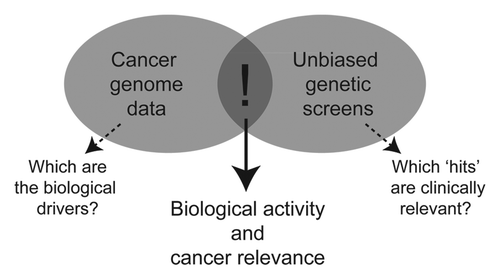
The approach our lab has pursued is to cross-compare cancer genome data with the results of genetic screens (). The rationale is that these are complementary tools: the genetic screen provides a functional filter for complex genome data and indicates potential biological roles. Conversely, evidence that a screen “hit” is also a target of genomic change in cancer provides a first indication of disease relevance. This “functional genomics” approach provides a rapid means to prioritize candidate genes form complex genomic data sets.
In our recent work, we applied a functional genomics approach to follicular lymphoma. Briefly, arrayCGH data revealed large and hemizygous chromosomal losses affecting chromosome 6q12–27 in ∼25% of FL cases. Moreover, patients carrying 6q deletions have a bad prognosis, indicating that this region contributes to lymphoma development and clinical outcome. To identify target genes of 6q loss, we built an RNAi library and performed in vitro screens followed by validation in a new mosaic mouse model of FL based on the vavPBcl2 transgene.Citation17 In short, we confirmed TNFAIP3/A20 as a tumor suppressor in lymphoma; we also identified several additional candidates that localized to hotspots within the 6q region. The presence of multiple target genes within this region had been anticipated by prior cytogenetic studies in reference Citation7 and may illustrate a biological principle behind large-scale chromosomal changes. We were especially intrigued by the ephrin receptor A7 gene (EPHA7) that produced only a short splice variant, and that can be shed form normal B cells but is lost in the tumors.
Ephrin receptors and their ligands form a large family of receptor tyrosine kinases that are involved in several physiological and pathological processes.Citation18,Citation19 Briefly, members of the ephrin A ligand family bind EPH-A receptors, while ephrin B ligands interact with EPH-B receptors. There is a great diversity of ligands and receptors that have tissue-specific expression patterns and overlapping ligand-receptor specificities. A key feature of these interactions is that their bi-directional signaling is triggered by ligand-receptor interactions and consequent receptor dimerization and the formation of higher-order clusters of activated receptors. Physiological roles of this cell-cell signaling pathway are established in axon guidance, angiogenesis and in developmental contexts, such as neural tube closure.Citation20–Citation22 One may speculate that bi-directional EPH signals have even broader effects in cell-cell contact and tissue organization. EPHA7 is remarkable for its expression of short splice variants that have been reported to act as dominant-negative inhibitors of the full-length receptors.Citation23,Citation24 The role of EPH signaling in cancer is quite unclear, and some reports suggest oncogenic activity for certain receptors, while others may have tumor-suppressive properties.Citation19 Methylation studies have revealed silencing of EPHA7 in B-ALL and also colon and prostate cancers.Citation25–Citation27 Other EPHA receptors, e.g., EPHA2 and EPHA3, have emerged as mutational targets lung cancer and melanoma.Citation28,Citation29 The functional consequence of these mutations has not been explored, although it is noteworthy that overexpression of EPHA2 has been observed in ovarian and breast cancer.Citation30,Citation31 Hence, EPH receptors are recurrent targets in cancer, and both oncogenic and tumor-suppressive functions have been discussed.
The truncated form of EPHA7 (EPHA7TR) is a soluble tumor suppressor against lymphoma.Citation1 Our detailed studies on EPHA7TR revealed that its knockdown accelerated lymphoma development in murine model of FL and also Burkitt lymphoma. Concordantly, re-expression or administration of purified EPHA7TR protein caused regression of xenografted human lymphomas via binding to EPHA2, inhibition of EPHA2 phosphorylation and its downstream ERK and SRC signals. These findings indicate a potential therapeutic application for EPHA7TR against lymphoma.
Studies in genetically engineered models indicate that restoration of a tumor suppressor can have dramatic therapeutic effects.Citation32,Citation33 The concept has been named tumor suppressor hypersensitivity in analogy to dependence of cancer cells on the continued activity of an oncogene.Citation34,Citation35 While several drugs that can block oncogenic kinases are now available and show great promise in the clinic, the restoration of a tumor suppressor that has been inactivated in cancer cells remains a challenge. Soluble tumor suppressors offer a unique opportunity, because their auto- and paracrine activities can be achieved, at least in principle, by exogenous administration.
The antitumor and signaling properties imply that soluble EPHA7TR has drug-like and anti-lymphoma activities.Citation1 These effects were readily detected upon local injection into xenografted tumors and could even be elicited by systemic (intravenous administration). To enhance its antitumor activity even further, we fused EPHA7TR to the anti-CD20 (Rituximab®) antibody that is already used in lymphoma therapy. This fusion readily localized to CD20-expressing lymphoma cells in vivo, caused a block in EPHA2 and ERK signaling with subsequent growth arrest and cell death induction. Overall, the fusion had superior activity compared with anti-CD20 alone.Citation1 Finally, we observed no frank toxicity either with the pure EPHA7TR protein or the fusion antibody. Hence, EPHA7TR-anti-CD20 is an example of a bi-functional therapeutic based on the antibody-guided delivery of a soluble tumor suppressor.
Ephrin signaling is an emerging area in cancer, and many important questions remain. EPHA7TR acts as a dominant inhibitor by binding to EPHA2 and likely prevents receptor dimerization and formation of active clusters of EPH receptors.Citation1 Hence, soluble EPHA7TR is a valuable reagent to explore signals emanating from EPH receptors in healthy and cancerous tissues. Ongoing mass spectrometric studies are designed to identify potential additional receptors of EPHA7TR and shed light on the downstream events triggered by EPHA7TR binding. The structural details of EPHA7TR binding to other EPH receptors are unclear, and reconstitution of EPHA7TR-EPHA2 complexes with rationally designed mutant and truncated alleles will provide new insight. These mechanistic studies will enhance the development of EPHA7TR-based peptide therapeutics, for example, by defining the minimum peptide sequence required and optimizing structural assembly of antibody fusions.
Does EPHA7TR have activity against other cancers? EPHA7TR is a dominant inhibitor of EPHA2 and -A3 receptors. This implies that EPHA7TR could produce therapeutic effects against tumors that inactivate EPHA7 and those that have increased expression/activity of EPHA2 and -A3. In lymphoma we observed loss of EPHA7 expression by deletion and/or epigenetic silencing in ∼70% of cases. Specifically, EPHA7 is inactivated in FL, in in marginal zone B-cell lymphomas,Citation24 in diffuse large B-cell (DLBCL) and in HIV-associated Burkitt lymphoma (BL).Citation1 Epigenetic silencing of EPHA7 has been reported in adult B-ALL and in solid tumors, including colorectal and prostate carcinoma.Citation25–Citation27 Further, EPHA2 and EPHA3 are targets of genomic change in lung and ovarian cancerCitation28,Citation29 and over-expressed in breast and ovarian cancers. These data provide a rationale to test EPHA7TR against a spectrum of cancers and derived cell lines.
Outlook—Beyond EPHA7
What determines the actual gene dosage of a tumor suppressor in cancer? Deletions of chromosome 6q (and most other genomic losses) are heterozygous and contain small or no regions of bi-allelic loss. Accordingly, we find that EPHA7 is hemizygously deleted in ∼20% of FLs. However, epigenetic silencing contributes to loss of EPHA7 expression in a cooperative manner in cases with deletions and also occurs in non-6q-deleted cases. The end result is low and near-absent expression of EPHA7 in most FL samples. The genetic concepts of hemi- and homozygosity do not adequately capture these quantitative aspects and suggest that tumor suppressor inactivation comes in three flavors: all, half and zero. Epigenetic silencing and genomic loss are non-mutually exclusive mechanisms of gene inactivation that contribute quantitatively to the final dosage of a tumor suppressor.
What shapes the organization of chromosomal aberrations in cancer? In distinction from somatic mutations the loss of larger genomic regions allows for the disruption of multiple target genes. We identify several known and candidate tumor suppressors that are targeted in a variably overlapping and hemizygous pattern in FL patients. These genes include TNFAIP3, PRDM1 and EPHA7 and others.Citation36–Citation38 Each loss is typically hemizygous, and none is absolutely required for lymphoma development. However, initial studies indicate cooperation between multiple tumor suppressors that are often jointly affected by deletions. Apparently, genes encoded in coherent genomic regions encode facultatively cooperating tumor suppressors. Conversely, the pattern of genomic loss along chromosome 6q reveals areas that are relatively spared from deletion. In part, these regions encode genes essential for cell survival. Hence, in addition to structural features encoded in the DNA sequence,Citation39 the patterns of genomic gains/losses in cancer likely reflect selective pressures for and against specific gene products.
Is there more to be discovered? With over 30,000 publications written about some tumor suppressor genes, one might wonder if there is anything new to be found. The data on various cancer genomes indicate a complex landscape of changes that has occurred in tumor evolution. Functional annotation has, in this case, revealed a surprising new soluble tumor suppressor. Importantly, while the deletion affects multiple tumor suppressor genes, restoration of EPHA7TR is sufficient to produce therapeutic responses. This indicates that important genetic modifiers of the malignant phenotype are still hidden in the genomic data. Hence, mining the cancer genome has only just begun, and there is great potential for actionable insight and new therapeutic reagents.
Figures and Tables
Figure 1 Cancer genomic analysis and unbiased genetic screens are complimentary tools in cancer gene discovery. Genomic analyses reveal many differences between cancer cells and their normal counterparts. Defining the key “drivers” remains a challenge. On the other hand, genetic screens identify modifiers of biological processes; however, their relevance to cancer is often unclear. Cross-referencing the results from these studies provides a functional filter for genomic data and a first indication of cancer relevance.
Acknowledgements
This work is supported by grants from the NCI (R01-CA142798-01), and a P30 supplemental award (H.G.W.), the Leukemia Research Foundation (H.G.W.), the Louis V. Gerstner Foundation (H.G.W.), the WLBH Foundation (H.G.W.), the Society of MSKCC (H.G.W.), the Starr Cancer Consortium grant I4-A410 (H.G.W.), Lymphoma Research Foundation Fellowship (E.O.).
Related Research Data
References
- Oricchio E, Nanjangud G, Wolfe AL, Schatz JH, Mavrakis KJ, Jiang M, et al. The Eph-receptor A7 is a soluble tumor suppressor for follicular lymphoma. Cell 2011; 147:554 - 564; PMID: 22036564; http://dx.doi.org/10.1016/j.cell.2011.09.035
- Oricchio E, Wolfe AL, Schatz JH, Mavrakis KJ, Wendel HG. Mouse models of cancer as biological filters for complex genomic data. Dis Model Mech 2010; 3:701 - 704; PMID: 20876355; http://dx.doi.org/10.1242/dmm.006296
- Bende RJ, Smit LA, van Noesel CJ. Molecular pathways in follicular lymphoma. Leukemia 2007; 21:18 - 29; PMID: 17039231; http://dx.doi.org/10.1038/sj.leu.2404426
- Staudt LM, Wilson WH. Focus on lymphomas. Cancer Cell 2002; 2:363 - 366; PMID: 12450791; http://dx.doi.org/10.1016/S1535-6108(02)00178-2
- Gaidano G, Hauptschein RS, Parsa NZ, Offit K, Rao PH, Lenoir G, et al. Deletions involving two distinct regions of 6q in B-cell non-Hodgkin lymphoma. Blood 1992; 80:1781 - 1787; PMID: 1356511
- Offit K, Parsa NZ, Gaidano G, Filippa DA, Louie D, Pan D, et al. 6q deletions define distinct clinico-pathologic subsets of non-Hodgkin lymphoma. Blood 1993; 82:2157 - 2162; PMID: 8104536
- Nanjangud G, Rao PH, Teruya-Feldstein J, Donnelly G, Qin J, Mehra S, et al. Molecular cytogenetic analysis of follicular lymphoma (FL) provides detailed characterization of chromosomal instability associated with the t(14;18)(q32;q21) positive and negative subsets and histologic progression. Cytogenet Genome Res 2007; 118:337 - 344; PMID: 18000388; http://dx.doi.org/10.1159/000108318
- Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011; 476:298 - 303; PMID: 21796119; http://dx.doi.org/10.1038/nature10351
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 2010; 42:181 - 185; PMID: 20081860; http://dx.doi.org/10.1038/ng.518
- McCaffrey AP, Meuse L, Pham TT, Conklin DS, Hannon GJ, Kay MA. RNA interference in adult mice. Nature 2002; 418:38 - 39; PMID: 12097900; http://dx.doi.org/10.1038/418038a
- Dickins RA, Hemann MT, Zilfou JT, Simpson DR, Ibarra I, Hannon GJ, et al. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet 2005; 37:1289 - 1295; PMID: 16200064
- Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006; 125:1253 - 1267; PMID: 16814713; http://dx.doi.org/10.1016/j.cell.2006.05.030
- Schlabach MR, Luo J, Solimini NL, Hu G, Xu Q, Li MZ, et al. Cancer proliferation gene discovery through functional genomics. Science 2008; 319:620 - 624; PMID: 18239126; http://dx.doi.org/10.1126/science.1149200
- Silva JM, Marran K, Parker JS, Silva J, Golding M, Schlabach MR, et al. Profiling essential genes in human mammary cells by multiplex RNAi screening. Science 2008; 319:617 - 620; PMID: 18239125; http://dx.doi.org/10.1126/science.1149185
- Zender L, Xue W, Zuber J, Semighini CP, Krasnitz A, Ma B, et al. An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell 2008; 135:852 - 864; PMID: 19012953; http://dx.doi.org/10.1016/j.cell.2008.09.061
- Bric A, Miething C, Bialucha CU, Scuoppo C, Zender L, Krasnitz A, et al. Functional identification of tumor-suppressor genes through an in vivo RNA interference screen in a mouse lymphoma model. Cancer Cell 2009; 16:324 - 335; PMID: 19800577; http://dx.doi.org/10.1016/j.ccr.2009.08.015
- Egle A, Harris AW, Bath ML, O'Reilly L, Cory S. VavP-Bcl2 transgenic mice develop follicular lymphoma preceded by germinal center hyperplasia. Blood 2004; 103:2276 - 2283; PMID: 14630790; http://dx.doi.org/10.1182/blood-2003-07-2469
- Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell 2008; 133:38 - 52; PMID: 18394988; http://dx.doi.org/10.1016/j.cell.2008.03.011
- Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer 2010; 10:165 - 180; PMID: 20179713; http://dx.doi.org/10.1038/nrc2806
- Depaepe V, Suarez-Gonzalez N, Dufour A, Passante L, Gorski JA, Jones KR, et al. Ephrin signalling controls brain size by regulating apoptosis of neural progenitors. Nature 2005; 435:1244 - 1250; PMID: 15902206; http://dx.doi.org/10.1038/nature03651
- Nie D, Di Nardo A, Han JM, Baharanyi H, Kramvis I, Huynh T, et al. Tsc2-Rheb signaling regulates EphA-mediated axon guidance. Nat Neurosci 2010; 13:163 - 172; PMID: 20062052; http://dx.doi.org/10.1038/nn.2477
- Sawamiphak S, Seidel S, Essmann CL, Wilkinson GA, Pitulescu ME, Acker T, et al. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature 2010; 465:487 - 491; PMID: 20445540; http://dx.doi.org/10.1038/nature08995
- Holmberg J, Clarke DL, Frisén J. Regulation of repulsion versus adhesion by different splice forms of an Eph receptor. Nature 2000; 408:203 - 206; PMID: 11089974; http://dx.doi.org/10.1038/35041577
- Dawson DW, Hong JS, Shen RR, French SW, Troke JJ, Wu YZ, et al. Global DNA methylation profiling reveals silencing of a secreted form of Epha7 in mouse and human germinal center B-cell lymphomas. Oncogene 2007; 26:4243 - 4252; PMID: 17260020; http://dx.doi.org/10.1038/sj.onc.1210211
- Kuang SQ, Bai H, Fang ZH, Lopez G, Yang H, Tong W, et al. Aberrant DNA methylation and epigenetic inactivation of Eph receptor tyrosine kinases and ephrin ligands in acute lymphoblastic leukemia. Blood 2010; 115:2412 - 2419; PMID: 20061560; http://dx.doi.org/10.1182/blood-2009-05-222208
- Wang J, Kataoka H, Suzuki M, Sato N, Nakamura R, Tao H, et al. Downregulation of EphA7 by hypermethylation in colorectal cancer. Oncogene 2005; 24:5637 - 5647; PMID: 16007213; http://dx.doi.org/10.1038/sj.onc.1208720
- Guan M, Xu C, Zhang F, Ye C. Aberrant methylation of EphA7 in human prostate cancer and its relation to clinicopathologic features. Int J Cancer 2009; 124:88 - 94; PMID: 18821581; http://dx.doi.org/10.1002/ijc.23890
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008; 455:1069 - 1075; PMID: 18948947; http://dx.doi.org/10.1038/nature07423
- Lisabeth EM, Fernandez C, Pasquale EB. Cancer Somatic Mutations Disrupt Functions of the EphA3 Receptor Tyrosine Kinase Through Multiple Mechanisms. Biochemistry 2012; PMID: 22242939; http://dx.doi.org/10.1021/bi2014079
- Zelinski DP, Zantek ND, Stewart JC, Irizarry AR, Kinch MS. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res 2001; 61:2301 - 2306; PMID: 11280802
- Lu C, Shahzad MM, Wang H, Landen CN, Kim SW, Allen J, et al. EphA2 overexpression promotes ovarian cancer growth. Cancer Biol Ther 2008; 7:1098 - 1103; PMID: 18443431; http://dx.doi.org/10.4161/cbt.7.7.6168
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445:656 - 660; PMID: 17251933; http://dx.doi.org/10.1038/nature05529
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature 2007; 445:661 - 665; PMID: 17251932; http://dx.doi.org/10.1038/nature05541
- Weinstein IB. Cancer. Addiction to oncogenes—the Achilles heal of cancer. Science 2002; 297:63 - 64; PMID: 12098689; http://dx.doi.org/10.1126/science.1073096
- Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev 2007; 21:3214 - 3231; PMID: 18079171; http://dx.doi.org/10.1101/gad.1609907
- Kato M, Sanada M, Kato I, Sato Y, Takita J, Takeuchi K, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature 2009; 459:712 - 716; PMID: 19412163; http://dx.doi.org/10.1038/nature07969
- Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, et al. Mutations of multiple genes cause deregulation of NFkappaB in diffuse large B-cell lymphoma. Nature 2009; 459:717 - 721; PMID: 19412164; http://dx.doi.org/10.1038/nature07968
- Mandelbaum J, Bhagat G, Tang H, Mo T, Brahmachary M, Shen Q, et al. BLIMP1 is a tumor suppressor gene frequently disrupted in activated B cell-like diffuse large B cell lymphoma. Cancer Cell 2010; 18:568 - 579; PMID: 21156281; http://dx.doi.org/10.1016/j.ccr.2010.10.030
- De S, Michor F. DNA replication timing and long-range DNA interactions predict mutational landscapes of cancer genomes. Nat Biotechnol 2011; 29:1103 - 1108; PMID: 22101487; http://dx.doi.org/10.1038/nbt.2030