Abstract
Glioblastoma multiforme (GBM) is notoriously resistant to treatment. Therefore, new treatment strategies are urgently needed. ATM elicits the DNA damage response (DDR), which confers cellular radioresistance; thus, targeting the DDR with an ATM inhibitior (ATMi) is very attractive. Herein, we show that dynamic ATM kinase inhibition in the nanomolar range results in potent radiosensitization of human glioma cells, inhibits growth and does not conflict with temozolomide (TMZ) treatment. The second generation ATMi analog KU-60019 provided quick, reversible and complete inhibition of the DDR at sub-micromolar concentrations in human glioblastoma cells. KU-60019 inhibited the phosphorylation of the major DNA damage effectors p53, H2AX and KAP1 as well as AKT. Colony-forming radiosurvival showed that continuous exposure to nanomolar concentrations of KU-60019 effectively radiosensitized glioblastoma cell lines. When cells were co-treated with KU-60019 and TMZ, a slight increase in radiation-induced cell killing was noted, although TMZ alone was unable to radiosensitize these cells. In addition, without radiation, KU-60019 with or without TMZ reduced glioma cell growth but had no significant effect on the survival of human embryonic stem cell (hESC)-derived astrocytes. Altogether, transient inhibition of the ATM kinase provides a promising strategy for radiosensitizing GBM in combination with standard treatment. In addition, without radiation, KU-60019 limits growth of glioma cells in co-culture with human astrocytes that seem unaffected by the same treatment. Thus, inter-fraction growth inhibition could perhaps be achieved in vivo with minor adverse effects to the brain.
Key words:
Introduction
Glioblastoma multiforme (GBM) represents one of the most aggressive human cancers. The current standard of care is surgical debulking and subsequent irradiation concomitant with temozolomide (TMZ).Citation1 While this regimen currently provides the largest increase in patient survival, more effective treatment resulting in improved survival is urgently needed. The blood-brain barrier (BBB) precludes many chemotherapeutic options, leaving radiation therapy (RT) as an important treatment modality. Although high-grade gliomas initially respond to RT, recurrence is almost certain.Citation2,Citation3 Recurring tumors tend to be more resistant to therapy; surgery is not always a treatment option for recurrence and re-irradiation of the brain must be finely balanced against radionecrosis and patient quality of life.Citation2,Citation4 Therefore, the use of potent radiosensitizers targeting the tumor is an attractive idea.Citation1,Citation5
Cells from ataxia telangiectasia (A–T) patients are exquisitely radiosensitive due to a profound inability to elicit the DNA damage response (DDR).Citation6 Thus, A–T mutated (ATM) kinase offers itself as a potential therapeutic target. AstraZeneca's second-generation ATM inhibitor (ATMi) KU-60019 is significantly more potent than its predecessor.Citation7,Citation8 In addition to radiosensitizing glioma cells, KU-60019 also inhibits migration, invasion and growth of glioma cells in vitro, perhaps by inhibiting prosurvival pathways.Citation7 Therefore, the combined actions of KU-60019 as a radiosensitizer and, in-between fractions, as an inhibitor of tumor growth and spread are very attractive.
The neurological defects seen in A–T patients imply an important role for ATM in the brain; however, this has only been investigated to some extent in mice.Citation9 Thus, it is critical to determine any deleterious effects of an ATMi on human brain. Herein, we report on the further characterization of KU-60019 and show that ATM is transiently and potently inhibited with nano-molar concentrations resulting in radiosensitization of several glioma cell lines. Additionally, inhibition is not antagonistic with standard treatment, and we establish that without radiation, the toxicity to normal human astrocytes is limited with or without co-treatment with TMZ. Our results suggest that inhibiting the ATM kinase to achieve substantial radiosensitization and reduction of glioma growth could be a viable therapeutic approach for the treatment of glioblastomas.
Results
KU-60019 inhibits ATm kinase activity at sub-micromolar concentrations.
The second generation ATMi KU-60019 was shown to be at least 10 times more effective than its predecessor KU-55933 with little to no nonspecific target effects at 1 µmol/L against a panel of 229 protein kinases.Citation7 In an expansion of those findings, we now show that in U1242 glioma cells, IR-induced p53 (S15) and H2AX (S139) phosphorylation was completely inhibited by as little as 300 nM KU-60019, with partial inhibition seen at 100 nM (p53, 80%; H2AX, 50% inhibition) (). While ATM is the primary kinase responsible for these phosphorylation events, the influence of other PIKKs (phosphoinositide-3-kinase-related protein kinase) should not be ignored.Citation7,Citation10,Citation11 When cells were incubated with KU-60019 under serum-free conditions, inhibition of p53 (S15) phosphorylation was seen at a dose as low as 10 nM (Fig. S1), suggesting that serum reduces the bioavailability of KU-60019. Our data shows that KU-60019 is 30–100 times more potent than its predecessor KU-55933, bringing the doses needed for effective signaling blockage in vitro into the nanomolar range.
KU-60019 acts quickly, reversibly, and inhibition remains effective for days.
KU-60019 at 300 nM completely inhibited p53 and H2AX phosphorylation as quickly as 15 min after application (). As p53 and H2AX have been reported to be phosphorylated by other kinases, we also confirmed specificity by monitoring phosphorylation of KAP1 at Ser 824.Citation12 As a permanent block of ATM kinase in cells would likely result in increased genomic instability,Citation6 the ATMi should ideally be reversible. Indeed, when U1242 cells were treated with KU-60019 for 1 h, washed to remove KU-60019 and then challenged with IR, the inhibitory effects of KU-60019 were reversed as early as 15 min after washout (). KU-60019 was stable in tissue culture up to 72 h, as demonstrated by a 95% and 70% blockage of p53 and H2AX phosphorylation, respectively (). We also established that the prolonged effect of KU-60019 was due to active drug and not cellular adaptation, as we observed a complete lack of IR-induced p53 phosphorylation in U1242 cells treated with conditioned KU-60019-containing media after several days (). Thus, KU-60019 is a specific, fast, reversible, long-lasting and highly potent inhibitor of ATM kinase activity in vitro.
Low doses of KU-60019 compromise pro-survival signaling.
Supporting our previous report in reference Citation7, even these low doses of KU-60019 inhibit pro-survival signaling by reducing the phosphorylation of AKT at S473. We noted a clear dose-dependent decrease in AKT phosphorylation, with 600 nM able to compromise AKT signaling by 50% (); this effect was again completely reversible (). These results suggest that even low concentrations of ATMi prevent pro-survival signaling and may, in turn, reduce glioma cell growth and migration as we previously reported in reference Citation7.
Nanomolar concentrations of KU-60019 radiosensitize glioma cells.
In our previous study, KU-60019 was applied for a 16 h period (-1 h to +15 h post-IR), at which point the media was removed, cells washed and fresh media applied, providing DERs of ∼3.0.Citation7 In vivo, it is possible that the drug might be present longer. Therefore, we tested the effects of continuous, low level KU-60019 exposure on the radiosurvival of glioma cells. U1242 cells were treated with KU-60019 (3-0.3 µM) for 1 h prior to IR with the drug remaining in the media throughout the course of the experiment. Interestingly, we found that the highest dose was so effective that we failed to produce any colonies. However, continuous exposure of KU-60019 at 300 and 600 nM effectively radiosensitized the cells with DERs of 1.8 and 2.1, respectively (). Similar effects were seen with U373 glioma cells, where 300 nM killed 98% of cells after 2 Gy (). In fact, we observed slight but statistically significant sensitization with only 150 nM where 30% of the cells were killed. Previously, we showed that KU-60019 slowed U1242 cell growth by 50% over 5 d.Citation7 Here, we show that 600 nM reduced U1242 and U373 glioma colony formation by 30% ( and S2). It should be noted that colonies displayed a dose-dependent decrease in size as concentrations of KU-60019 increased, further solidifying the idea that KU-60019 alone may be sufficient to slow tumor growth (Fig. S3).
In order for an ATMi to move forward as a potential treatment of GBM, it needs to be co-administered with TMZ prior to radiation.Citation1 Both U1242 and U373 cells displayed dose-dependent sensitivity to TMZ in a growth assay (Fig. S4). In line with a recent report in reference Citation13, we noticed a small but non-significant increase in U1242 radiosensitization with the combination treatment (). These results establish that KU-60019 in the nanomolar range is capable of providing potent radiosensitization in vitro and is not antagonistic with TMZ in a selection of glioma cell lines.
KU-60019 is stable in aqueous solution.
Thus far, KU-60019 was dissolved in DMSO followed by dilution in PBS or media. To determine if this concentrated stock could be diluted in PBS and remain soluble and active at relatively high concentrations, the DMSO stock was diluted to make 30 and 100 µM solutions. These were left at room temperature for 15 min prior to centrifugation at high speed to remove any insoluble (precipitated) material. The supernatants were then further diluted with tissue culture media to 1 or 3 µM. No precipitation of KU-60019 occurred, and these stocks effectively blocked ATM signaling after dilution (, compare lanes 3–6 with lane 7). To determine the stability of KU-60019, we diluted the DMSO stock in saline to a final concentration of 100 µM, which was left at room temperature for 3 d or 3 weeks. KU-60019 remained stable in saline at room temperature for up to 3 weeks, as seen by the complete inhibition of p53 and H2AX phosphorylation after dilution (, compare lanes 2 and 3). These results suggest that KU-60019 remains soluble and stable in PBS or saline for long periods of time.
KU-60019 is not toxic to human astrocytes.
Normal fibroblasts are equally radiosensitized by KU-60019 as are glioma cells.Citation7 Thus, the tumor specificity of this ATMi in vivo would have to depend on the precision of RT, with the effects of the ATMi only observed within the radiation field. Drug delivery is nowhere near as precise. Whether ATMi is delivered systemically or locally, normal tissue will certainly be exposed. To determine the effects of ATMi on normal human CNS cells is technically challenging due to the scarcity of testing material. Using hESCs and a defined differentiation protocol, we routinely generate human astrocytes.Citation14 The terminally differentiated astrocytes are not actively dividing, so survival cannot be determined by colony-forming assays. Therefore, we utilized a trypan blue-flow cytometry assay, assuming that metabolically active cells will efflux the dye.Citation7 In an untreated culture of GFAP+ astrocytes (Fig. S5A), ∼15% of the cells stained with trypan blue, confirming that live yet terminally differentiated astrocytes can efflux the dye. Neither KU-60019, TMZ nor the combination of the two increased cell killing, making us confident that short-term ATMi exposure would not be unacceptably toxic to normal brain outside of the radiation field (Fig. S5B).
Combining KU-60019 and TMZ selectively reduces glioma cell growth in co-cultures with human astrocytes.
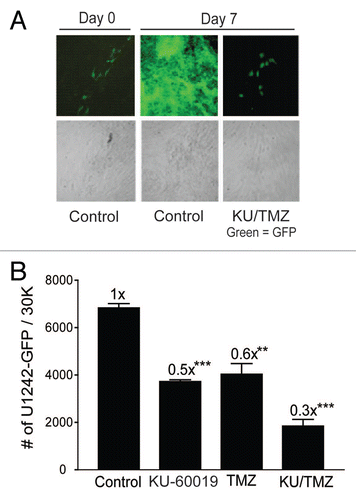
The hESC-derived astrocytes offer a unique system for determining the relative toxicity of KU-60019 in co-culture with glioma cells. Using GFP as marker for U1242 cells in co-cultures with astrocytes, we were able to monitor glioma cell growth in an environment more akin to in vivo conditions. The tumor cell-specific action of TMZ is achieved by exploiting the enhanced growth rate of tumor cells over surrounding fully differentiated non-growing tissue. Therefore, when considering adding KU-60019 to a potential regimen, it is important to consider the interactions between KU-60019 and TMZ outside of the radiation field. By treating co-cultured astrocytes and U1242 glioma cells, we showed that when KU-60019 is combined with TMZ, a decrease in glioma cell growth by 70% was observed, whereas KU-60019 or TMZ alone reduced glioma cell growth by 40–50% (). Altogether, this result suggests that KU-60019 could potentially limit glioma growth outside of the radiation field without causing harm to normal brain.
Discussion
The need for more effective treatments for GBM is exceedingly urgent. While short-term survival gains are seen, the 5-year survival rate remains abysmal. An issue often overlooked is the quality of life during and after treatment.Citation4 Surgery and chemoradiation significantly slow the progression of GBM disease but at what cost to the patient? The use of radiosensitizers may not reduce recurrence, but the reduction in total radiation dose to the brain is expected to go a long way to improve patient cognition. This study outlines a strategy to radiosensitize GBM, and we document cell killing with mere nanomolar concentrations of KU-60019 combined with low-dose radiation. Similar fast-acting and reversible properties have been reported previously on this and a different, less potent ATMi.Citation15,Citation16 KU-60019 is stable and reversible and has little to no toxic effect on brain (astrocytes) in the absence of radiation.
The support of combining a cell signaling inhibitor with the current treatment regimen for so called “trimodal therapy” is gaining ground. Work exploring inhibitors targeting the TGFβ receptor 1 kinase, VEGF, EGFR, PI3K, AKT and CHK1 is under way.Citation5,Citation17–Citation19 Curiously, ATM is increasingly linked with the modulation of several, if not all, of these factors, painting an emerging picture that the regulation of the DDR may be only one aspect of the intricate signaling networks that ATM controls.Citation20 Therefore, to consider an ATMi purely as a radiosensitizer may underestimate its therapeutic potential.Citation21 We have clearly documented the ability of KU-60019 to reduce AKT phosphorylation as well as inhibiting glioma cell migration and invasion.Citation7 Further, in this report, we show significant decreases in glioma cell growth by treatment with KU-60019 alone or in combination with TMZ, which has little to no effect on human astrocytes. While the major therapeutic strength of an ATMi is not accomplished until IR is administered, subtle drug-alone effects should not be ignored. GBMs are aggressive tumors, with a cell doubling time in vitro under 24 h. Therefore, intra-fractional growth is likely. Additionally, sub-lethal doses of radiation can increase glioma growth and migration.Citation22 Altogether, the decreases in tumor growth and migration we observe with KU-60019 could translate into increased tumor control outside of the radiation field. This finding is very exciting but will require extensive testing in vivo, which is already underway.
The TMZ-resistant subset of GBMs has been identified as MGMT+.Citation5,Citation17 It is doubtful that these MGMT+ tumors would be any less receptive to KU-60019, as cell killing is likely achieved by different mechanisms. Our finding that combining ATMi with TMZ does not cause increased radiosensitization in vitro over ATMi alone is not surprising.Citation13 However, since we did not extensively investigate the response to TMZ in a range of glioma cell lines with parallel MGMT assays, it is possible that in some cases, KU-60019 and TMZ may synergize. Regardless, with U1242 cells, we noticed a small increase in toxicity/growth inhibition when these drugs were combined without radiation. This effect was observed over 7 d but would likely be cummulative, as the toxic effects of TMZ take longer to accumulate.Citation13 Therefore, in vivo synergistic effects could not be ruled out; however, our data suggest it is less likely—at least with the glioma cell lines tested here.
ATM is required for p53-mediated apoptosis of neurons after exposure to radiation, which is distinct from the response in astrocytes.Citation9,Citation23 Whether transient inhibition of ATM would delay apoptosis for a short time and result in damage or not to normal brain remains to be determined. While the contribution of cancer stem cells to the formation and recurrence of glioblastoma is controversial, their importance should not be ignored. Specifically, CD133+ GBM stem cells (GSCs) show enhanced DDR signaling and are primed to respond to DNA damage effectively via constitutively upregulated ATM signaling.Citation24 Thus, the challenge is to specifically target ATM in GSCs and minimize the toxicity to normal brain.
Altogether, and with an increased body of literature tying ATM to the regulation of prosurvival signaling,Citation20,Citation25–Citation27 we outline an expanded role for ATMi kinase inhibitors as cancer therapeutics. We show that in conjunction with current standard of care, KU-60019 increases glioma cell death combined with radiation, as expected. In addition, the drug has excellent dynamic properties, permitting the fine-tuning of inhibitory responses and allowing for quick drug withdrawal to limit potential toxicity to the brain. We also show that without radiation, KU-60019 inhibits glioma cell growth in co-cultures with normal human astrocytes. Importantly, this occurs when little to no toxicity to astrocytes is observed. It remains to be determined whether these advantageous effects would translate to the in vivo situation.
Materials and Methods
Reagents.
The following antibodies were used: anti-p(S15)-p53, -p(S473)-AKT, -AKT (Cell Signaling Technology, Inc.), anti-p(S824)-KAP1 (Bethyl Laboratories Inc.), anti-γH2AX (Millipore), anti-β-actin (Santa Cruz Biotechnology) and anti-GFAP (AbCam). KU-60019 was from AstraZeneca.Citation7 Temozolomide was from Sigma-Aldrich.
Cell culture and treatments.
U1242, U1242/luc-GFP, U373 and U87 human malignant glioma cells were cultured in DMEM (10% FBS and antibiotics).Citation7 Glioma cells were obtained from the ATCC and Dr. Allan Yates, Ohio State University, respectively. Routine characterization includes the ability to form intra-cranial tumors in nude mice and qRT-PCR (U1242) expression profiling. Human astrocytes were derived from BG01V human embryonic stem cells (hESC) obtained from ATCC.Citation14 Astrocytes were cultured in DMEM and grown to quiescence. The cells used in this study have not been tested and authenticated by an external service provider. Irradiations were performed using an MDS Nordion Gammacell 40 137-Cs source (1.05 Gy/min).
Western blotting.
Proteins were separated by SDS-PAGE and transferred onto PVDF membranes (BioRad) for western blotting.Citation7
Cell survival assays.
Glioma radiosurvival and surviving fractions (astrocytes) were determined by colony-forming assay and trypan blue/flow cytometry, respectively.Citation7 Relative radiosensitization was calculated by comparing dose enhancement ratios (D37 dose of control cells/D37 dose of treated cells = DER). A DER greater than 1 indicates radiosensitization.
Statistics.
Unpaired one- or two-tailed t tests were performed on triplicate (or more) data sets using GraphPad Prism 3.0 (GraphPad Software, Inc.). p values are indicated as; * < 0.05, ** < 0.01, *** < 0.001.
Disclosure of potential Conflicts of Interest
Commercial research grant from KuDOS/AstraZeneca (K.V.). The other authors disclose no conflicts of interest.
Financial Support
Supported by R01NS064593, R21ES016636 (K.V.), T32CA085159 and the American Brain Tumor Association (S.E.G.).
Figures and Tables
Figure 1 KU-60019 blocks ATM kinase activity at low concentrations. U1242 glioma cells were exposed to KU-60019 (3, 1, 0.3, 0.1, 0.03 or 0.01 µM) for 1 h, irradiated with 5 Gy and collected for western blotting after 5 min. Fold depicts phospho-protein levels normalized to β-actin levels.
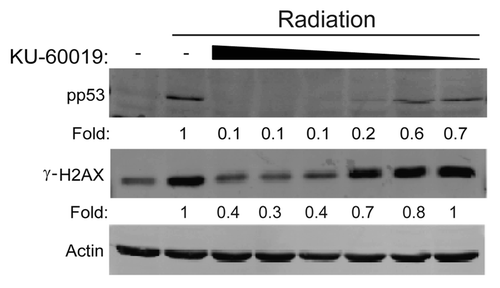
Figure 2 KU-60019 reversibly inhibits ATM kinase activity 15 min after treatment and remains effective for 72 h. U1242 glioma cells were exposed to KU-60019 at; (A) 3 or 0.3 µM for 15, 30, 45 or 60 min, (B) 3 µM for 1 h. Indicated cells were then washed four times, fresh media was applied, and cells were incubated for a further 0, 15, 30 or 45 min prior to irradiation, (C) 3 µM for 24, 48 or 72 h. Cells were then irradiated (5 Gy) and collected for western blotting after 5 min or (D) 3 µM for 1, 18, 24 or 48 h, then irradiated (5 Gy), and cells collected after 5 min for western blotting. Conditioned media (CM) was removed from the tissue culture dishes, reapplied to fresh cells, incubated for 1 h, then exposed to 5 Gy, and cells collected after 5 min for western blotting. Fold depicts phospho-protein levels normalized to β-actin levels.-indicates phosphorylation levels below the level of detection (< 0.1x of control treatment).
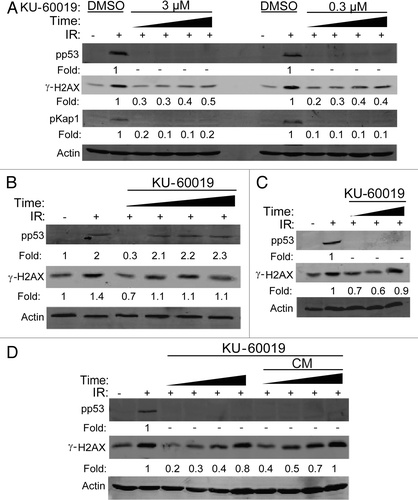
Figure 3 KU-60019 reversibly inhibits basal AKT phosphorylation in a dose-dependent manner. U1242 glioma cells were exposed to KU-60019 at (A) 3, 1, 0.6, 0.3 or 0.1 µM for 1 h and then collected for western blotting or (B) 3 µM for 1 h. Cells were then washed four times with fresh media and incubated for a further 1, 2, 3 or 4 h before collecting the cells for western blotting. Fold depicts phospho-protein levels normalized to β-actin levels.
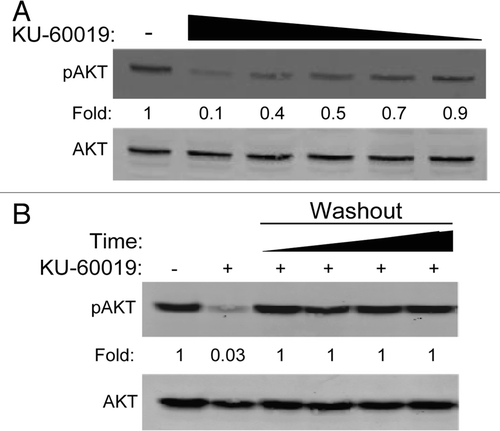
Figure 4 KU-60019 potently radiosensitizes glioma cells at low concentrations. U1242 or U373 glioma cells were exposed to KU-60019. (A) U1242 at 0.6 or 0.3 µM, (B) U373 at 0.6, 0.3 or 0.15 µM or (C) U1242 at 0.6 µM of KU-60019 and TMZ at 100 µM or the combination of both for 1 h prior to IR. Surviving fractions were determined by crystal violet staining and colony counting. Data points, surviving cells plotted as a fraction of control (no IR); error bars; SEM; n = 3. Where error bars are not seen they are obscured by symbols. DER, dose enhancement ratios of D37 values compared with irradiation only control.
Figure 5 KU-60019 remains stable in buffer and saline solutions at room temperature. (A) KU-60019 (6 mM in DMSO) was diluted in PBS to either 100 or 30 µM. The stocks were left at room temperature for 15 min, spun and insoluble material removed. The supernatants were then further diluted with tissue culture media to either 1 or 3 µM. U1242 cells were exposed to either 1 or 3 µM of these dilutions for 1 h and exposed to 5 Gy and then collected for western blotting after 5 min. (B) KU-60019 (6 mM in DMSO) was diluted to 100 µM in saline and stored at room temperature for 3 d (3d) or 3 weeks (3w) followed by dilution and exposure of U1242 cells to 1 or 3 µM, or 3 µM freshly diluted (from DMSO) for 1 h, exposed to 5 Gy and collected for western blotting after 5 min.
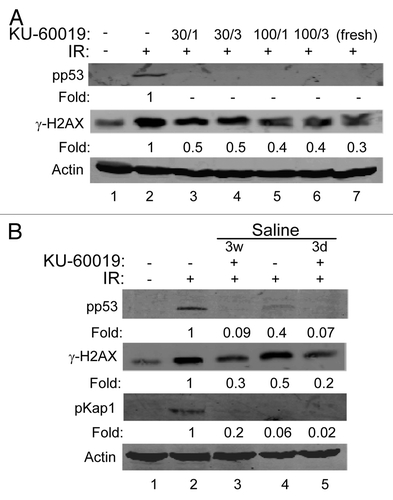
Figure 6 Combining KU-60019 and TMZ reduces glioma cell growth in the absence of radiation. U1242/luc-GFP glioma cells were seeded onto a monolayer of normal human astrocytes. Cells were exposed to TMZ (100 µM) and/or KU-60019 (3 µM). Growth was determined by (A) fluorescence microscopy and (B) GFP flow cytometry 7 d later. Data points, number of U1242/luc-GFP cells/30K cells counted. Error bars; SEM; n = 4.
Additional material
Download Zip (98.8 KB)Acknowledgements
The VCU Flow Cytometry Core Facility is supported by NIHP30CA16059. The VCU Microscope Facility is supported by P30NS047463.
Related Research Data
References
- Stupp R, Hegi ME, Gilbert MR, Chakravarti A. Chemoradiotherapy in malignant glioma: standard of care and future directions. J Clin Oncol 2007; 25:4127 - 4136; PMID: 17827463; http://dx.doi.org/10.1200/JCO.2007.11.8554
- Niyazi M, Siefert A, Schwarz SB, Ganswindt U, Kreth FW, Tonn JC, et al. Therapeutic options for recurrent malignant glioma. Radiother Oncol 2011; 98:1 - 14; PMID: 21159396; http://dx.doi.org/10.1016/j.radonc.2010.11.006
- Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med 2008; 359:492 - 507; PMID: 18669428; http://dx.doi.org/10.1056/NEJMra0708126
- Taphoorn MJ, Sizoo EM, Bottomley A. Review on quality of life issues in patients with primary brain tumors. Oncologist 2010; 15:618 - 626; PMID: 20507891; http://dx.doi.org/10.1634/theoncologist.2009-0291
- Squatrito M, Holland EC. DNA damage response and growth factor signaling pathways in gliomagenesis and therapeutic resistance. Cancer Res 2011; 71:5945 - 5949; PMID: 21917735; http://dx.doi.org/10.1158/0008-5472.CAN-11-1245
- Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol 2008; 9:759 - 769; PMID: 18813293; http://dx.doi.org/10.1038/nrm2514
- Golding SE, Rosenberg E, Valerie N, Hussaini I, Frigerio M, Cockcroft XF, et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol Cancer Ther 2009; 8:2894 - 2902; PMID: 19808981; http://dx.doi.org/10.1158/1535-7163.MCT-09-0519
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 2004; 64:9152 - 9159; PMID: 15604286; http://dx.doi.org/10.1158/0008-5472.CAN-04-2727
- Herzog KH, Chong MJ, Kapsetaki M, Morgan JI, McKinnon PJ. Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science 1998; 280:1089 - 1091; PMID: 9582124; http://dx.doi.org/10.1126/science.280.5366.1089
- Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, et al. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev 1999; 13:152 - 157; PMID: 9925639; http://dx.doi.org/10.1101/gad.13.2.152
- Stiff T, O'Driscoll M, Rief N, Iwabuchi K, Löbrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res 2004; 64:2390 - 2396; PMID: 15059890; http://dx.doi.org/10.1158/0008-5472.CAN-03-3207
- Li X, Lee YK, Jeng JC, Yen Y, Schultz DC, Shih HM, et al. Role for KAP1 serine 824 phosphorylation and sumoylation/desumoylation switch in regulating KAP1-mediated transcriptional repression. J Biol Chem 2007; 282:36177 - 36189; PMID: 17942393; http://dx.doi.org/10.1074/jbc.M706912200
- Chalmers AJ, Ruff EM, Martindale C, Lovegrove N, Short SC. Cytotoxic effects of temozolomide and radiation are additive- and schedule-dependent. Int J Radiat Oncol Biol Phys 2009; 75:1511 - 1519; PMID: 19931733; http://dx.doi.org/10.1016/j.ijrobp.2009.07.1703
- Adams BR, Golding SE, Rao RR, Valerie K. Dynamic dependence on ATR and ATM for double-strand break repair in human embryonic stem cells and neural descendants. PLoS One 2010; 5:10001; PMID: 20368801; http://dx.doi.org/10.1371/journal.pone.0010001
- White JS, Choi S, Bakkenist CJ. Transient ATM kinase inhibition disrupts DNA damage-induced sister chromatid exchange. Sci Signal 2010; 3:44; PMID: 20516478; http://dx.doi.org/10.1126/scisignal.2000758
- Rainey MD, Charlton ME, Stanton RV, Kastan MB. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res 2008; 68:7466 - 7474; PMID: 18794134; http://dx.doi.org/10.1158/0008-5472.CAN-08-0763
- Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, et al. Malignant astrocytic glioma: genetics, biology and paths to treatment. Genes Dev 2007; 21:2683 - 2710; PMID: 17974913; http://dx.doi.org/10.1101/gad.1596707
- Sathornsumetee S, Reardon DA, Desjardins A, Quinn JA, Vredenburgh JJ, Rich JN. Molecularly targeted therapy for malignant glioma. Cancer 2007; 110:13 - 24; PMID: 17520692; http://dx.doi.org/10.1002/cncr.22741
- Weber GL, Parat MO, Binder ZA, Gallia GL, Riggins GJ. Abrogation of PIK3CA or PIK3R1 reduces proliferation, migration and invasion in glioblastoma multiforme cells. Oncotarget 2011; 2:833 - 849; PMID: 22064833
- Hawkins AJ, Golding SE, Khalil A, Valerie K. DNA double-strand break-induced pro-survival signaling. Radiother Oncol 2011; 101:13 - 17; PMID: 21726915; http://dx.doi.org/10.1016/j.radonc.2011.05.074
- Choi S, Gamper AM, White JS, Bakkenist CJ. Inhibition of ATM kinase activity does not phenocopy ATM protein disruption: implications for the clinical utility of ATM kinase inhibitors. Cell Cycle 2010; 9:4052 - 4057; PMID: 20953138; http://dx.doi.org/10.4161/cc.9.20.13471
- Wild-Bode C, Weller M, Rimner A, Dichgans J, Wick W. Sublethal irradiation promotes migration and invasiveness of glioma cells: implications for radiotherapy of human glioblastoma. Cancer Res 2001; 61:2744 - 2750; PMID: 11289157
- Gosink EC, Chong MJ, McKinnon PJ. Ataxia telangiectasia mutated deficiency affects astrocyte growth but not radiosensitivity. Cancer Res 1999; 59:5294 - 5298; PMID: 10537312
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006; 444:756 - 760; PMID: 17051156; http://dx.doi.org/10.1038/nature05236
- Khalil A, Morgan RN, Adams BR, Golding SE, Dever SM, Rosenberg E, et al. ATM-dependent ERK signaling via AKT in response to DNA double-strand breaks. Cell Cycle 2011; 10:481 - 491; PMID: 21263216; http://dx.doi.org/10.4161/cc.10.3.14713
- Golding SE, Valerie K. MRE11 and ATM AKTivate pro-survival signaling. Cell Cycle 2011; 10:3227 - 3236; PMID: 21946569; http://dx.doi.org/10.4161/cc.10.19.17048
- Fraser M, Harding SM, Zhao H, Coackley C, Durocher D, Bristow RG. MRE11 promotes AKT phosphorylation in direct response to DNA double-strand breaks. Cell Cycle 2011; 10:2218 - 2232; PMID: 21623170; http://dx.doi.org/10.4161/cc.10.13.16305