Abstract
Cisplatin was shown to induce the ataxia telangiectasia mutated (ATM)-dependent phosphorylation of tumor protein p63 isoform, (ΔNp63α), leading to a transcriptional regulation of specific genes implicated in the control of cell death of squamous cell carcinoma (SCC) cells. We previously observed that the cisplatin-induced phosphorylated (p)-ΔNp63α transcriptionally regulates the expression of specific microRNAs (miRNAs) in SCC cells. We found here that cisplatin exposure of SCC cells led to modulation of the members of the autophagic pathway, such as Atg1/Ulk1, Atg3, Atg4A, Atg5, Atg6/Becn1, Atg7, Atg9A and Atg10, by a direct p-ΔNp63α-dependent transcriptional regulation. We further found that specific miRNAs (miR-181a, miR-519a, miR-374a and miR-630), which are critical downstream targets of the p-ΔNp63α, modulated the protein levels of ATG5, ATG6/BECN1, ATG10, ATG12, ATG16L1 and UVRAG, adding another level of expression control for autophagic pathways in SCC cells upon cisplatin exposure. Our data support the notion that the cisplatin-induced p-ΔNp63α could regulate key pathways implicated in response of cancer cells to chemotherapeutics.
Introduction
Cisplatin is the most often used drug in the treatment of human epithelial cancers because of its ability to dramatically induce the programmed cell death (apoptosis) of neoplastic cells.Citation1,Citation2 However, almost all tumors eventually become resistant to platinum chemotherapy.Citation1,Citation2 At the molecular level, cisplatin induces DNA damage and leads to the accumulation of activated members of the p53 family.Citation3–Citation6 Tumor protein (TP)-53 transcriptional factor members (TP53, TP63 and TP73) respond to DNA damage through the induction of cell cycle arrest, apoptosis or cellular senescence.Citation4–Citation6 When induced, TP53, TP63 and TP73 alter the expression of a large set of downstream target genes encoding mRNAs and microRNAs (miRNAs), leading, in turn, to cell cycle arrest, cell death and increased DNA repair.Citation4–Citation10 MiRNAs acting through a RNA interference pathway repress target gene expression at the posttranscriptional and translational levels, and their altered expression has been found in a variety of tumor types, and several functional studies have shown the oncogenic, tumor-suppressive or apoptotic potential of specific miRNAs.Citation8–Citation14
Emerging evidence supports the notion that a cisplatin-induced autophagy plays a central role in tumor cell resistance to platinum-based therapy.Citation15–Citation23 A dose- and time-dependent induction of autophagy observed in tumor cells following cisplatin treatment is evidenced by upregulation of BECN1 and cisplatin-triggered activation of AMPK pathway leading to a subsequent suppression of mTOR activity.Citation16–Citation23 Accumulating evidence suggests that induction of autophagy leads to an adaptive cell response, which either preceds or delays cisplatin-induced apoptosis and contributes to chemoresistance in various pathophysiologic conditions, including cancer.Citation18–Citation20,Citation23–Citation32 Furthermore, several oncogenes and tumor suppressor genes, including Tp53 family members were shown to be involved in pathways leading to an autophagic outcome.Citation33–Citation37 Although the essential genes for autophagy (Atg) have been identified, the molecular mechanisms through which ATG proteins control autophagy in mammalian cells remain poorly understood.Citation36–Citation40
We previously observed that ΔNp63α is dynamically phosphorylated (p-ΔNp63α) following exposure of squamous cell carcinoma (SCC) cells to cisplatin.Citation41–Citation43 We further found that the S385G mutation targeting the TA2 domain in ΔNp63α negatively affected the phosphorylation of this protein.Citation41 We then showed that a large number of genes that are involved in apoptosis, cell cycle arrest, DNA damage response and endoplasmic reticulum stress are specifically targeted by the p-ΔNp63α transcriptional factor in ΔNp63α-wt SCC cells upon cisplatin exposure.Citation42 However, the S385G mutation in the ΔNp63α protein dramatically abolished the transcriptional regulation of downstream genes and binding of the non-p-ΔNp63α protein to the specific promoters of mRNAs and miRNAs in ΔNp63α-S385G SCC cells upon cisplatin exposure.Citation8,Citation41–Citation43 These observations led us to suggest that ΔNp63α phosphorylation resulted in a decrease of its lifespan, which, in turn, activated its transcriptional activity within 12–24 h followed by its degradation.Citation41,Citation42 Coordinated phosphorylation-dependent degradation of transcription factors was shown to connect activity of transcription factor (oncogenic or tumor suppressive), and its destruction playing an important role in controlling cell growth (reviewed in ref. Citation42).
Here, we present evidence that the endogenous p-ΔNp63α transcription factor regulates autophagic gene expression in cisplatin-sensitive SCC (ΔNp63α-wt) cells through both transcriptional and posttranscriptional mechanisms, while it fails to perform these functions in cisplatin-resistant SCC (ΔNp63α-S385G) cells.
Results
Transcriptional regulation of autophagic genes by p-ΔNp63α.
In this study, we again employed the SCC JHU-029 stable clones ΔNp63α-wt and ΔNp63α-S385G, which exclusively produce endogenous wild-type (wt)-ΔNp63α or ΔNp63α-S385G, respectively.Citation41–Citation43 The idea behind the S385G mutation is that the S385 residue of the ΔNp63α protein is phosphorylated by ATM kinase in SCC cells exposed to cisplatin treatment.Citation41 However, changing the “serine” residue for “glycine” residue alters the ability for ΔNp63α-S385G to be phosphorylated by ATM kinase.Citation41,Citation42 Although both TAp63α and ΔNp63α proteins showed many potential and active phosphorylation sites for serine/threonine and tyrosine kinases, S385 was reported to function as a target for ATM kinase serving as a master switch for protein kinase pathways induced by cisplatin exposure.Citation41 The S385G change was shown to subsequently lead to cisplatin resistance, per se, for SCC cells.Citation41–Citation43 We found that the p-ΔNp63α transcriptional factor is potentially implicated in fine regulation of specific target genes involved in cell death through cell cycle arrest and apoptosis.Citation42,Citation43 We further showed that the endogenous p-ΔNp63α protein forms complexes with the NF-Y transcriptional regulator while binding the TP63-responsive elements in the gene promoters.Citation42,Citation43
Here, we extend our studies to further examine the role for endogenous p-ΔNp63α in transcriptional regulation of the specific autophagic gene promoters. First, we analyzed the specific autophagic promoters (Atg1/Ulk1, Atg3, Atg4A, Atg5, Atg6/Becn1, Atg7, Atg9A and Atg10) using TFSEARCH and TRANSFAC databases and defined the putative consensus elements that potentially bind several transcriptional factors (Figs. S1-8). TP63-responsive element was defined according to the previous report in reference Citation44. We further exposed ΔNp63α-wt cells and ΔNp63α-S385G cells to control medium (without cisplatin) or medium with 10 µg/ml cisplatin for 12 h and applied the ChIP analysis with anti-ΔNp63 antibody to determine whether ΔNp63α preferentially binds to specific autophagic promoters (Atg1/Ulk1, Atg3, Atg4A, Atg5, Atg6/Becn1, Atg7, Atg9A and Atg10) upon cisplatin exposure. As positive controls, we used input DNA-derived PCR of specific sequences from the various autophagic promoters containing TP63-responsive elements (as underlined in Figs. S1–8). As negative controls, we used ChiP-PCR from non-specific sequences (without TP63-responsive elements as underlined in Figs. S1–8). We showed that endogenous ΔNp63α bound to the specific regions of the tested promoters in ΔNp63α-wt cells upon cisplatin exposure (, left parts); however, no significant binding was observed for the specific sequences under control conditions without cisplatin (, left parts) or for the non-specific sequences under both control (without cisplatin) and cisplatin-induced conditions (, right parts). We further found that S385G mutation blocked the binding of the endogenous ΔNp63α protein to the tested autophagic gene promoters even under cisplatin-induced conditions ().
We further examined whether endogenous p-ΔNp63α affects the functionality of the autophagic gene promoters using the LightSwitch Pro Reporter System, which allows monitoring the Renilla luciferase reporter activity driven by the tested autophagic gene promoters. We thus found that cisplatin treatment (10 µg/ml, 12 h) dramatically induced the luciferase activity controlled by the tested autophagic promoters (Atg1/Ulk1, Atg3, Atg4A, Atg5, Atg6/Becn1, Atg7, Atg9A and Atg10) in ΔNp63α-wt cells (). However, no significant changes were observed in ΔNp63α-S385G cells under cisplatin exposure compared with control conditions without cisplatin (). We next examined whether protein levels of these specific autophagic markers changed in ΔNp63α-wt cells upon cisplatin exposure (10 µg/ml, 16 h) and found an increase in the ATG1/ULK1, ATG3, ATG4A, ATG5, ATG6/BECN1, ATG7, ATG9A and ATG10 protein levels in ΔNp63α-wt cells ( and B), while no such changes were observed in ΔNp63α-S385G cells ( and D) after cisplatin treatment (10 µg/ml, 16 h). Altogether, these results strongly suggest that the endogenous p-ΔNp63α protein transcriptionally regulates the expression of specific autophagic genes.
Regulation of autophagic gene expression by p-ΔNp63-dependent miRNAs.
We previously found that cisplatin dramatically downregulated three miRNAs (miR-519a, miR-181a, miR-374a) and upregulated one miRNA (miR-630) in SCC cells in a p-ΔNp63α-dependent manner, as shown in Tables S1 and 2 and reported in detail in reference Citation8. Using the following databases: microRNA.org, Targets and Expression; TargetScan:, Prediction of microRNA targets and miRDB, MicroRNA Target Prediction And Functional Study Database, we found a number of the miRNA-bound mRNA targets (that identified by miRanda with mirSVR scores from -0.1256 to -0.5491, PhastCons scores 0.4775–0.6217 and target prediction scores from 73 to 85) involved in autophagic processes. They include the 3′-untranslated region (UTR) “seed” sequences for miR-181a and miR-374a in Atg5, for miR-519a in Atg6 and Atg10, for miR-630 in Atg12, for miR-519a in Atg16L1, and for miR-374a and miR-630 in Uvrag (Fig. S9). We suggest that these miRNA targets could potentially be affected by the cisplatin-induced p-ΔNp63α-dependent miRNAs in SCC cells upon cisplatin exposure.
To validate the predicted miRNA/3′-UTR relationships, we examined whether the specific miRNAs modulate the reporter activity fused to the 3′-UTR “seed” sequences of target mRNA in SCC cells, and whether cisplatin exposure affects this process. ΔNp63α-wt cells were transfected with the LightSwitch UTR plasmids for Atg5, Atg6/Becn1, Atg10, Atg12, Atg16L1 and Uvrag along with control (scrambled), mimic and inhibitor miRNAs (miR-181a, miR-519a, miR-374a and miR-630) for 36 h as indicated (). Cells with the scrambled miRNA were exposed to control medium without cisplatin or medium with 10µg/ml cisplatin for 12 h, while cells with mimic or inhibitors for specific miRNAs were exclusively exposed to control medium without cisplatin. We showed that cisplatin exposure induced the luciferase reporter activity driven by the 3′-UTR-derived from Atg5, Atg6/Becn1, Atg10 and Atg16L1 (). At the same time, mimics for miR-181a, miR-374a and miR-519a dramatically modulated these activities (), while inhibitors for miR181a, miR-374a and miR-519a mirrored the cisplatin-induced effect (), supporting the notion that cisplatin exposure downregulates levels of miR-181a, miR-374a and miR-519a. Although cisplatin and mimic for miR-630 inhibited the 3′-UTR-driven luciferase activity for Atg12, supporting the previous observation of the cisplatin-dependent upregulation of miR-630 level (), miR-630 inhibitor showed no changes compared with control medium without cisplatin (). Intriguingly, two miRNA mimics (miR-374a and miR-630) displayed the inhibitory effect on the Uvrag 3′-UTR-driven luciferase activity, while both inhibitors activated this activity (). Since cisplatin was shown to downregulate miR-374a and upregulate miR-630 in SCC cells at a similar degree (Table S1), the effect of these miRNAs appears to be mutually exclusive for the Uvrag 3′-UTR luciferase activity upon cisplatin treatment ().
We next tested the protein levels of the autophagic markers (ATG5, ATG6/BECN1, ATG10, ATG12, ATG16L1 and UVRAG) in total lysates obtained from ΔNp63α-wt cells transfected with the indicated miRNA mimics and inhibitors for 24 h and subsequently treated with control medium without cisplatin or medium with 10 µg/ml cisplatin for 16 h. We showed that ATG5, ATG6, ATG10, ATG12 and ATG16L1 levels upregulated by cisplatin or miRNA inhibitors compared with control conditions without cisplatin ( and E). However, ATG12 levels were downregulated by cisplatin and miR-630 mimic compared with control conditions without cisplatin (). Interestingly, UVRAG levels changed slightly after cisplatin exposure, since the effects of miR-374a and miR-630 were likely to be mutually cancelled ().
Cisplatin induced autophagy in SCC cells through a ΔNp63α-dependent pathway.
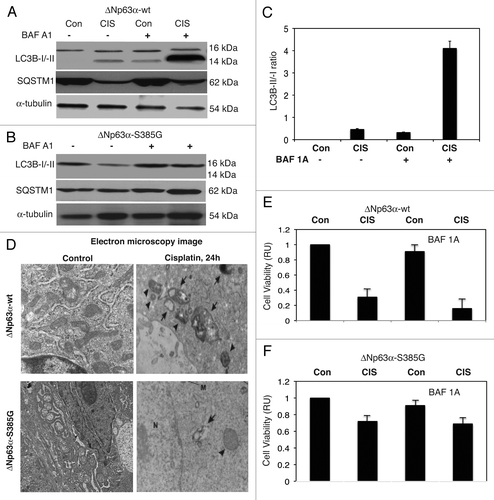
Our prior study showed that ΔNp63α functions as pro-survival regulator contributing to cisplatin resistance by activating Akt1 transcription in SCC and ovarian cancer cells.Citation44 However, the endogenous p-ΔNp63α was shown to induce several cell death mRNAs and miRNAs.Citation8,Citation42,Citation43 Because cisplatin's effect on the autophagy flux is well documented in several pathophysiologic conditions by immunofluorescent/electron microscopy, LC3B-I/-II conversion and SQSTM1 degradation,Citation15–Citation23,Citation45,Citation46 we therefore examined the effect of cisplatin exposure on ΔNp63α-wt cells and ΔNp63α-S385G cells. Since S385G mutation in endogenous ΔNp63α abolished its ability to be phosphorylated by ATM kinase upon cisplatin exposure, we evaluated whether this impaired protein modification would affect ΔNp63α activity in regard to apoptotic flux pathway. Both ΔNp63α-wt cells and ΔNp63α-S385G cells were exposed to control medium without cisplatin or medium with 10 µg/ml cisplatin. Cisplatin was shown to induce the conversion of cytosolic LC3B-I into a phosphatidyl-ethanolamine-conjugated LC3B-II as well as degradation of SQSTM1 (p62) in ΔNp63α-wt cells () but not in ΔNp63α-S385G cells (). We then grew cells in the presence of bafilomycin A1 (BAF A1), the inhibitor of certain steps of autophagy flux/lysosomal degradation pathway, which was shown to enhance DNA fragmentation and apoptosis.Citation20,Citation47–Citation49 We found that BAF A1 significantly increased the LC3B-II protein levels and LC3B-II/-I ratio in ΔNp63α-wt cells upon cisplatin exposure (), while it had no effect on ΔNp63α-S385G cells (). Quantitative analysis showed that the LC3B-I/-II conversion was exclusively induced in ΔNp63α-wt cells by cisplatin and substantially increased in the presence of BAF A1 (). We further found that cisplatin exposure induced the formation of autophagosomes in ΔNp63α-wt cells (, upper part), while only one or two appeared in ΔNp63α-S385G cells (, lower part). We next showed that sensitive ΔNp63α-wt cells exposed to cisplatin displayed a low viability compared with control conditions without cisplatin (). However, resistant ΔNp63α-S385G cells showed much greater ability to survive under cisplatin pressure (). We finally found that BAF A1 dramatically enhanced the cell death of ΔNp63α-wt cells, while it only slightly affected ΔNp63α-S385G cells ( and F), supporting the notion that the autophagic pathway (which is induced by endogenous p-ΔNp63α but not non-p-ΔNp63α) is delaying or even inhibiting a cisplatin-mediated apoptotic cell death.
Discussion
Mounting evidence supports that autophagic pathways, which can be stimulated by multiple forms of cellular stress, including nutrient or growth factor deprivation, hypoxia, reactive oxygen species, DNA damage, protein aggregates, damaged organelles or intracellular pathogens, are thereby central components of the integrated cellular response to stress.Citation25–Citation35 Autophagy can serve as a cell survival pathway by suppressing apoptosis or just delaying apoptotic cell death.Citation50,Citation51
Pharmacological or genetic inhibition of autophagy was shown to result in an increased cell death, suggesting a protective role of autophagy induced by chemotherapeutic agents (doxorubicin, cisplatin, paclitaxel and tamoxifen) that might increase cell survival in response to DNA damage via BECN1 induction, PARP1 activation or HMGB1 release.Citation15–Citation24,Citation27,Citation31,Citation47–Citation53 Other studies uncovered that the reactive oxygen species induced ATM-TSC2-mTORC1-mediated autophagy, thereby supporting the crosstalk between the cellular damage response and key pathways involved in metabolism, protein synthesis and cell survival.Citation54,Citation55 Prior reports showed that cisplatin leads to an autophagic flux in various pathophysiologic conditions, including tumor cell chemoresistance, nephrotoxicity and tubular cell injury, as shown by immunofluorescent and electron microscopy, LC3BI/-II conversion and SQSTM1 degradation.Citation15–Citation24,Citation46,Citation47,Citation52
As in our previous reports, we employed the unique cellular models of SCC cells expressing endogenous ΔNp63α-wt or ΔNp63α-S385G proteins.Citation8,Citation41–Citation43 The advantage of these SCC clones is that upon cisplatin exposure, endogenous ΔNp63α-wt protein undergoes ATM-dependent phosphorylation at the S385, leading to its subsequent degradation, while mutated ΔNp63α-S385G protein fails to be phosphorylated and remains fairly stable under cisplatin pressure.Citation41 In our experimental settings, endogenous p-ΔNp63α was shown to activate expression of numerous genes involved in apoptosis and cell cycle arrest, while endogenous non-p-ΔNp63α displayed the characteristics of a pro-survival regulator, contributing to tumorigenesis and chemoresistance, and failed to induce either apoptosis or autophagy.Citation42,Citation43 We previously showed that cisplatin exposure of sensitive ΔNp63α-wt cells leads to a formation of endogenous p-ΔNp63α, which binds to a proteasomic regulator, Rpn13, thereby protecting and enhancing the LKB1-induced autophagic flux assessed by immunofluorescent microscopy and LC3B-I/-II conversion in the presence of a lysosomal inhibitor.Citation43 However, resistant ΔNp63α-S385G cells (that do not support the ATM-dependent phosphorylation of ΔNp63α and produced non-pΔNp63α) failed to display autophagic characteristics in response to cisplatin treatment.Citation43 We re-examined this process in more detail, showing that the cisplatin exposure induced autophagic flux in ΔNp63α-wt cells evidenced by electron microscopy, LC3B-I/LC3B-II conversion and SQSTM1 degradation in the presence and absence of autophagosome/lysosomal inhibitor. We observed that the endogenous p-ΔNp63α expressed in ΔNp63α-wt cells upon cisplatin exposure induced autophagosome flux, as evidenced by LC3B-I/-II conversion, p62 (SQSTM1) downregulation and formation of autophagosomes. However, the endogenous non-p-ΔNp63α protein expressed in ΔNp63α-S385G cells failed to induce these autophagic processes after cisplatin exposure.
Using a systems biology approach, previous studies emphasized the role for E2F1, TP53, SREBP1, USF, AP-1, FOXO1, NFE2 and NAC1 in the regulation of the autophagic gene promoters, while miR-10b, 30a, 98, 101, 124, 130, 142 and 204 were shown to act as putative posttranscriptional regulators of the autophagic-lysosomal genes.Citation28,Citation33–Citation40,Citation46,Citation56–Citation59 E2F1 transcription factor was shown to induce expression of specific autophagic genes (e.g., Map1Lc3, Atg1 and Dram1) through a direct binding to the E2F1 cis-elements encompassing the promoters of these genes.Citation36 E2F1 activation was shown to enhance autophagy while reducing endogenous E2F1 expression inhibited DNA damage-induced autophagy.Citation34,Citation36
Accumulating evidence supports that Tp53 family transcription factors regulate a network of genes implicated in cell death and may enhance or fine-tune the autophagic response upon stress stimuli.Citation33–Citation40 Tp53 can modulate autophagy in a dual fashion, depending on its subcellular localization.Citation33–Citation38 Tp53 functions as a nuclear transcription factor transactivating pro-apoptotic, cell cycle-arrest and pro-autophagic genes. However, cytoplasmic Tp53 can trigger a mitochondrial mechanism to promote cell death and can repress autophagy. Genotoxic stress can induce autophagy in a TP53-dependent fashion through transcriptional activation of the autophagy-inducing genes.Citation60–Citation62 Two genes known to be critical regulators of autophagy damage-regulated autophagy modulator, Dram1 and autophagy-initiation kinase (Atg1/Ulk1), were shown to be transcriptionally activated by Tp53, leading to a subsequent induction of autophagic pathways.Citation60
Other members of the Tp53 family, notably, TP73, were also shown to modulate the autophagic process, however, through a DRAM-independent mechanism.Citation63,Citation64 Using ChIP analysis, Rosenbluth and Pietenpol demonstrated that endogenous TP73 binds the regulatory regions of specific autophagic genes, such as Atg5, Atg7 and Uvrag.Citation64 Although TP73 was found to induce expression of Atg5 and Atg7, the Tp73 knockdown increased Uvrag expression levels, suggesting that by binding to a region upstream of the Uvrag transcriptional start site, TP73 represses its expression via recruitment of histone deacetylases.Citation62,Citation66 ΔNp63α was shown to activate Atm transcription,Citation65 thereby contributing to ATM-TSC2-mTORC1-dependent autophagic pathway.Citation55 We have previously reported that the endogenous p-ΔNp63α, but not non-p-ΔNp63α, induced some autophagic features through an indirect stabilization of LKB1 levels and was likely to activate the ATM-TSC2-mTORC1 signaling in SCC cells upon cisplatin exposure.Citation43 However, a direct evidence of TP63 involvement in regulation of autophagic flux markers was missing.
To fill this gap, we examined the role for p-ΔNp63α vs. nonp-ΔNp63α in transcriptional and posttranscriptional regulation of autophagic genes, such as Atg1/Ulk1, Atg3, Atg4A, Atg5, Atg6/Becn1, Atg7, Atg9A and Atg10 in SCC cells upon cisplatin exposure. In contrast to endogenous non-p-ΔNp63α, endogenous p-ΔNp63α was shown to activate transcription of the specific autophagic promoters and increased the protein levels of tested autophagic proteins after cisplatin treatment. Our previous report showed that a set of specific miRNAs was also affected by cisplatin-induced p-ΔNp63α transcription factor, leading to a downregulation of miR-181a, miR-519a and miR374a and upregulation of miR-630 in SCC cells.Citation8 In this study, we further found that mimics for these miRNAs (miR-181a, miR-519a, miR-374a and miR-630) downregulated the ATG5, ATG6/BECN1, ATG10, ATG12, ATG16L1 and UVRAG protein levels, while tested miRNA inhibitors upregulated protein levels of specific autophagic proteins, thereby adding another level of expression control for autophagic pathways in SCC cells upon cisplatin exposure. Interestingly, some autophagic proteins (e.g., UVRAG) appeared to be targeted by several miRNAs, ones that are downregulated upon cisplatin exposure (e.g., miR-374a) and ones that upregulated by cisplatin treatment (e.g., miR-630). Elucidation of the functional implications of this bivalent regulation may lead to a better understanding of the miRNA-mediated cellular stress response to cisplatin exposure specifically and to chemotherapeutic drugs in general.Citation66–Citation73
Distinct members of autophagic flux exert their specific roles through physical and functional interactions between each other and components of the apoptotic and cell cycle arrest machinery; thereby, the final outcome of cellular response to stress might lead to a variety of choices.Citation51,Citation54,Citation74 During chemotherapy- and UV (UV)-induced apoptosis, the UV-resistanceassociated gene product, UVRAG, activates the BECN1/phosphatidylinositol-3-kinase III (PI3K III) complex, which promotes autophagosome formation. UVRAG also interacts with BAX, thereby inhibiting mitochondrial translocation of the BAX, reducing cytochrome c release, caspase 9 and -3 activation and, ultimately, suppressing apoptosis.Citation75–Citation78 BECN1 was shown to interact with several cell death regulators, including ATG14L, UVRAG, BIF1, RUBICON, AMBRA1, HMGB1 and others.Citation54 BECN1 binds to BIF1 through UVRAG while activating autophagy.Citation76,Citation78 However, BECN1 binds to either ATG14L or UVRAG while forming complexes with PI3K III (reviewed in refs. Citation78 and Citation79). Interestingly, Becn1-specific miR30a was shown to inhibit BECN1 protein expression and leads to reduced autophagic flux.Citation80 SQSTM1 was also found to associate with ATG8 and LC3B and facilitate degradation of ubiquitinated protein aggregates by autophagy, while cisplatin-resistant tumor cells expressed much higher levels of SQSTM1 than did cisplatin-sensitive tumor cells.Citation47,Citation81 The switch from autophagy to apoptosis was shown by downregulation of autophagic proteins, BECN1 and ATG7, calpain-mediated cleavage of ATG5 and caspase 8-mediated cleavage of BECN1 (reviewed in refs. Citation82–Citation84). This functional network supports an idea of intricate interplay between autophagic and apoptotic pathways, between cell life and death.Citation50,Citation54,Citation56,Citation57
Cisplatin treatment of ΔNp63α-wt cells was previously shown to reduce levels of p-ΔNp63α-dependent miR-181a, miR519a and miR-374a and to concomitantly induce the miR-630 levels, thereby leading to drastic changes in survival of SCC cells.Citation8 Interestingly, p-ΔNp63α induced transcription of specific autophagic proteins, while p-ΔNp63α-dependent miRNA's expression was activated (as miR-630) or reduced (as miR181a, miR-519a and miR-374a). As a result, the protein levels of specific autophagic proteins were modulated by a dual transcriptional and posttranscriptional mechanism involving the p-ΔNp63α transcriptional factor and miRNAs regulated by p-ΔNp63α. We found that cisplatin-sensitive ΔNp63α-wt cells displayed a low viability while treated with cisplatin. However, resistant ΔNp63α-S385G cells showed much greater ability to survive under cisplatin exposure. When the cisplatin pressure was applied to SCC cells that were treated in the presence of BAF A1, ΔNp63α-wt cells, but not ΔNp63α-S385G cells, displayed the enhanced cell death phenotype, further supporting the notion that autophagy induced by cisplatin in this experimental model is likely to protect SCC cells against apoptosis. Our prior study showed that miR-181a, miR-519a, miR-374a and miR-630 mimics dramatically reduced, while their inhibitors induced, levels of cell cycle arrest and pro-apoptotic regulators, such as CDKN1A and 1B, PARP1 and 2, TP73, YES and anti-apoptotic regulators, such as BCL2 and BCL2L2 (reviewed in ref. Citation8). We further report that in addition to the targets regulating cell cycle arrest and apoptosis, p-ΔNp63α but not non-p-ΔNp63α modulated the expression of the specific autophagic signaling intermediates (Atg1/Ulk1, Atg3, Atg4A, Atg5, Atg6/Becn1, Atg7, Atg9A and Atg10) through a p-ΔNp63α-mediated transcription and (ATG5, ATG6/BECN1, ATG10, ATG12, ATG16L1 and UVRAG) via a miRNA-dependent modulation.
Materials and Methods
Cells and reagents.
The squamous cell carcinoma (SCC) cell line JHU-029 (expressing wt-TP53, wt-TP63 is amplified and ΔNp63α is overexpressed) was isolated from primary tissue at the Department of Otolaryngology/Head and Neck Surgery (JHMI) then obtained from the Head and Neck Cancer Research Division Tissue Bank (characterized, tested and provided as a gift by Dr. Joseph A. Califano (JHMI), as described in references Citation45 and Citation85). The original cell line was authenticated by a short tandem repeat profiling analysis using the AmpFISTR Identifiler PCR Amplification Lit (Applied Biosystems) with the following 16 markers: amelogenin, CSF1PO, D12S317, D16S539, D18S51, D19S433, D21S11, D2S1338, D3S1358, D5S818, D7S820, D8S1179, FGA, THO1, TPOX and VWA at the JHMI Fragment Analysis Facility. The stable SCC cell lines expressing ΔNp63α-wt or ΔNp63α-S385G were generated using Flp-In technology.Citation41,Citation42 Cells were maintained in RPMI medium 1640 and 10% fetal bovine serum and incubated with control medium without cis-diamminedichloroplatinum (cisplatin, CIS, Sigma) or medium with 10 µg/ml cisplatin (Sigma) for the indicated time periods.Citation41,Citation42 Cells were lysed with 50 mM Tris, pH 7.5, 100 mM NaCl, 2 mM EDTA, 0.5% Triton X-100, 0.5% Brij-50, 1 mM PMSF, 0.5 mM NaF, 0.1 mM Na3VO4, 2x complete pro-tease inhibitor cocktail, sonicated for 10 sec intervals and spun for 30 min at 15,000x g.Citation41,Citation42 Supernatants were analyzed by immunoblotting, and the levels of tested proteins were normalized against β-actin or α-tubulin levels.Citation41–Citation43
Antibodies.
We used a monoclonal antibody against human β-actin (Sigma), a rabbit polyclonal antibody against ΔNp63 [anti-p40, PC373, residues 5–17 epitope (EMD/Calbiochem), polyclonal antibodies against ATG1/ULK1 (ab71059), ATG3 (ab66287), ATG4A (ab64738), ATG5 (ab77580), ATG6/BECN1 (ab62472), ATG7 (ab89775), ATG9A (ab54500), ATG10 (ab64125), ATG12 (ab64142) and UVRAG (ab70807)] purchased from Abcam and a monoclonal antibody against ATG16L1 (ST1525) purchased from EMD Corporation. Antibodies to MAP1LC3B (#3868), α-tubulin (#2125) and SQSTM1 (#8025) were obtained from Cell Signaling. A custom rabbit polyclonal antibody against a phosphorylated pep-tide encompassing the ΔNp63α protein sequence (ATM motif, NKLPSV-pS-QLINPQQ, residues 379–392) was prepared.Citation41,Citation42
Chromatin immunoprecipitation (ChIp).
First, 5 × 106 cell equivalents of chromatin (2–2.5 kb in size) were immunoprecipitated (IP) with 10 µg of anti-ΔNp63 antibody as described in references Citation8, Citation42 and Citation43. After reversal of formaldehyde cross-linking, RNA-ase A and proteinase K treatments, IP-enriched DNAs were used for PCR amplification. PCR consisted of 40 cycles of 94°C for 30 s, 60°C for 30 s and 72°C for 30 s using Taq DNA polymerase (Invitrogen). Although the tested promoters contain multiple potential TP63 binding sites, the regions for PCR were selected based on the efficiency of amplification, choosing the highest PCR outcome. The following PCR primers were used to amplify specific regions of the autophagic promoters: Atg1/Ulk1, sense, (-700)
5′-TTC TAA GAA TTA ATT CTG CG-3′ (-681) and antisense (-520)
5′-GGG GAA GGC ATG GGA GGG GG-3′ (-501); Atg3, sense, (-800)
5′-ATA GCG CAT AGA GCC TGC GG-3′ (-781) and antisense, (-620)
5′-GCA GGC GCA ATT CCT TGC CC-3′ (-601); Atg4A, sense (-1,350)
5′-GCT GAA TAG TTC CTT TCC CT-3′ (-1,331) and antisense, (-1,170)
5′-AAA GTT CCA GAA AAG AGA AA-3′ (1,151); Atg5, sense, (-990)
5′-TCC TGT GGC TAC TAC TAG AA-3′ (-961) and antisense, (-800)
5′-TAC TAT TTT TTT TTT GAG AC-3′ (-781); Atg6/Becn1, sense, (-760)
5′-TGC AAC CTC CGC CTC CGG G-3′ (-741) and antisense, (-580)
5′-CAA AGT GCT GGA ATT ACA AG-3′ (-561); Atg7, sense, (-1,180)
5′-TTT GAG ACG GAG TCT CGC TC-3′ (-1,161), and antisense, (-1,000)
5′-TTT CAC CGT TTT AGC CGG GA-3′ (-981); Atg9A, sense, (-1,010)
5′-GTT AAC GTG AGG AGA TAC AT-3′ (-991) and antisense, (-830)
5′-CTG TCA CGT TTC ACA CGA AA-3′ (-811); and Atg10, sense, (-1,070)
5′-ATA ATC TAA ATT GGC AGC TA-3′ (-1,051) and antisense, (-880)
5′-AGC TCC TTT TCA GTC ACC CC-3′ (-861) yielding the 200–209 bp PCR products.
The non-specific regions for the autophagic promoters were amplified using the following primers: for Atg1/Ulk1, sense, (-1,350) 5′-TGC ACG TGG TGA AAG CCA TT-3′ (-1,331) and antisense, (-1,220) 5′-TGG CTG CCG GCG GTG TGA CT-3′ (-1,201); for Atg3, sense, (-600)
5′-GCG GGG CGC GGT TAC CTC CC-3′ (-581) and antisense, (-470)
5′-AGG TAT AGG CGA AAA AGC CC-3′ (-451); for Atg4A, sense, (-1,950)
5′-ACC CGG GAG GCA GAG GTT GC-3′ (-1,931) and antisense, (-1,820)
5′-ATT ATT TGT CAA AGG GAA TC-3′ (-1,801); for Atg5, sense, (-1,500)
5′-CTA CCA TTA TAT TTT ACT AT-3′ (-1,531) and antisense, (-1,370)
5′-GCA CCT TAA TCC CAC AAG CT-3′ (-1,351); for Atg6/Becn1, sense, (-1,950)
5′-CTG ACA CAC TCT TAA TTC TG-3′ (-1,931) and antisense, (-1,820)
5′-GGC AGT ACA TTT AGT GTA GT-3′ (-1,801); for Atg7, sense, (-500)
5′-GAA TAA CTT TAT CTC ACT GA-3′ (-481) and antisense, (-370)
5′-GCC ACC CTG ATG GCC CCT GT-3′ (-351); for Atg9A, sense, (-300)
5′-AGA CGC ACC AAA CGT CCC GG-3′ (-281) and antisense, (-170)
5′-CCC GCC CCA CGT AGA GCA CA-3′ (-151); for Atg10, sense, (-1,550)
5′-CAT TCT TTG CCA TGA GGG AT-3′ (-1,531) and antisense, (-1,420)
5′-TTT AAA TTA ACA TGG TGG TT-3′ (-1,401) yielding the 150 bp PCR products. The enrichment binding of endogenous p-ΔNp63α to the specific promoter regions was also assessed by quantitative PCR (qPCR) using the above-mentioned primers, as previously described in references Citation8 and Citation42. ChIP-PCR values (relative units, RU) were normalized by the glycerladehyde3-phospho-dehydrogenase (GAPDH) values and presented as fold changes over Input PCR divided by 100. Experiments were performed in triplicate.
Luciferase reporter assay.
We used the promoter-reporter plasmids for Atg1/Ulk1 (#S707592), Atg3 (#S708396), Atg4A (#S704488), Atg5 (#719105), Atg6/Becn1 (#719414), Atg7 (#S703125), Atg9A (#S707572) and Atg10 (#S707535) purchased from SwitchGear Genomics, and the 3′-UTR luciferase reporter plasmids for Atg5 (#S813215), Atg6/Becn1 (#S805083), Atg10 (#S806774), Atg12 (#S810024), Atg16L1 (#S809093) and Uvrag (#S812965) were also obtained from SwitchGear Genomics. For the promoter-mediated luciferase activity assay and 3′-UTR-mediated luciferase activity assay, a total of 5 × 104 cells/well in a 24-well plate were transfected with the control (empty) pLightSwitch_Prom vector (#S707592) or with the empty pLightSwitch_3UTR vector (#S890005), respectively using Fugene HD reagent (Roche) as recommended by the manufacturer. Both vectors represent a fully optimized reporter system that includes an improved luminescent reporter gene (RenSP). In addition, cells were transfected with the selected autophagic promoter or 3′-UTR plasmids as listed above. The LightSwitch Luciferase Assay Reagent (SwitchGear Genomics) enables the monitoring of luciferase reporter signal according to the manufacturer's protocol. For the 3′-UTR assays, cells were also transfected with 100 ng of the mimics or inhibitors of tested miRNAs. At 36 h, cells were also treated with 10 µg/ml cisplatin or control medium without cisplatin for an additional 12 h. The RenSP Renilla luciferase activity was measured at 480 nm using a luminometer.
miRNA mimics and inhibitors and transfection.
Individual miRNA mimics (precursors) [hsa-miR-181a (PM10381), hsa-miR519a (PM12922), hsa-miR-374a (PM12702) and hsa-miR-630 (PM11552)] and inhibitors [hsa-miR-181a (AM10381), hsa-miR519a (AM12922), hsa-miR-374a (AM12702) and hsa-miR-630 (AM11552)] were purchased from Ambion/Applied Biosystems.Citation8 As a negative (scrambled) control, we used the AllStars Negative Control siRNA for mammalian species (#1027281, Qiagen). Cells were transfected with 10–50 pmol of the mimic, inhibitor or scrambled control in 50–200 µl serum-free media with 5 µl Lipofectamine-2000 reagent (Invitrogen) for 36 h.
Transmission electron microscopy.
Cells were cultured in the presence or absence of the 10 µg/ml of cisplatin for 24 h. Cells were trypsinized, fixed and embedded in spur resin. The 90-nm thick sections were cut and examined with a JEOL 1200EX transmission electron microscope.Citation46
Cell viability assay.
104 cells/well in 96-well plates were then incubated in serum-free medium with 5 µg/ml of the 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (American Tissue Culture Collection) in the dark for 4 h at 37°C. Cells were lysed and incubated for 2 h at 37°C, and the measurements (A570 nm to A650 nm) were obtained on a Spectra Max 250 plate reader (Molecular Devices), as described in reference Citation44. Each assay was repeated three times in triplicate. Diagrams indicated the extent of cell viability expressed as a portion of control (cells grown without cisplatin) represented as 1. The bars are the mean ± SD of triplicate; p < 0.05, t-test.
Statistical analysis.
Results were expressed as means ± SD from at least three independent experiments in triplicate. Differences in measured variables between experimental and control group were assessed by using the Student t-test. Statistically significant difference was accepted at p < 0.05.
Bioinformatics.
To define the putative responsive elements we used the TFSEARCH (www.cbrc.jp/research/db/TFSEARCH.html) and TRANSFAC (v7.4, www.generegulation.com/pub/databases.html) databases. For prediction of the miRNA-specific “seed” sequences in target 3′-UTRs, we used the following databases: microRNA.org-Targets and Expression (August 2010, Computational Biology Center, Memorial Sloan-Kettering Cancer Center, www.microrna.org), TargetScan: Prediction of microRNA targets, v5.2 (June 2011, www.targetscan.org) and miRDB-MicroRNA Target Prediction And Functional Study Database, v3.0 (April 2009, www.mirdb.org). All the targets were selected based on mirSVR scores (from -0.1256 to -0.5491), PhastCons scores (0.4775–0.6217), and target prediction scores (from 73 to 85) were assigned by the computational target prediction algorithm.Citation52,Citation86
Concluding Remarks
Our observations suggest that p-ΔNp63α (but not non-pΔNp63α), along with Tp53 and TP73, positively regulates autophagic flux in SCC cells upon cisplatin exposure, while nonp-ΔNp63α plays the role of pro-survival regulator, activating Akt1 transcription and the β-catenin pathway.Citation44,Citation87,Citation88 Through regulation of miRNA expression and processing,Citation8 p-ΔNp63α also contributes to regulation of cell cycle arrest, apoptosis and autophagy, ultimately leading to a specific tumor cell response to chemotherapeutic stress. In contrast to non-p-ΔNp63α, the cisplatin-induced p-ΔNp63α activates key components that lead to autophagic flux and are subsequently implicated in the response of cancer cells to chemotherapeutic drugs. Although the execution of cell death is usually attributed to apoptosis, cell cycle arrest and necrosis, the autophagic flux signaling plays a critical role in delaying cell death, thereby generating the conditions for temporary cell survival under stress pressure.Citation26,Citation28,Citation30,Citation50
Disclosure of potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Dedication
I wish to dedicate this work to a memory of my father, Abraham G. Ratovitsky (on his centennial anniversary), whose life and principles always guided me through my life experiences.
Abbreviations
| ATG | = | autophagy-like gene |
| AMPK | = | AMP-activated protein kinase |
| ATM | = | ataxia telangiectasia mutated |
| BECN1 | = | beclin 1 |
| ChIP | = | chromatin immunoprecipitation |
| DRAM | = | damage-regulated autophagy modulator |
| GAPDH | = | glyceraldehyde3-phosphate dehydrogenase |
| HMG | = | high mobility group protein |
| NF | = | nuclear factor |
| PARP | = | poly-ADP ribose polymerase |
| RU | = | relative units |
| RLU | = | relative luciferase units |
| SCC | = | squamous cell carcinoma |
| SQSTM | = | sequestrome |
| TP | = | tumor protein |
| UTR | = | untranslated region |
| UVRAG | = | ultraviolet radiation resistance-associated gene |
Figures and Tables
Figure 1 P-ΔNp63α bound to the autophagic gene promoters upon cisplatin treatment. ΔNp63α-wt cells were exposed to control medium without cisplatin or medium with10 µg/ml of cisplatin for 12 h. (A) ChIP analysis of the p-ΔNp63α protein binding to the specific region (left parts) and non-specific region (right parts) of the indicated autophagic gene promoter sequences. The amplification of the Input DNA (upper parts) and ChiP-precipitated DNA from untreated cells (middle parts) and cisplatin-treated cells (lower parts) was shown with primers for the specific region and control (non-specific) region. (B) qPCR analysis of the ΔNp63α-enriched binding to the autophagic promoters in ΔNp63α-wt cells and ΔNp63α-S385G cells upon cisplatin exposure. qPCR experiments were performed in triplicate with ±SD as indicated (<0.05). The amount of immunoprecipitated-enriched DNA in each sample (ChIP) is represented as signal relative to the total amount of input chromatin DNA (Input) using the same primers multiplied by 100.
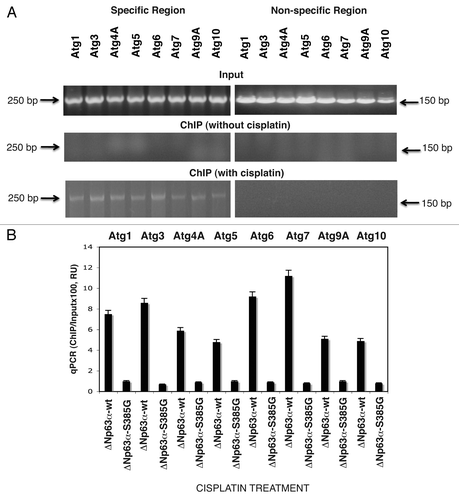
Figure 2 P-ΔNp63α regulated the luciferase activity driven by the autophagic gene promoters upon cisplatin treatment. ΔNp63α-wt cells (A) and ΔNp63α-S385G cells (B) were transfected for 24 h with 100 ng of the LightSwitch_Pro reporter plasmids containing the indicated autophagic promoters or with 100 ng of the control promoter-less reporter plasmid. Cells were exposed to control medium without cisplatin (Con) and medium with 10 µg/ml cisplatin (CIS) for 12 h. RenSP Renilla luciferase reporter activity assays were conducted in triplicate (±SD are indicated, p < 0.05). Data presented as relative to data obtained from the control untreated cells containing the promoter-less reporter plasmid designated as 1.
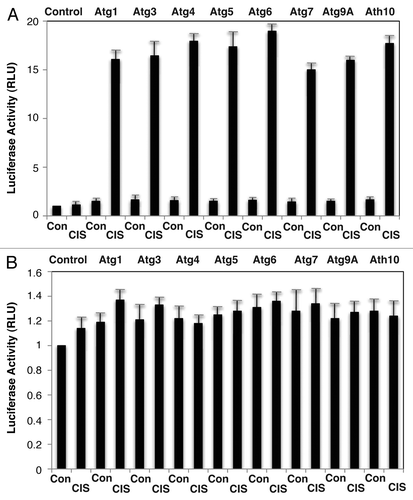
Figure 3 Cisplatin induced specific autophagic proteins. Cells were treated with control medium without cisplatin (Con) or medium with 10 µg/ml cisplatin (CIS) for 16 h. Protein levels were analyzed by immunoblotting with indicated antibodies. The loading levels were tested with an antibody to β-actin. (A and B) ΔNp63α-wt cells. (C and D) ΔNp63α-S385G cells.
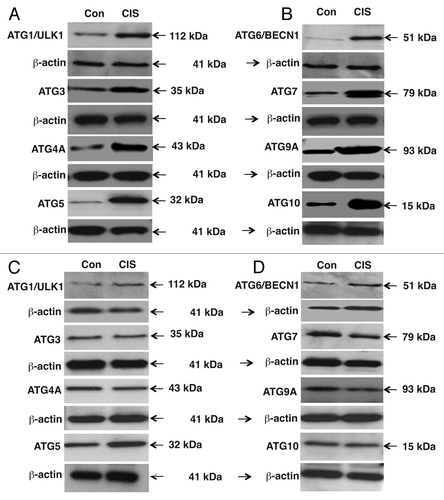
Figure 4 The 3′-UTR sequences for the target autophagic mRNA bound to specific miRNAs upon cisplatin exposure. ΔNp63α-wt cells were transfected with the LightSwitch_3UTR vectors for the specific 3′-UTR sequences as indicated ((A) Atg5/miR-181a and Atg5/miR-374a; (B) Atg6/miR-519a and Atg10/miR-519a; (C) Atg16L/miR-519a and Atg12/miR-630, (D) Uvrag/miR-630 and Uvrag/miR-374a. Cells were also transfected with mock (scrambled) miRNA, and specific miRNA mimics and inhibitors for 36 h. Cells were treated with control medium without cisplatin (Con) or medium with 10 µg/ml cisplatin (CIS) for additional 12 h and then tested for the RenSP Renilla luciferase reporter activity. Measurements (in triplicate) for the luciferase activity (RLU).
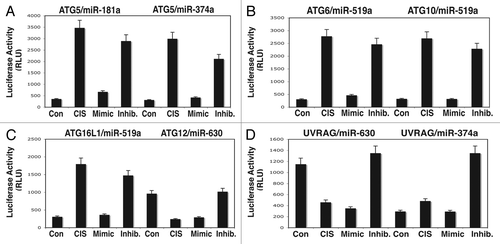
Figure 5 Cisplatin treatment modulates protein levels of autophagic markers through miRNA-dependent regulation. ΔNp63α-wt cells were transfected with the mock (scrambled) miRNA and specific miRNA mimics or inhibitors for 24 h. Cells were treated with control medium without cisplatin (Con) or medium with 10 µg/ml cisplatin (CIS) for additional 16 h and then tested for protein levels of the indicated endogenous autophagic markers. Loading levels were tested using an antibody against β-actin.
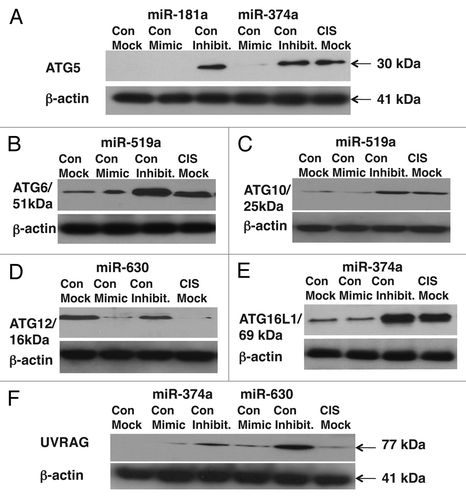
Figure 6 Cisplatin induced autophagic flux in SCC cells. ΔNp63α-wt (A) and ΔNp63α-S385G (B) cells were cultured in the presence (CIS) or absence (Con) of the 10 µg/ml of cisplatin for 16 h and protein levels for LC3B-I, -II, SQSTM1 (p62) and α-tubulin (loading control) were examined using immunoblotting with antibodies to MAP1LC3B (#3868), α-tubulin (#2125) and SQSTM1 (#8025) from Cell Signaling. In some samples, the 100 nM of bafilomycin A1 (BAF A1, #B1793, Sigma) was added for 12 h. (C) Quantitative analysis of LC3B-I/-II conversion. Immunoblots were scanned using Phosphorimager (Molecular Dynamics) and quantified by ImageQuant software version 3.3 (Molecular Dynamics). Values of LC3B-II were expressed as a portion of LC3B-I values defined as 1. The LC3B-II/LC3B-I ratios were plotted as bars using the Microsoft Excel software with standard deviations (±SD) resulting from three independent experiments and three individual measurements of each experiment. (p < 0.05, t-test). (D) ΔNp63α-wt cells (upper parts) and ΔNp63α-S385G cells (lower parts) were cultured in the presence (Cisplatin) or absence (Control) of the 10 µg/ml of cisplatin for 16 h. Cells were trypsinized, fixed and embedded in spur resin. the 90-nm thick sections were cut and examined with a JEOL 1200EX transmission electron microscope (46). Arrows indicate autophagic vacuoles. (E and F) Cell viability assay. ΔNp63α-wt cells (E) and ΔNp63α-S385G cells (F) were cultured in the presence (CIS) or absence (Con) of the 10 µg/ml of cisplatin for 72 h in the presence or absence of BAF A1 (100 nMm 12 h). 104 cells/well in 96-well plates were then incubated in serum-free medium with 5 µg/ml of the 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (American Tissue Culture Collection) in the dark for 4 h at 37°C. Cells were lysed and incubated for 2 h at 37°C, and the measurements (A570 nm to A650 nm) were obtained on a Spectra Max-250 plate reader (Molecular Devices), as described in reference Citation44. Each assay was repeated at three times in triplicate. Diagrams indicated the extent of cell viability expressed as a portion of control represented as 1. The bars are the mean ± SD of triplicate; p < 0.05, t-test.
Additional material
Download Zip (208.5 KB)Acknowledgements
This work is partly supported by the K01 grant CA164092 from NCI (R.G.P.) and by the grant #892469 (E.A.R.) from Flight Attendant Research Institutions.
Related Research Data
References
- Helmbach H, Kern MA, Rossmann E, Renz K, Kissel C, Gschwendt B, et al. Drug resistance towards etoposide and cisplatin in human melanoma cells is associated with drug-dependent apoptosis deficiency. J Invest Dermatol 2002; 118:923 - 932; PMID: 12060385; http://dx.doi.org/10.1046/j.1523-747.2002.01786.x
- Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer 2007; 7:573 - 584; PMID: 17625587; http://dx.doi.org/10.1038/nrc2167
- Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 2002; 416:560 - 564; PMID: 11932750; http://dx.doi.org/10.1038/416560a
- Oren M. Decision making by p53: life, death and cancer. Cell Death Differ 2003; 10:431 - 442; PMID: 12719720; http://dx.doi.org/10.1038/sj.cdd.4401183
- Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 2006; 9:45 - 56; PMID: 16413471; http://dx.doi.org/10.1016/j.ccr.2005.12.013
- Murray-Zmijewski F, Lane DP, Bourdon JC. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ 2006; 13:962 - 972; PMID: 16601753; http://dx.doi.org/10.1038/sj.cdd.4401914
- Yang C, Kaushal V, Haun RS, Seth R, Shah SV, Kaushal GP. Transcriptional activation of caspase 6 and -7 genes by cisplatin-induced p53 and its functional significance in cisplatin nephrotoxicity. Cell Death Differ 2008; 15:530 - 544; PMID: 18064040; http://dx.doi.org/10.1038/sj.cdd.4402287
- Huang Y, Chuang A, Hao H, Talbot C, Sen T, Trink B, et al. Phospho-ΔNp63α is a key regulator of the cisplatin-induced microRNAome in cancer cells. Cell Death Differ 2011; 18:1220 - 1230; PMID: 21274007; http://dx.doi.org/10.1038/cdd.2010.188
- Ory B, Ramsey MR, Wilson C, Vadysirisack DD, Forster N, Rocco JW, et al. A microRNA-dependent program controls p53-independent survival and chemosensitivity in human and murine squamous cell carcinoma. J Clin Invest 2011; 121:809 - 820; PMID: 21293058; http://dx.doi.org/10.1172/JCI43897
- Zhang X, Lu X. Posttranscriptional regulation of miR-NAs in the DNA damage response. RNA Biol 2011; 8:960 - 963; PMID: 21941125; http://dx.doi.org/10.4161/rna.8.6.17337
- Chen K, Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet 2007; 8:93 - 103; PMID: 17230196; http://dx.doi.org/10.1038/nrg1990
- Pratt AJ, MacRae IJ. The RNA-induced silencing complex: a versatile gene-silencing machine. J Biol Chem 2009; 284:17897 - 17901; PMID: 19342379; http://dx.doi.org/10.1074/jbc.R900012200
- Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA 2006; 103:2257 - 2261; PMID: 16461460; http://dx.doi.org/10.1073/pnas.0510565103
- Yang H, Kong W, He L, Zhao JJ, O'Donnell JD, Wang J, et al. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res 2008; 68:425 - 433; PMID: 18199536; http://dx.doi.org/10.1158/0008-5472.CAN-07-2488
- Claerhout S, Verschooten L, Van Kelst S, De Vos R, Proby C, Agostinis P, et al. Concomitant inhibition of AKT and autophagy is required for efficient cisplatin-induced apoptosis of metastatic skin carcinoma. Int J Cancer 2010; 127:2790 - 2803; PMID: 21351258; http://dx.doi.org/10.1002/ijc.25300
- O'Donovan TR, O'Sullivan GC, McKenna SL. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 2011; 7:509 - 524; PMID: 21325880; http://dx.doi.org/10.4161/auto.7.5.15066
- Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu Y, et al. DAMP-mediated autophagy contributes to drug resistance. Autophagy 2011; 7:112 - 114; PMID: 21068541; http://dx.doi.org/10.4161/auto.7.1.14005
- Yang C, Kaushal V, Shah SV, Kaushal GP. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am J Physiol Renal Physiol 2008; 294:777 - 787; PMID: 18256309; http://dx.doi.org/10.1152/ajprenal.00590.2007
- Kaushal GP, Kaushal V, Herzog C, Yang C. Autophagy delays apoptosis in renal tubular epithelial cells in cisplatin cytotoxicity. Autophagy 2008; 4:710 - 712; PMID: 18497570
- Harhaji-Trajkovic L, Vilimanovich U, Kravic-Stevovic T, Bumbasirevic V, Trajkovic V. AMPK-mediated autophagy inhibits apoptosis in cisplatin-treated tumour cells. J Cell Mol Med 2009; 13:3644 - 3654; PMID: 20196784; http://dx.doi.org/10.1111/j.1582-4934.2009.00663.x
- Ren JH, He WS, Nong L, Zhu QY, Hu K, Zhang RG, et al. Acquired cisplatin resistance in human lung adenocarcinoma cells is associated with enhanced autophagy. Cancer Biother Radiopharm 2010; 25:75 - 80; PMID: 20187799; http://dx.doi.org/10.1089/cbr.2009.0701
- Furuya D, Tsuji N, Yagihashi A, Watanabe N. Beclin 1 augmented cis-diamminedichloroplatinum induced apoptosis via enhancing caspase 9 activity. Exp Cell Res 2005; 307:26 - 40; PMID: 15922724; http://dx.doi.org/10.1016/j.yexcr.2005.02.023
- Inoue K, Kuwana H, Shimamura Y, Ogata K, Taniguchi Y, Kagawa T, et al. Cisplatin-induced macroautophagy occurs prior to apoptosis in proximal tubules in vivo. Clin Exp Nephrol 2010; 14:112 - 122; PMID: 20013139; http://dx.doi.org/10.1007/s10157-009-0254-7
- Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ 2007; 14:500 - 510; PMID: 16990848; http://dx.doi.org/10.1038/sj.cdd.4402039
- Berry DL, Baehrecke EH. Autophagy functions in programmed cell death. Autophagy 2008; 4:359 - 360; PMID: 18212526
- Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ 2009; 16:966 - 975; PMID: 19325568; http://dx.doi.org/10.1038/cdd.2009.33
- Hou W, Han J, Lu C, Goldstein LA, Rabinowich H. Autophagic degradation of active caspase 8: a crosstalk mechanism between autophagy and apoptosis. Autophagy 2010; 6:891 - 900; PMID: 20724831; http://dx.doi.org/10.4161/auto.6.7.13038
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280 - 293; PMID: 20965422; http://dx.doi.org/10.1016/j.molcel.2010.09.023
- Muñoz-Gámez JA, Rodríguez-Vargas JM, Quiles-Pérez R, Aguilar-Quesada R, Martín-Oliva D, de Murcia G, et al. PARP-1 is involved in autophagy induced by DNA damage. Autophagy 2009; 5:61 - 74; PMID: 19001878; http://dx.doi.org/10.4161/auto.5.1.7272
- Chen S, Rehman SK, Zhang W, Wen A, Yao L, Zhang J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim Biophys Acta 2010; 1806:220 - 229; PMID: 20637264
- Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE, et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010; 29:5299 - 5310; PMID: 20622903; http://dx.doi.org/10.1038/onc.2010.261
- Sun Y, Liu JH, Jin L, Lin SM, Yang Y, Sui YX, et al. Overexpression of the Beclin1 gene upregulates chemosensitivity to anti-cancer drugs by enhancing therapy-induced apoptosis in cervix squamous carcinoma CaSki cells. Cancer Lett 2010; 294:204 - 210; PMID: 20207475; http://dx.doi.org/10.1016/j.canlet.2010.02.001
- Tasdemir E, Chiara Maiuri M, Morselli E, Criollo A, D'Amelio M, Djavaheri-Mergny M, et al. A dual role of p53 in the control of autophagy. Autophagy 2008; 4:810 - 814; PMID: 18604159
- Maiuri MC, Tasdemir E, Criollo A, Morselli E, Vicencio JM, Carnuccio R, et al. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ 2009; 16:87 - 93; PMID: 18806760; http://dx.doi.org/10.1038/cdd.2008.131
- Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy 2008; 4:600 - 606; PMID: 18497563
- Polager S, Ofir M, Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 2008; 27:4860 - 4864; PMID: 18408756; http://dx.doi.org/10.1038/onc.2008.117
- Bitomsky N, Hofmann TG. Apoptosis and autophagy: Regulation of apoptosis by DNA damage signaling—roles of p53, p73 and HIPK2. FEBS J 2009; 276:6074 - 6083; PMID: 19788416; http://dx.doi.org/10.1111/j.1742-4658.2009.07331.x
- Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol 2009; 335:1 - 32; PMID: 19802558; http://dx.doi.org/10.1007/978-3-642-00302-8_1
- Kusama Y, Sato K, Kimura N, Mitamura J, Ohdaira H, Yoshida K. Comprehensive analysis of expression pattern and promoter regulation of human autophagy-related genes. Apoptosis 2009; 14:1165 - 1175; PMID: 19657746; http://dx.doi.org/10.1007/s10495-009-0390-2
- Liang C, Jung JU. Autophagy genes as tumor suppressors. Curr Opin Cell Biol 2010; 22:226 - 233; PMID: 19945837; http://dx.doi.org/10.1016/j.ceb.2009.11.003
- Huang Y, Sen T, Nagpal J, Upadhyay S, Trink B, Ratovitski E, et al. ATM kinase is a master switch for the ΔNp63α phosphorylation/degradation in human head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle 2008; 7:2846 - 2855; PMID: 18769144; http://dx.doi.org/10.4161/cc.7.18.6627
- Huang Y, Chuang AY, Romano RA, Liégeois NJ, Sinha S, Trink B, et al. Phospho-DeltaNp63α/NF-Y protein complex transcriptionally regulates DDIT3 expression in squamous cell carcinoma cells upon cisplatin exposure. Cell Cycle 2010; 9:328 - 338; PMID: 20023394; http://dx.doi.org/10.4161/cc.9.2.10432
- Huang Y, Ratovitski EA. Phospho-ΔNp63α/Rpn13dependent regulation of LKB1 degradation modulates autophagy in cancer cells. Aging (Albany NY) 2010; 2:959 - 968; PMID: 21191146
- Sen T, Sen N, Brait M, Begum S, Chatterjee A, Hoque MO, et al. DeltaNp63α confers tumor cell resistance to cisplatin through the AKT1 transcriptional regulation. Cancer Res 2011; 71:1167 - 1176; PMID: 21266360; http://dx.doi.org/10.1158/0008-5472.CAN-10-1481
- Zhao Y, Hao Y, Ji H, Fang Y, Guo Y, Sha W, et al. Combination effects of salvianolic acid B with low-dose celecoxib on inhibition of head and neck squamous cell carcinoma growth in vitro and in vivo. Cancer Prev Res (Phila) 2010; 3:787 - 796; PMID: 20501859; http://dx.doi.org/10.1158/1940-6207.CAPR-09-0243
- Zhang Y, Cheng Y, Ren X, Zhang L, Yap KL, Wu H, et al. NAC1 modulates sensitivity of ovarian cancer cells to cisplatin by altering the HMGB1-mediated autophagic response. Oncogene 2012; In press PMID: 21743489
- Yu H, Su J, Xu Y, Kang J, Li H, Zhang L, et al. p62/SQSTM1 involved in cisplatin resistance in human ovarian cancer cells by clearing ubiquitinated proteins. Eur J Cancer 2011; 47:1585 - 1594; PMID: 21371883; http://dx.doi.org/10.1016/j.ejca.2011.01.019
- Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes?. Autophagy 2008; 4:849 - 950; PMID: 18758232
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542 - 545; PMID: 17611390
- Shen S, Kepp O, Kroemer G. The end of autophagic cell death?. Autophagy 2012; 8:1 - 3; PMID: 22082964; http://dx.doi.org/10.4161/auto.8.1.16618
- Meschini S, Condello M, Lista P, Arancia G. Autophagy: Molecular mechanisms and their implications for anticancer therapies. Curr Cancer Drug Targets 2011; 11:357 - 379; PMID: 21247381; http://dx.doi.org/10.2174/156800911794519707
- Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 2007; 27:91 - 105; PMID: 17612493; http://dx.doi.org/10.1016/j.molcel.2007.06.017
- Xi G, Hu X, Wu B, Jiang H, Young CY, Pang Y, et al. Autophagy inhibition promotes paclitaxel-induced apoptosis in cancer cells. Cancer Lett 2011; 307:141 - 148; PMID: 21511395; http://dx.doi.org/10.1016/j.canlet.2011.03.026
- Maiuri MC, Criollo A, Kroemer G. Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J 2010; 29:515 - 516; PMID: 20125189; http://dx.doi.org/10.1038/emboj.2009.377
- Alexander A, Kim J, Walker CL. ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy 2010; 6:672 - 673; PMID: 20581436; http://dx.doi.org/10.4161/auto.6.5.12509
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466:68 - 76; PMID: 20562859; http://dx.doi.org/10.1038/nature09204
- Jegga AG, Schneider L, Ouyang X, Zhang J. Systems biology of the autophagy-lysosomal pathway. Autophagy 2011; 7:477 - 489; PMID: 21293178; http://dx.doi.org/10.4161/auto.7.5.14811
- Zhao J, Brault JJ, Schild A, Goldberg AL. Coordinate activation of autophagy and the proteasome pathway by FoxO transcription factor. Autophagy 2008; 4:378 - 380; PMID: 18227643
- Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 2007; 6:472 - 483; PMID: 18054316; http://dx.doi.org/10.1016/j.cmet.2007.11.004
- Crighton D, Wilkinson S, Ryan KM. DRAM links autophagy to p53 and programmed cell death. Autophagy 2007; 3:72 - 74; PMID: 17102582
- Scherz-Shouval R, Weidberg H, Gonen C, Wilder S, Elazar Z, Oren M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci USA 2010; 107:18511 - 18516; PMID: 20937856; http://dx.doi.org/10.1073/pnas.1006124107
- Gao W, Shen Z, Shang L, Wang X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damageinduced cell death. Cell Death Differ 2011; 18:1598 - 1607; PMID: 21475306; http://dx.doi.org/10.1038/cdd.2011.33
- Crighton D, O'Prey J, Bell HS, Ryan KM. p73 regulates DRAM-independent autophagy that does not contribute to programmed cell death. Cell Death Differ 2007; 14:1071 - 1079; PMID: 17304243; http://dx.doi.org/10.1038/sj.cdd.4402108
- Rosenbluth JM, Pietenpol JA. mTOR regulates autophagy-associated genes downstream of p73. Autophagy 2009; 5:114 - 116; PMID: 19001857; http://dx.doi.org/10.4161/auto.5.1.7294
- Craig AL, Holcakova J, Finlan LE, Nekulova M, Hrstka R, Gueven N, et al. DeltaNp63 transcriptionally regulates ATM to control p53 Serine-15 phosphorylation. Mol Cancer 2010; 9:195 - 201; PMID: 20663147; http://dx.doi.org/10.1186/1476-4598-9-195
- D'agostino L, Giordano A. NSP 5a3a: a potential novel cancer target in head and neck carcinoma. Oncotarget 2010; 1:423 - 435; PMID: 21311098
- Sen T, Chang X, Sidransky D, Chatterjee A. Regulation of ΔNp63α by NFκB. Cell Cycle 2010; 9:4841 - 4847; PMID: 21088498; http://dx.doi.org/10.4161/cc.9.24.14093
- Sunwoo JB. A cisplatin-resistant subpopulation of mesenchymal-like cells in head and neck squamous cell carcinoma. Cell Cycle 2011; 10:2834 - 2835; PMID: 21964299; http://dx.doi.org/10.4161/cc.10.17.16750
- Luchenko VL, Salcido CD, Zhang Y, Agama K, Komlodi-Pasztor E, Murphy RF, et al. Schedule-dependent synergy of histone deacetylase inhibitors with DNA damaging agents in small cell lung cancer. Cell Cycle 2011; 10:3119 - 3128; PMID: 21900747; http://dx.doi.org/10.4161/cc.10.18.17190
- Hoellein A, Pickhard A, von Keitz F, Schoeffmann S, Piontek G, Rudelius M, et al. Aurora kinase inhibition overcomes cetuximab resistance in squamous cell cancer of the head and neck. Oncotarget 2011; 2:599 - 609; PMID: 21865609
- Ory B, Ellisen LW. A microRNA-dependent circuit controlling p63/p73 homeostasis: p53 family crosstalk meets therapeutic opportunity. Oncotarget 2011; 2:259 - 264; PMID: 21436470
- Nohata N, Sone Y, Hanazawa T, Fuse M, Kikkawa N, Yoshino H, et al. miR-1 as a tumor suppressive microRNA targeting TAGLN2 in head and neck squamous cell carcinoma. Oncotarget 2011; 2:29 - 42; PMID: 21378409
- Nijwening JH, Kuiken HJ, Beijersbergen RL. Screening for modulators of cisplatin sensitivity: unbiased screens reveal common themes. Cell Cycle 2011; 10:380 - 386; PMID: 21239890; http://dx.doi.org/10.4161/cc.10.3.14642
- Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010; 6:764 - 776; PMID: 20639694; http://dx.doi.org/10.4161/auto.6.6.12709
- Yin X, Cao L, Kang R, Yang M, Wang Z, Peng Y, et al. UV irradiation resistance-associated gene suppresses apoptosis by interfering with BAX activation. EMBO Rep 2011; 12:727 - 734; PMID: 21597469; http://dx.doi.org/10.1038/embor.2011.79
- Itakura E, Mizushima N. Atg14 and UVRAG: mutually exclusive subunits of mammalian Beclin 1-PI3K complexes. Autophagy 2009; 5:534 - 536; PMID: 19223761; http://dx.doi.org/10.4161/auto.5.4.8062
- Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 2007; 9:1142 - 1151; PMID: 17891140; http://dx.doi.org/10.1038/ncb1634
- Liang C, Feng P, Ku B, Oh BH, Jung JU. UVRAG: a new player in autophagy and tumor cell growth. Autophagy 2007; 3:69 - 71; PMID: 17106237
- Yin XC, Cao L, Peng YH, Tan YF, Xie M, Kang R, et al. A critical role for UVRAG in apoptosis. Autophagy 2011; 7:1242 - 1244; PMID: 21606679; http://dx.doi.org/10.4161/auto.7.10.16507
- Zhu H, Wu H, Liu X, Li B, Chen Y, Ren X, et al. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy 2009; 5:816 - 823; PMID: 19535919
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 24145; PMID: 17580304; http://dx.doi.org/10.1074/jbc.M702824200
- Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase 8. Science 2004; 304:1500 - 1502; PMID: 15131264; http://dx.doi.org/10.1126/science.1096645
- Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 2006; 8:1124 - 1132; PMID: 16998475; http://dx.doi.org/10.1038/ncb1482
- Li H, Wang P, Yu J, Zhang L. Cleaving Beclin 1 to suppress autophagy in chemotherapy-induced apoptosis. Autophagy 2011; 7:1239 - 1241; PMID: 21610315; http://dx.doi.org/10.4161/auto.7.10.16490
- Gu X, Song X, Dong Y, Cai H, Walters E, Zhang R, et al. Vitamin E succinate induces ceramide-mediated apoptosis in head and neck squamous cell carcinoma in vitro and in vivo. Clin Cancer Res 2008; 14:1840 - 1848; PMID: 18347187; http://dx.doi.org/10.1158/1078-0432.CCR-07-1811
- Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res 2008; 36:149 - 153; microRNA.org PMID: 18158296; http://dx.doi.org/10.1093/nar/gkm995
- Patturajan M, Nomoto S, Sommer M, Fomenkov A, Hibi K, Zangen R, et al. ΔNp63 induces β-catenin nuclear accumulation and signaling. Cancer Cell 2002; 1:359 - 367; http://dx.doi.org/10.1016/S1535-6108(02)00057-0
- Huang Y, Chuang AY, Ratovitski EA. PhosphoΔNp63α/miR-885-3p axis in tumor cell life and cell death upon cisplatin exposure. Cell Cycle 2011; 10:3938 - 3947; PMID: 22071691; http://dx.doi.org/10.4161/cc.10.22.18107