Abstract
Wip1 is a stress-response phosphatase that negatively regulates several tumor suppressors, including p53. In a sizeable fraction of tumors, overexpression or amplification of Wip1 compromises p53 functions; inhibition of Wip1 activity is an attractive strategy for improving treatment of these tumors. However, over half of human tumors contain mutations in the p53 gene or have lost both alleles. Recently, we observed that in cancer cells lacking wild type p53, reduction of Wip1 expression was ineffective, whereas, surprisingly, overexpression of Wip1 increased anticancer drug sensitivity. The increased sensitivity resulted from activation of the intrinsic pathway of apoptosis through increased levels of the pro-apoptotic protein Bax and decreased levels of the anti-apoptotic protein Bcl-xL. We showed that interaction of Wip1 and the transcription factor RUNX2, specifically through dephosphorylation of RUNX2 phospho-S432, resulted in increased expression of Bax. Interestingly, overexpression of Wip1 increased drug sensitivity only in the p53-negative tumor cells while protecting the wild type p53-containing normal cells from drug-induced collateral injury. Here, we provide evidence that Wip1 overexpression decreases expression of Bcl-xL through negative regulation of NFκB activity. Thus, Wip1 overexpression increases the sensitivity of p53-negative cancer cells to anticancer drugs by separately affecting Bax and Bcl-xL protein levels.
Introduction
As a monotherapy or in combination with other methods, chemotherapy is a method of choice for treatment of a variety of malignancies. The use of chemotherapeutic drugs such as doxorubicin, 5-fluorouracil or cisplatin analogs is directed toward triggering tumor cell death and eliminating tumor cells from the body. By damaging DNA, these antitumor agents activate several signaling pathways that control cell cycle checkpoints and induce programmed cell death (apoptosis) in tumor cells. A serious challenge for oncologists is tumor drug resistance. Several of the mechanisms used by tumors to evade anticancer drug-induced cell death involve mutation or functional inactivation of the tumor suppressor p53, which generally alters the balance between pro-apoptotic and anti-apoptotic proteins.Citation1,Citation2
p53 is a major regulator of cellular stress responses and induces genes involved in cell cycle arrest, DNA repair, senescence and apoptosis.Citation3 The tumor suppressor function of p53 results primarily from its ability to promote apoptosis through a combination of transcription-dependent and -independent mechanisms.Citation2 A portion of the complex p53 pathway is depicted schematically in . Following exposure to an activating stress, such as excessive oncogene activity or DNA damaging drugs, p53 acts as a sequence-specific transcription factor to induce the transcription of a large number of genes, including the pro-apoptotic proteins Puma, Noxa, BaxCitation4 and two of its negative regulators, the E3 ubiquitin ligase Mdm2Citation5 and the serine-threonine protein phosphatase Wip1.Citation6,Citation7 As direct targets of p53, Mdm2 and Wip1 function in negative feedback loops to limit p53 activity by decreasing its stability and activity, respectively. p53 represses transcription of the pro-apoptotic proteins Bcl-2 and Bcl-xL through incompletely defined mechanisms.Citation2 In addition, p53 can suppress the anti-apoptotic functions of Bcl-2 and Bcl-xL proteins through direct protein-protein interactions. Finally, wild-type p53 and the pro-inflammatory transcription factor NFκB generally exhibit mutual antagonism through direct and indirect mechanisms.Citation8,Citation9
Figure 1. Schematic representation of a selected portion of the p53 pathway regulating apoptosis.
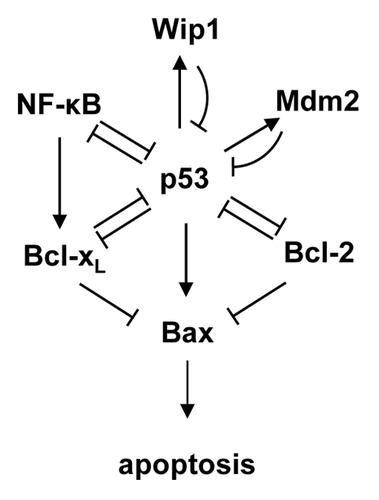
Wild-type p53 can become functionally inactivated through overexpression of its negative regulators or through enhanced degradation, leading to increased resistance to anticancer therapies.Citation10,Citation11 For example, increased expression of Mdm2 in adult medulloblastoma was associated with resistance to radiotherapy and reduced survival time.Citation12 Amplification or overexpression of Wip1 has been detected in several different cancers and is usually associated with a poor prognosis.Citation13 Wip1 negatively regulates upstream signaling from damaged DNA toward p53.Citation14,Citation15 It can dephosphorylate critical serine and threonine phosphorylations, thus inhibiting the functions of p53 itself and those of several important kinases upstream of p53, such ATM, Chk1, Chk2 and p38 MAPK.16‑19 Thus, in tumors with functional p53, Wip1 functions as a survival factor by negatively regulating p53-dependent proapoptotic signaling. Inhibition of Wip1 activity remains an attractive target for the development of new therapies directed against tumors retaining wild-type p53.Citation20
Mutation of p53 can lead to resistance to apoptosis.Citation21,Citation22 Several strategies have been proposed to overcome the increased resistance to apoptosis exhibited by p53-negative tumors.Citation23,Citation24 For example, inactivation of Chk1 in p53-negative tumors compromises G2 arrest in response to anticancer therapy and induces mitotic catastrophe, thus eliminating the tumor cells.Citation25 Unfortunately, Chk1 inhibition can be highly toxic to normal tissues and may induce severe side effects.Citation26
We recently reported that elevated levels of Wip1 phosphatase increased the sensitivity of p53-negative tumor cells to chemotherapeutic agents through increasing the Bax/Bcl-xL ratio, a critical factor regulating execution of the apoptotic program.Citation27 Here, we provide additional evidence that Wip1 overexpression individually affects Bax and Bcl-xL levels by distinct mechanisms. These findings suggest that the biological properties of Wip1 as a stress-responsive phosphatase may provide the basis for improved anticancer therapy of p53-negative tumors.
Wip1-Dependent Sensitization of p53-Negative Cells to Anticancer Treatment
In tumors lacking functional p53, inhibition of Wip1 is ineffective, whereas overexpression of Wip1 surprisingly sensitized cells to anticancer drugs through significantly increased levels of apoptosis.Citation27 This sensitization required the enzymatic activity of Wip1, as the phosphatase-deficient mutant of Wip1 was unable to increase tumor cell lethality after treatment with the chemotherapeutic drug cisplatin. Thus, the tumor cells with elevated levels of Wip1 readily underwent caspase-dependent apoptosis, the process that is characteristically compromised in tumors lacking wild-type p53.
Previously, we and others showed that one potential strategy to overcome the resistance to therapy that characterizes tumors lacking wild-type p53 is through inhibition of Chk1.Citation25,Citation28,Citation29 Cells negative for both p53 and Chk1 were unable to maintain arrest of the cell cycle at the G1/S and G2/M checkpoints, failed to finish mitosis with unrepaired DNA and died.Citation25,Citation30 Since it had been reported previously that Wip1 specifically dephosphorylated Chk1 and inhibited its functions,Citation16 we expected that overexpression of Wip1 in a p53-negative background would lead to defective G1/S and G2/M checkpoints and subsequent mitotic cell death in response to DNA damage. Although we observed some reduction in Chk1 phosphorylation upon overexpression of Wip1, the Chk1-regulated G2/M checkpoint was not compromised, and the treated cells failed to reach mitosis.Citation27 Thus, under our experimental conditions, the increased dephosphorylation of Chk1 by Wip1 did not promote mitotic cell death.
Overexpression of Wip1 Affects Bax and Bcl-xL Levels by Distinct Mechanisms
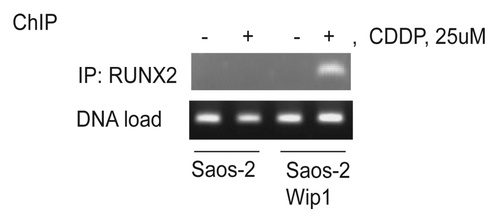
In our recent report, we showed that a high level of Wip1 together with cytotoxic drug treatment launches caspase-9 and -3-dependent apoptosis. To identify mechanisms leading to the increased sensitivity, we examined the expression levels of several pro- and anti-apoptotic proteins.Citation27 We noted that Bax protein levels were dramatically higher following cisplatin treatment in Wip1-overexpressing cells compared with control cells.Citation27 The best-characterized transcriptional factor inducing Bax transcription after the DNA damage is p53,Citation31 which is absent in Saos-2 cells. It has been shown, however, that after Bone morphogenetic protein stimulation or etoposide treatment, Bax transcription was induced by another transcriptional factor, RUNX2.Citation32 RUNX2 belongs to the runt domain-containing family of transcription factors. The RUNX transcriptional factors exhibit tissue-specific expression and regulate distinct processes. RUNX1 is mainly expressed in hematopoietic cells, RUNX2 is essential for osteoblast differentiation, and RUNX3 controls neurogenesis and thymopoiesis as well as gastric epithelia proliferation.Citation33 Depending on molecular context, RUNX proteins can function as transcriptional activators or repressors, and their activity can be regulated both on the transcriptional level and by posttranslational modification. RUNX2 activity was shown to be regulated by p38 MAPK, ERK1/2,Citation34 cdc2Citation35 and sequentially by Cdk1/cyclinB and PP2A phosphatase.Citation36 Phosphorylation of S104 and S451 inhibits the activity of RUNX2 by preventing association with the co-factor Core-binding factor, β subunit.Citation37 We found that Wip1 phosphatase can dephosphorylate S432 of RUNX2, another inhibitory site.Citation27 In our recent report, we showed that, of the several potential sites for Wip1 phosphatase activity, the RUNX2 variant bearing the serine 432-to-alanine mutation led to the greatest activation of the Bax promoter driving luciferase expression.Citation27 Several mechanisms could be proposed to explain activation of transcriptional activity of RUNX2 by Wip1. For example, dephosphorylation of Runx2 on Ser432 may lead to better interaction with necessary co-factors and/or may stimulate RUNX2 binding to DNA. To provide further support for the involvement of Wip1 in the induction of Bax following cisplatin treatment, we examined the association of RUNX2 with the Bax promoter by chromatin immunoprecipitation. As shown in , association of RUNX2 with Bax promoter chromatin was detected only in cells overexpressing Wip1 and only after cisplatin treatment. The importance of RUNX2 in apoptotic response was confirmed by a siRNA experiment.Citation27 Silencing of RUNX2 expression decreased cell death after cisplatin treatment in Saos-2 Wip1-on cells.
Figure 2. Wip1 overexpression increased RUNX2 binding to Bax promoter chromatin in Saos-2 Wip1-ON cells following treatment with cisplatin (CDDP). Wip1 was induced by doxycycline for 24 h and then cells were treated with cisplatin for 6 h and processed for chromatin immunoprecipitation (ChIP) assay of RUNX2 on Bax promoter as described previously.Citation25 To precipitate RUNX2 we used anti-RUNX2 antibodies M-70x from Santa-Cruz Biotechnology. Primers for Bax promoter were 5'-CCC GGG AAT TCC AGA CTG CAG-3' and 5'-GAG CTC TCC CCA GCG CAG AAG-3'.Citation38
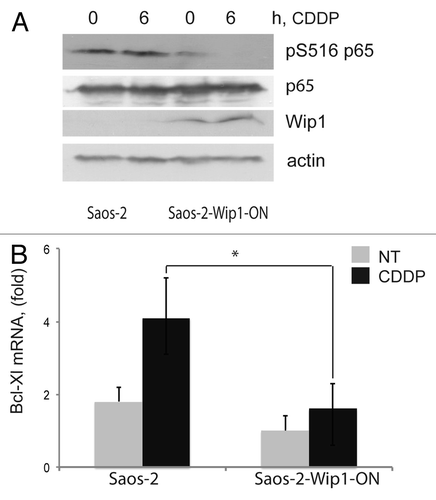
Regulation of apoptosis is complex and characterized by multiple redundancies. Among the changes in the levels of pro- and anti-apoptotic proteins, we noted decreased levels of the anti-apoptotic Bcl-xL protein in cells overexpressing Wip1, both before and during the course of cisplatin treatment.Citation27 Bcl-xL is often elevated in human tumors, and its overexpression is generally associated with resistance to therapy. Bcl-xL expression is positively regulated by the NFκB pathway.Citation39,Citation40 Previously, it was reported that Wip1 could dephosphorylate p65 (RELA) and thus inhibit the most prevalent form of the NFκB complex.Citation41 Furthermore, Wip1 expression is positively regulated by NFκB, thus forming a negative feedback loop downregulating NFκB function following exposure to an inflammatory stress.Citation42,Citation43 To test whether negative regulation of NFκB function by overexpressed Wip1 contributed to the increased sensitivity to cisplatin, we determined the levels of p65 S536 phosphorylation in our system. As shown in , we observed that in tumor cells with elevated levels of Wip1, the levels of activating S536 phosphorylation of p65 were lower both before and after cisplatin treatment compared with control cells, while the levels of total p65 protein remained constant. To test whether the observed decrease in Bcl-xL protein levels reflected transcriptional regulation, we determined relative Bcl-xL mRNA levels by quantitative PCR. As shown in , the levels of Bcl-xL mRNA were significantly lower in Wip1-overexpressing cells than in control cells. These results suggest that reduced levels of Bcl-xL mRNA and protein, probably due to downregulation of NFκB activity, contributed to the increased sensitivity to cisplatin observed in the Wip1-overexpressing tumor cells.
Figure 3. Decreased levels of Bcl-xL expression and NF|B p65 phosphorylation in cells with Wip1 overexpression. (A) Decreased levels of activating Ser536 phosphorylation of NF|B p65 in Saos-2 Wip1-ON cells after Wip1 induction. Wip1 was induced by doxycycline for 24 h and then cells were treated with cisplatin (CDDP) for 6 h and harvested. Whole cell lysates containing 70 μg of protein were analyzed by western blot using the following primary antibodies: anti-phospho-p65 Ser536, anti-p65 (Cell Signaling Technologies), anti-Wip1 (H-300) (Santa Cruz Biotechnologies) and anti-®-actin antibody (A 2103; Sigma). (B) Lower levels of Bcl-xL mRNA in Saos-2 Wip1-ON cells after Wip1 induction. Total RNA was purified and reverse-transcribed into cDNA using SuperScript II (Invitrogen) and oligo-dT primers. Real-time PCR was performed using the following primer pairs: Bcl-xL (5'-GAT CCC CAT GGC AGC AGT AAA GCA AG-3', 5'-CCC CAT CCC GGA AGA GTT CAT TCA CT-3') and GAPDH (5'-GAA GGT GAA GGT CGG AGT C-3', 5'-GAA GAT GGT GAT GGG ATT TC-3'). The expression of Bcl-xL was normalized to that of GAPDH.
Potential Benefits of Elevated Levels of Wip1 in Normal Tissue during Anticancer Therapy
An interesting implication of our findings is that transient activation of Wip1 would direct the toxicity of chemotherapeutic agents toward p53-defective tumor cells while protecting normal tissues that preserve wild-type functional p53. Many of the undesirable side effects of chemotherapy result from p53-dependent responses of normal cells, especially in sensitive tissues. In these circumstances, the transient activation of Wip1 as a negative regulator of p53 activity could protect normal cells from the toxicity of anticancer drugs and thereby decrease the side effects of anticancer therapy. Indeed, in mice that ubiquitously overexpressed Wip1, the intestinal epithelium and testes, which are sensitive tissues that usually exhibit high levels of cell death during anticancer treatment, exhibited much lower levels of apoptosis.Citation27
Concluding Remarks
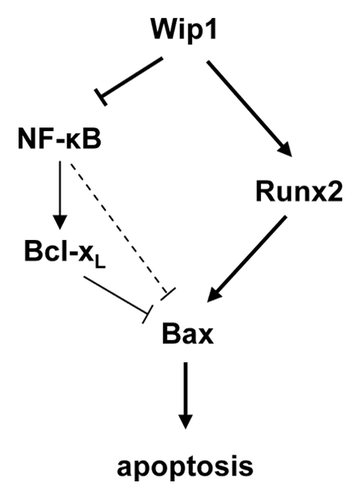
The novel proapoptotic functions of Wip1 activity revealed in a p53-negative environment, which is characteristic of a large proportion of malignancies, could provide a basis for the development of new methods of anticancer treatment. Our proposed mechanism of Wip1 proapoptotic activity through regulation of the Bax/Bcl-xL ratio is presented in . In the context of treatment with an anticancer drug, overexpression of Wip1 relieves Runx2 of the repressive effects of Ser432 phosphorylation, activating it as a transcription factor and inducing expression of its target gene Bax.Citation27 Concomitantly, Wip1 inhibits the activity of the NFκB complex through dephosphorylation of Ser536 of the p65 RelA subunit.Citation41 The ensuing downregulation of NFκB activity results in reduced levels of the NFκB target gene Bcl-xL. In addition, the inhibition of NFκB activity also reduces possible direct or indirect repression of Bax expression by NFκB.Citation38,Citation44 The strategy specifically directed to the transient activation of Wip1 in patients presenting tumors negative for p53 could increase the specificity of the current anticancer therapies and simultaneously provide protection from negative side effects.
Figure 4. Schematic representation of Wip1 regulation of the Bax/Bcl-xL ratio during treatment with chemotherapeutic agents.
Acknowledgments
This work was supported by the “Ligue Contre le Cancer Interrégionale du Grand Est,” the “Conseil Regional de Bourgogne,” C. Garrido group has the label “La Ligue Contre le Cancer.”This work was supported in part by the Intramural Research Program of the National Cancer Institute, National Institutes of Health.
References
- Basu A, Haldar S. The relationship between BcI2, Bax and p53: consequences for cell cycle progression and cell death. Mol Hum Reprod 1998; 4:1099 - 109; http://dx.doi.org/10.1093/molehr/4.12.1099; PMID: 9872359
- Hemann MT, Lowe SW. The p53-Bcl-2 connection. Cell Death Differ 2006; 13:1256 - 9; http://dx.doi.org/10.1038/sj.cdd.4401962; PMID: 16710363
- Kruse JP, Gu W. Modes of p53 regulation. Cell 2009; 137:609 - 22; http://dx.doi.org/10.1016/j.cell.2009.04.050; PMID: 19450511
- Nakamura Y. Isolation of p53-target genes and their functional analysis. Cancer Sci 2004; 95:7 - 11; http://dx.doi.org/10.1111/j.1349-7006.2004.tb03163.x; PMID: 14720320
- Barak Y, Juven T, Haffner R, Oren M. mdm2 expression is induced by wild type p53 activity. EMBO J 1993; 12:461 - 8; PMID: 8440237
- Fiscella M, Zhang H, Fan S, Sakaguchi K, Shen S, Mercer WE, et al. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci U S A 1997; 94:6048 - 53; http://dx.doi.org/10.1073/pnas.94.12.6048; PMID: 9177166
- Rossi M, Demidov ON, Anderson CW, Appella E, Mazur SJ. Induction of PPM1D following DNA-damaging treatments through a conserved p53 response element coincides with a shift in the use of transcription initiation sites. Nucleic Acids Res 2008; 36:7168 - 80; http://dx.doi.org/10.1093/nar/gkn888; PMID: 19015127
- Ak P, Levine AJ. p53 and NF-κB: different strategies for responding to stress lead to a functional antagonism. FASEB J 2010; 24:3643 - 52; http://dx.doi.org/10.1096/fj.10-160549; PMID: 20530750
- Gudkov AV, Gurova KV, Komarova EA. Inflammation and p53: A Tale of Two Stresses. Genes Cancer 2011; 2:503 - 16; http://dx.doi.org/10.1177/1947601911409747; PMID: 21779518
- Lain S, Lane D. Improving cancer therapy by non-genotoxic activation of p53. Eur J Cancer 2003; 39:1053 - 60; http://dx.doi.org/10.1016/S0959-8049(03)00063-7; PMID: 12736103
- Martinez-Rivera M, Siddik ZH. Resistance and gain-of-resistance phenotypes in cancers harboring wild type p53. Biochem Pharmacol 2012; 83:1049 - 62; http://dx.doi.org/10.1016/j.bcp.2011.12.026; PMID: 22227014
- Giordana MT, Duó D, Gasverde S, Trevisan E, Boghi A, Morra I, et al. MDM2 overexpression is associated with short survival in adults with medulloblastoma. Neuro Oncol 2002; 4:115 - 22; PMID: 11916503
- Lowe J, Cha H, Lee MO, Mazur SJ, Appella E, Fornace AJ Jr.. Regulation of the Wip1 phosphatase and its effects on the stress response. Front Biosci 2012; 17:1480 - 98; http://dx.doi.org/10.2741/3999; PMID: 22201816
- Lu X, Nguyen TA, Moon SH, Darlington Y, Sommer M, Donehower LA. The type 2C phosphatase Wip1: an oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev 2008; 27:123 - 35; http://dx.doi.org/10.1007/s10555-008-9127-x; PMID: 18265945
- Moon SH, Nguyen TA, Darlington Y, Lu X, Donehower LA. Dephosphorylation of γ-H2AX by WIP1: an important homeostatic regulatory event in DNA repair and cell cycle control. Cell Cycle 2010; 9:2092 - 6; http://dx.doi.org/10.4161/cc.9.11.11810; PMID: 20495376
- Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev 2005; 19:1162 - 74; http://dx.doi.org/10.1101/gad.1291305; PMID: 15870257
- Shreeram S, Demidov ON, Hee WK, Yamaguchi H, Onishi N, Kek C, et al. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol Cell 2006; 23:757 - 64; http://dx.doi.org/10.1016/j.molcel.2006.07.010; PMID: 16949371
- Takekawa M, Adachi M, Nakahata A, Nakayama I, Itoh F, Tsukuda H, et al. p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J 2000; 19:6517 - 26; http://dx.doi.org/10.1093/emboj/19.23.6517; PMID: 11101524
- Lu X, Nguyen TA, Donehower LA. Reversal of the ATM/ATR-mediated DNA damage response by the oncogenic phosphatase PPM1D. Cell Cycle 2005; 4:1060 - 4; http://dx.doi.org/10.4161/cc.4.8.1876; PMID: 15970689
- McConnell JL, Wadzinski BE. Targeting protein serine/threonine phosphatases for drug development. Mol Pharmacol 2009; 75:1249 - 61; http://dx.doi.org/10.1124/mol.108.053140; PMID: 19299564
- Solomon H, Madar S, Rotter V. Mutant p53 gain of function is interwoven into the hallmarks of cancer. J Pathol 2011; 225:475 - 8; http://dx.doi.org/10.1002/path.2988; PMID: 22025211
- Sturm I, Bosanquet AG, Hermann S, Güner D, Dörken B, Daniel PT. Mutation of p53 and consecutive selective drug resistance in B-CLL occurs as a consequence of prior DNA-damaging chemotherapy. Cell Death Differ 2003; 10:477 - 84; http://dx.doi.org/10.1038/sj.cdd.4401194; PMID: 12719725
- Bouchet BP, de Fromentel CC, Puisieux A, Galmarini CM. p53 as a target for anti-cancer drug development. Crit Rev Oncol Hematol 2006; 58:190 - 207; http://dx.doi.org/10.1016/j.critrevonc.2005.10.005; PMID: 16690321
- Mandinova A, Lee SW. The p53 pathway as a target in cancer therapeutics: obstacles and promise. Sci Transl Med 2011; 3:rv1; http://dx.doi.org/10.1126/scitranslmed.3001366; PMID: 21209413
- Demidova AR, Aau MY, Zhuang L, Yu Q. Dual regulation of Cdc25A by Chk1 and p53-ATF3 in DNA replication checkpoint control. J Biol Chem 2009; 284:4132 - 9; http://dx.doi.org/10.1074/jbc.M808118200; PMID: 19060337
- Greenow KR, Clarke AR, Jones RH. Chk1 deficiency in the mouse small intestine results in p53-independent crypt death and subsequent intestinal compensation. Oncogene 2009; 28:1443 - 53; http://dx.doi.org/10.1038/onc.2008.482; PMID: 19169280
- Goloudina AR, Tanoue K, Hammann A, Fourmaux E, Le Guezennec X, Bulavin DV, et al. Wip1 promotes RUNX2-dependent apoptosis in p53-negative tumors and protects normal tissues during treatment with anticancer agents. Proc Natl Acad Sci U S A 2012; 109:E68 - 75; http://dx.doi.org/10.1073/pnas.1107017108; PMID: 22065775
- Jurvansuu J, Fragkos M, Ingemarsdotter C, Beard P. Chk1 instability is coupled to mitotic cell death of p53-deficient cells in response to virus-induced DNA damage signaling. J Mol Biol 2007; 372:397 - 406; http://dx.doi.org/10.1016/j.jmb.2007.06.077; PMID: 17663993
- Koniaras K, Cuddihy AR, Christopoulos H, Hogg A, O’Connell MJ. Inhibition of Chk1-dependent G2 DNA damage checkpoint radiosensitizes p53 mutant human cells. Oncogene 2001; 20:7453 - 63; http://dx.doi.org/10.1038/sj.onc.1204942; PMID: 11709716
- Goloudina A, Yamaguchi H, Chervyakova DB, Appella E, Fornace AJ Jr., Bulavin DV. Regulation of human Cdc25A stability by Serine 75 phosphorylation is not sufficient to activate a S phase checkpoint. Cell Cycle 2003; 2:473 - 8; http://dx.doi.org/10.4161/cc.2.5.482; PMID: 12963847
- Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA, et al. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene 1994; 9:1799 - 805; PMID: 8183579
- Eliseev RA, Dong YF, Sampson E, Zuscik MJ, Schwarz EM, O’Keefe RJ, et al. Runx2-mediated activation of the Bax gene increases osteosarcoma cell sensitivity to apoptosis. Oncogene 2008; 27:3605 - 14; http://dx.doi.org/10.1038/sj.onc.1211020; PMID: 18223689
- Ito Y. RUNX genes in development and cancer: regulation of viral gene expression and the discovery of RUNX family genes. Adv Cancer Res 2008; 99:33 - 76; http://dx.doi.org/10.1016/S0065-230X(07)99002-8; PMID: 18037406
- Ge C, Xiao G, Jiang D, Yang Q, Hatch NE, Roca H, et al. Identification and functional characterization of ERK/MAPK phosphorylation sites in the Runx2 transcription factor. J Biol Chem 2009; 284:32533 - 43; http://dx.doi.org/10.1074/jbc.M109.040980; PMID: 19801668
- Qiao M, Shapiro P, Fosbrink M, Rus H, Kumar R, Passaniti A. Cell cycle-dependent phosphorylation of the RUNX2 transcription factor by cdc2 regulates endothelial cell proliferation. J Biol Chem 2006; 281:7118 - 28; http://dx.doi.org/10.1074/jbc.M508162200; PMID: 16407259
- Rajgopal A, Young DW, Mujeeb KA, Stein JL, Lian JB, van Wijnen AJ, et al. Mitotic control of RUNX2 phosphorylation by both CDK1/cyclin B kinase and PP1/PP2A phosphatase in osteoblastic cells. J Cell Biochem 2007; 100:1509 - 17; http://dx.doi.org/10.1002/jcb.21137; PMID: 17171635
- Wee HJ, Huang G, Shigesada K, Ito Y. Serine phosphorylation of RUNX2 with novel potential functions as negative regulatory mechanisms. EMBO Rep 2002; 3:967 - 74; http://dx.doi.org/10.1093/embo-reports/kvf193; PMID: 12231506
- Cianfrocca R, Muscolini M, Marzano V, Annibaldi A, Marinari B, Levrero M, et al. RelA/NF-kappaB recruitment on the bax gene promoter antagonizes p73-dependent apoptosis in costimulated T cells. Cell Death Differ 2008; 15:354 - 63; http://dx.doi.org/10.1038/sj.cdd.4402264; PMID: 18034190
- Khoshnan A, Tindell C, Laux I, Bae D, Bennett B, Nel AE. The NFκB cascade is important in Bcl-xL expression and for the anti-apoptotic effects of the CD28 receptor in primary human CD4+ lymphocytes. J Immunol 2000; 165:1743 - 54; PMID: 10925251
- Marinari B, Costanzo A, Marzano V, Piccolella E, Tuosto L. CD28 delivers a unique signal leading to the selective recruitment of RelA and p52 NF-kappaB subunits on IL-8 and Bcl-xL gene promoters. Proc Natl Acad Sci U S A 2004; 101:6098 - 103; http://dx.doi.org/10.1073/pnas.0308688101; PMID: 15079071
- Chew J, Biswas S, Shreeram S, Humaidi M, Wong ET, Dhillion MK, et al. WIP1 phosphatase is a negative regulator of NF-kappaB signalling. Nat Cell Biol 2009; 11:659 - 66; http://dx.doi.org/10.1038/ncb1873; PMID: 19377466
- Lowe JM, Cha H, Yang Q, Fornace AJ Jr.. Nuclear factor-kappaB (NF-kappaB) is a novel positive transcriptional regulator of the oncogenic Wip1 phosphatase. J Biol Chem 2010; 285:5249 - 57; http://dx.doi.org/10.1074/jbc.M109.034579; PMID: 20007970
- Salminen A, Kaarniranta K. Control of p53 and NF-κB signaling by WIP1 and MIF: role in cellular senescence and organismal aging. Cell Signal 2011; 23:747 - 52; http://dx.doi.org/10.1016/j.cellsig.2010.10.012; PMID: 20940041
- Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol 1999; 19:3485 - 95; PMID: 10207072