Abstract
Many cancer cells are unable to maintain a numerically stable chromosome complement. It is well established that aberrant cell division can generate progeny with increased ploidy, but the genetic factors required for maintenance of diploidy are not well understood. Using an isogenic model system derived by gene targeting, we examined the role of Chk1 in p53-proficient and -deficient cancer cells. Targeted inactivation of a single CHK1 allele in stably diploid cells caused an elevated frequency of mitotic bypass if p53 was naturally mutated or experimentally disrupted by homologous recombination. CHK1-haploinsufficient, p53-deficient cells frequently underwent sequential rounds of DNA synthesis without an intervening mitosis. These aberrant cell cycles resulted in whole-genome endoreduplication and tetraploidization. The unscheduled bypass of mitosis could be suppressed by targeted reversion of a p53 mutation or by exogenous expression of Cdk1. In contrast, the number of tetraploid cells was not increased in isogenic cell populations that harbor hypomorphic ATR mutations, suggesting that suppression of unscheduled mitotic bypass is a distinct function of Chk1. These results are consistent with a recently described role for Chk1 in promoting the expression of genes that promote cell cycle transitions and demonstrate how Chk1 might prevent tetraploidization during the cancer cell cycle.
Keywords: :
Introduction
Loss of the normal diploid karyotype is a cardinal feature of the majority of human cancers.Citation1,Citation2 The defects that underlie the development of polyploidy and aneuploidy during tumorigenesis remain obscure, but these cellular phenotypes have been frequently attributed to losses of checkpoint control, which are similarly common to many cancers. Checkpoints monitor basic growth processes, such as chromosomal DNA replication and mitosis, and ensure that they occur at discrete, non-overlapping intervals during the cell cycle. Many of the most commonly studied checkpoint proteins are strongly activated by DNA damage. The precise relationship between DNA damage-activated cell cycle checkpoint proteins and the stabilization of cell ploidy remains a fundamental question in cancer biology.
To study the factors required for stable ploidy, we focused on proteins that are required for the G2/M checkpoint. After DNA damage, ATR, Chk1 and p53 function to arrest cells in G2 and thereby prevent them from entering mitosis and segregating damaged chromosomes. These proteins promote G2/M arrest via parallel inhibitory pathways that converge on the cyclin B1-Cdk1 complex.Citation3 Chk1 is a serine/threonine kinase that monitors the progress of DNA replication.Citation4 Upstream of Chk1, ATR signals in response to stalled replication forks, functioning to stabilize these structures and trigger downstream pathways that modulate DNA repair and trigger cell cycle arrest.Citation5 Chk1 is directly phosphorylated by ATRCitation6 and is a critical mediator of the ATR-dependent G2/M checkpoint pathway. ATR and CHK1 are both essential for cellular viability, which implies that the encoded proteins are required for normal cell proliferation.Citation7,Citation8 Interestingly, activation of the G2/M checkpoint appears to be a nonessential function of Chk1, as a phosphorylation site mutant of Chk1 defective for this activity is nonetheless able to support viability.Citation9,Citation10 The nature of Chk1’s essential contribution to cell proliferation remains incompletely understood.
To facilitate the study of chromosomal stability in proliferating human cells with uniformly reduced and stable levels of Chk1, we used gene-targeting techniques to create a panel of isogenic cancer cells that express only one functional CHK1 allele in the presence or absence of functional p53. Detailed characterization of this genetic system revealed an important role for Chk1 in maintaining a normal cell cycle, thereby preventing the growth of subclones with increased ploidy.
Results
Chk1 activity has most often been experimentally manipulated in cultured human cells by the use of RNAi or chemical inhibitors. As we wished to study the effect of Chk1 on chromosomal stability over a period of many generations, we instead chose to study cells with defined genetic alterations. In human somatic cell knockouts and knockins, phenotypes tend to be highly stable over many generations, and uniform cell populations harboring defined alterations can be readily expanded.Citation11
First, we examined isogenic derivatives of the colorectal cancer cell line DLD-1, in which one of two endogenous wild-type CHK1 alleles were disrupted by homologous recombination.Citation10 As expected, independently derived clones harboring a single copy of CHK1 (CHK1+/-) exhibited reduced expression of Chk1 protein compared with isogenic cells with two functional alleles (). Parental DLD-1 cells lack functional p53 protein due to a mutation in codon 241 (S241F); the remaining wild-type p53 allele is silenced.Citation12 This inactivating p53 mutation (genotype p53-/Sil) has been successfully reverted by the knockin of a wild-type exon (genotype p53+/Sil) which results in a functionally restored p53 pathway. Cytometric analysis of these wild-type p53 knockin cells has demonstrated restoration of cell cycle regulation and normal diploidy.Citation12 Clones harboring a functional p53 allele (p53+/ Sil) expressed decreased levels of p53, consistent with the reduced half-life of wild-type p53 protein and increased expression of the downstream transcriptional target CDKN1A, which encodes p21 (). We inactivated one copy of CHK1 in p53-/Sil and p53+/Sil cells (), thereby generating clonal populations of heterozygous CHK1 knockouts in both p53-deficient and -proficient backgrounds.
Figure 1. Tetraploidization is suppressed by CHK1 and p53. (A) Chk1, p53 and p21 proteins were assessed in derivatives of DLD-1 by immunoblot. α-tubulin was probed as a loading control. (B) Representative images of diploid (left) and tetraploid (right) metaphase chromosome spreads. (C) Representative images of chromosome 18 signals. Nuclei were counterstained with DAPI (blue). (D) The number of chromosome 18-specific FISH signals per nucleus was assessed in wild type DLD-1 cells (CHK1+/+; n=550) and in CHK1-haploinsufficient derivatives (CHK1+/-; n=350). (E) Mitotic cells stained with antibodies to γ-tubulin (red) to detect centrosomes and α-tubulin (green) to detect mitotic spindles. Bipolar (left) and multipolar spindles (right) were observed. Nuclei were counterstained with DAPI. (F) Chk1, p53 and α-tubulin (loading control) proteins were assessed in derivatives of HCT116. Bars,10 μm.
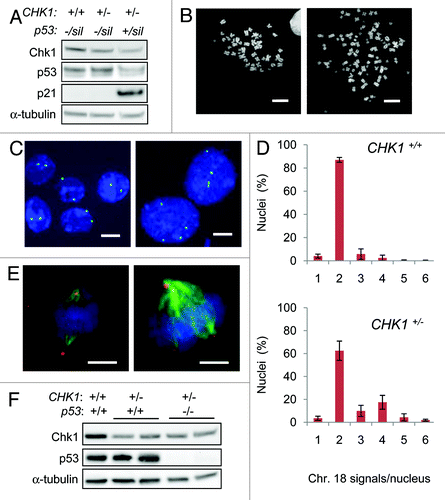
The maintenance of ploidy varied dramatically with genotype. DLD-1 cells have been shown to exhibit a near-diploid karyotype that is highly stable during extensive propagation in culture.Citation13 Accordingly, the majority of metaphase spreads from parental DLD-1 (CHK1+/+) cultures were composed of the expected near-diploid chromosome complement (), with ~5% of the cell population harboring elevated numbers of chromosomes (). In contrast, cell lines that harbored only a single wild-type CHK1 allele (CHK1+/-) exhibited a distinct subpopulation of mitotic cells (~30%) that was tetraploid ( and ); the tetraploid subpopulation was notably reduced in CHK1+/- cells with wild-type p53. Because of the technical limitations of counting chromosomes in metaphase spreads, we analyzed larger numbers of cells in interphase by fluorescence in situ hybridization (FISH) using a centromeric probe to chromosome 18 (). The distribution of FISH signals in the CHK1+/- cells was bimodal, consistent with an increased tetraploid population (). Multipolar mitotic spindles with extra centrosomes () were observed in 19% of CHK1+/- p53Sil/- cells and 6% of CHK1+/+ p53Sil/- cells. The increased proportion of mitotic cells with supernumerary centrosomes was thus consistent with the overall increase in ploidy.
Table 1. Analysis of metaphase spreads.
To test the generality of these findings in a distinct cell model, we used the same gene targeting approach to disrupt one CHK1 allele in HCT116 cells, which, like DLD-1, are derived from a mismatch repair-deficient colorectal cancer. HCT116 cells harbor wild-type p53 alleles and have been a widely used model system for studying the genetic basis of chromosomal stability.Citation14-Citation16 Previously, we demonstrated that approximately 9% of the p53‑/- HCT116 derivative were tetraploid, a small but statistically significant increase over p53+/+ HCT116.Citation17 We generated two independent CHK1+/- clones from both p53+/+ and p53-/- HCT116 (). As in DLD-1, the targeted alterations of CHK1 and p53 in HCT116 revealed cooperative effects of Chk1 and p53 in the suppression of tetraploidization. While no significant increase in tetraploidy was observed in HCT116 CHK1+/- cells with wild-type p53, greater than 30% of CHK1+/-p53-/- cells were tetraploid ().
To examine how tetraploidization might have occurred, we tracked the onset and completion of mitosis in asynchronous cell populations by time-lapse microscopy. Stable expression of GFP-tagged histone 2B in CHK1+/+ and CHK1+/- cells allowed the visualization of chromatin and nuclear morphology during unperturbed growth. The majority of cells that condensed chromatin and entered mitosis subsequently gave rise to daughter cells, irrespective of genotype (). While nearly all cells harboring two CHK1 alleles entered and then completed mitosis, many CHK1+/- nuclei failed to exhibit chromosome condensation or dissolution of the nuclear membrane over 72 h and, therefore, appeared to remain in interphase (). The defect in the CHK1-haploinsufficient population was restricted to mitotic entry, as there were no apparent increases in abnormal cell division, mitotic slippage or binucleate progeny. To confirm that the significant fraction of CHK1+/- cells that remained in interphase had bypassed mitosis and reentered S phase, we measured the incorporation of the nucleotide analog 5-ethynyl-2'-deoxyuridine (EdU) by flow cytometry. Analysis of asynchronous cells revealed active DNA synthesis in the > 4N subpopulation of cells with the CHK1+/- genotype; this fraction was more apparent when cells were synchronized in early S phase prior to incubation with EdU ().
Figure 2. CHK1-haploinsufficient, p53-mutant human cells bypass mitosis and undergo whole genome endoreduplication. (A) Sequential images of CHK1+/+ and CHK1+/- DLD-1 cells expressing a histone H2B-GFP fusion protein were captured by time lapse fluorescence microscopy. Black arrows indicate cells undergoing mitosis. White arrows track cells that fail to condense chromatin during the experiment. The fates of histone H2B-GFP expressing cells tracked over 72 h were scored (n > 90). (B) Incorporation of EdU in asynchronous cells or in cells synchronized in S-phase. (C) Representative image of diplochromosomes found in metaphase spreads of CHK1+/- cells. Bar, 10 μm.
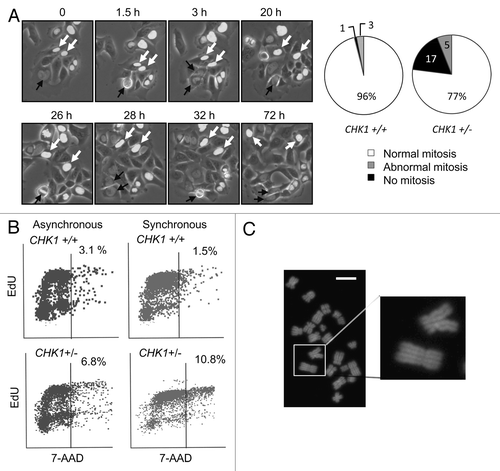
Diplochromosomes were observed in approximately 5% of metaphase spreads from CHK1+/- cells () but were not detected in isogenic cells harboring two functional copies of CHK1. Diplochromosomes are a cytological marker of re-replication caused by a failure to degrade cohesin, the centromeric component required to maintain the attachment of sister chromatids prior to anaphase.Citation18 The detection of diplochromosomes in tetraploid cell populations was another indication that mitotic bypass and whole-genome endoreduplication had occurred in these cells.
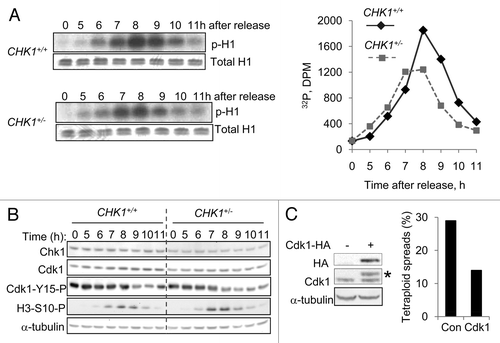
The entry of cells into mitosis is controlled by the progressive activation of Cdk1.Citation19 The frequent occurrence of mitotic bypass in CHK1+/- cells suggested that normal levels of Chk1 might be required for full Cdk1 activation, so we measured levels of Cdk1 activity in DLD-1 cells harboring one or two functional CHK1 alleles. Cells were transiently arrested in early S phase by double thymidine block and then released into drug-free medium. In both cell lines, Cdk1 kinase activity peaked at 8 h after release from the block; this peak level was lower in CHK1+/- cells (). As expected, inhibitory Cdk1 phosphorylation (Cdk1-Y15-P) decreased as phosphorylated histone H3 (H3-S10-P), a marker of mitosis, increased (). Like Chk1, the overall levels of Cdk1 and Cdk1-Y15-P appeared somewhat lower in CHK1+/- cells. This result suggested that moderately decreased levels of Cdk1 protein, rather than increased levels of Cdk1 inhibitory phosphorylation, might have accounted for the limited Cdk1 activity in CHK1+/- cells. To test this idea, we stably expressed an epitope-tagged Cdk1 protein in CHK1+/- cells and cultured these cells for 30 generations. Exogenous Cdk1 expression reduced the tetraploid fraction from 30% to 14% ().
Figure 3. Reduction in CHK1 gene dosage causes reduction in Cdk1 activity. (A) CHK1+/+ and CHK1+/- DLD-1 cells harboring mutant p53 were synchronized by double thymidine block, released, and harvested at the indicated time points. Proteins immunoprecipitated from non-denatured lysates with an anti-Cdk1 antibody were assessed for histone H1 kinase activity. 32P-labeled histone H1 was resolved by gel electrophoresis and assessed by autoradiography and scintillation counting. Total H1 protein was visualized by staining with Coomassie blue. (B) Chk1 and Cdk1 total protein, and Cdk1-Y15-P and H3-S10-P phosphoproteins were assessed in lysates from cells synchronized as in (A), by immunoblot. α-tubulin was assessed as a loading control. (C) Hemaglutinin (HA) epitope-tagged Cdk1 (Cdk1-HA) was stably expressed in pooled clones derived from CHK1+/- cells. The expression of exogenous Cdk1 protein was assessed by immunoblotting with an anti-HA antibody; endogenous and exogenous Cdk1 protein (indicated with an asterisk) were probed with an anti-Cdk1 antibody. Ploidy was assessed by numerical analysis of metaphase chromosomes (n = 50). Con, empty vector control.
A recent study by Davoli et al. has demonstrated that persistent activation of DNA damage signaling by telomeric dysfunction or by low doses of a DNA damaging chemical agent can cause the prolonged downregulation of Cdk1 activity, and thereby trigger mitotic bypass and tetraploidization in p53-null cells.Citation20 The near-complete knockdown of Chk1 has been shown to cause activation of ATR and the generation of DNA strand breaks during DNA replication.Citation21 Therefore, we wondered whether endogenous DNA damage might have contributed to the tetraploidization phenotypes that we observed in our system, which were similarly dependent on p53 genotype.
Might CHK1-haploinsufficiency have caused a level of endogenous DNA damage sufficient to cause the downregulation of Cdk1? Cdk1-Y15-P was not detectably increased in CHK1+/- cells (), suggesting that DNA damage was not significantly elevated. Examination of metaphase spreads revealed no increases in chromatid breaks in CHK1+/- cells (not shown). To detect low levels of DNA strand breaks with a high degree of sensitivity, we assessed DNA repair proteins 53BP1 and γH2AX, which rapidly accumulate at discrete sites of DNA damage. Endogenous DNA damage has been observed in many cancer cell lines.Citation22 Accordingly, both CHK1+/+ and CHK1+/- cells exhibited immunofluorescent staining patterns of 53BP1 and γH2AX that were consistent with low level activation of DNA damage signaling pathways (). The relatively modest reduction of CHK1 expression in heterozygous cells ( and ) did not appear to significantly alter this staining pattern, nor increase the numbers of foci. This result contrasts somewhat with studies of normal mouse cells, in which Chk1 haploinsufficiency caused a measurable increase in 53BP1 foci.Citation23 It is possible that the elevated level of DNA damage in human cancer cells precluded us from appreciating subtle differences between genotypes.
Figure 4. DNA damage and tetraploidization in p53-mutant cells with altered ATR-Chk1 signaling. (A) The localization and distribution of 53BP1 and γH2AX were assessed by immunofluorescence in untreated DLD-1 and derivatives with the indicated genotypes. Where indicated, cells were fixed 90 minutes after treatment with 4 Gy ionizing radiation. Nuclei were counterstained with DAPI. Bars, 20 um. (B) Levels of ATR, Chk1 and Chk1 S317-P phosphoprotein in cells treated with 10 Gy ionizing radiation were determined by immunoblot. (C) Ploidy was assessed in DLD-1 and the indicated derivatives by numerical analysis of metaphase chromosomes (n > 20).
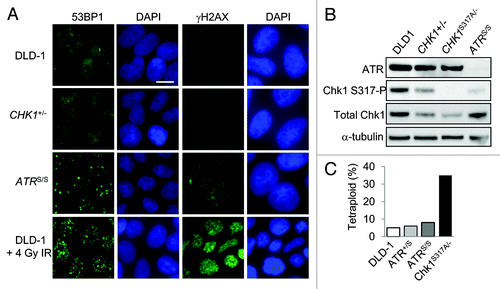
To assess the contribution of upstream DNA damage signals, we also examined isogenic cell lines that harbor one or two copies of the hypomorphic mutation in ATR that causes the autosomal recessive disorder ATR-Seckel syndrome (designated ATR+/S and ATRS/S respectively).Citation24 ATRS/S cells express markedly reduced levels of ATR protein and exhibit reduced signaling to Chk1 after DNA damage (). Like Chk1, ATR maintains replication fork stabilityCitation5 and is known to actively suppress DNA strand breakage.Citation25,Citation26 Consistent with this function, ATRS/S cells exhibited readily detectable 53BP1 and γH2AX foci (). The level of endogenous DNA damage in both CHK1+/+ and CHK1+/- cells appeared lower than in ATRS/S cells. For comparison, 53BP1 and γH2AX foci were visualized in unmodified DLD-1 cells after a single 4 Gy dose of ionizing radiation (), which has been previously shown to suppress their growth by approximately 50%.Citation24
To explore whether ATR might suppress tetraploidy, we examined the chromosome complement of ATR+/S and ATRS/S cells. We also examined another derivative of DLD-1 in which the single functional CHK1 allele has a point mutation that disrupts the conserved Chk1 S317 phosphorylation site (genotype CHK1S317A/-). The S317 site is phosphorylated by ATR and is required for the cooperative phosphorylation of neighboring sites.Citation9,Citation10 ATR signals to Chk1 S317 in response to DNA damage, and CHK1S317A/- cells exhibit a defective G2/M checkpoint as well as abnormalities in DNA replication fork dynamics.Citation10 The CHK1S317A allele, therefore, confers impaired signaling from ATR to Chk1 but nonetheless supports cellular viability.Citation9,Citation10 Like parental DLD-1 cells, the ATR+/S, ATRS/S and CHK1S317A/- cells express only mutant p53 protein.
Karyotype analysis revealed that ATR-deficient cells maintained a diploid chromosome complement (). Evidently, the DNA damage visualized by repair focus staining () did not significantly alter the ability of these cells to maintain diploidy. In contrast, 30% of CHK1S317A/- cells were tetraploid () and in this regard were similar to CHK1+/- cells (). These results suggest that the tendency toward tetraploidization was dependent only on reduced CHK1 gene dosage and was independent of upstream signals from ATR.
Irrespective of genotype, all cell populations studied here retained a predominantly diploid chromosome complement. The appearance of subpopulations of tetraploid cells implied that mitotic bypass occurred during some but not all cell cycles. This simple observation raised two fundamental questions. First, at what rate did cells bypass mitosis and become tetraploid? Second, did tetraploid cells continue to proliferate and contribute progeny to the overall population? Obviously, diploid cells can give rise to either diploid or tetraploid progeny, while tetraploid cells cannot conceivably give rise to diploid cells. If the proliferative capacity of diploid and tetraploid cells were the same, then one would expect that a cell line with a steady rate of mitotic bypass would eventually become completely tetraploid. Serial analyses of cell ploidy over many generations demonstrated that this was not the case.
To understand how steady-state levels of tetraploidy might relate to the dynamic process of tetraploidization, we analyzed single-cell clones derived from CHK1+/+ and CHK1+/- cells. DLD-1 cells of each genotype were plated in multiwell plates at limiting dilution. The resulting single cell-derived clones were then propagated in parallel for a total of 30 generations. Cytometric analysis showed that expanded clones were completely diploid, completely tetraploid or mixtures of diploid and tetraploid cells (). The proportion and composition of each subclone allowed us to estimate a rate of tetraploidization, which was calculated as (number of mixed clones)/[(total number of clones analyzed - number of tetraploid clones) x 30 generations]. The completely tetraploid clones were subtracted from the total clone pool, because these were presumably already tetraploid at the time of plating and therefore did not represent events that occurred during the generations of subsequent growth. We thus estimated the rate of spontaneous tetraploidization in CHK1+/- cells to be 7.5 x 10-3 per generation, while the rate in CHK1+/+ was 2.0 x 10-3 per generation. These calculations underestimate the actual rate of tetraploidization, because mitotic bypass events that occurred during later generations would have contributed fewer progeny to the total populations, and therefore would be increasingly unlikely to be detected. A lower rate of proliferation by tetraploid cells would also lead to the underestimation of the tetraploid fraction. Despite these limitations, this simple experiment illustrates how tetraploidization in the context of CHK1 haploinsufficiency is an ongoing process that reflects an underlying instability.
Table 2. Evolution of tetraploid populations.
The very identification of pure tetraploid clones indicated that some cells that had undergone bypass of mitosis were indeed viable and could continue to proliferate. Interestingly, while tetraploid cells comprised nearly 30% of the steady-state CHK1+/- cell population (), only 7% of the recovered clones were composed solely of tetraploid cells (). The relative underrepresentation of tetraploid clones after limiting dilution and expansion suggests that tetraploidization caused a survival defect. The observed equilibrium of the tetraploid fraction in mixed populations probably resulted from tetraploid cells arising at an increased rate but with reduced proliferative capacity.
Discussion
Our study provides genetic evidence that Chk1 is required for efficient entry into mitosis. In unperturbed cells, Chk1 binds to chromatin and directly phosphorylates histone H3 at T11, thereby promoting acetylation at H3 K7,Citation27 an activating mark. When DNA damage is sensed, Chk1 rapidly dissociates from chromatin, thereby causing local transcription to be repressed. In this way, Chk1 actively regulates transcription of many genes that promote progression of the cell cycle, including the pro-mitotic CCNB1 and CDK1.Citation27 The repression of transcription that follows Chk1 chromatin dissociation thus promotes DNA damage-induced cell cycle arrest. Our proposed role for chromatin-bound Chk1 as a suppressor of mitotic bypass is a logical extension of this checkpoint mechanism: chromatin-bound Chk1 promotes cell cycle progression, while release of Chk1 from the chromatin causes cell cycle arrest. In automotive terms, Chk1 has been primarily thought of as a “brake” designed to halt cell cycle progression. We suggest that Chk1 functions instead like a switch between “gas” and “brake” that both controls acceleration and deceleration. Further biochemical studies will be needed to test and refine this model.
Although Cdk1 activity and protein were modestly reduced in CHK1+/- cells (), CDK1 transcripts were not found to vary significantly with CHK1 genotype (not shown). It is possible that the ~50% reduction in Chk1 expression caused by reduced gene dosage in our system () caused subtle and therefore difficult-to-quantify variations in the expression of numerous pro-mitotic genes, the cumulative effects of which may have resulted in the observed decrease in Cdk1 activity. New pathways that control Cdk1 have recently been described.Citation28,Citation29 It will be interesting to determine whether Chk1 affects the expression or function of these regulators.
The mechanism by which CHK1+/- human cells acquire a tetraploid chromosome complement appears to be distinct from other pathways to tetraploidy that have recently been elucidated. Tetraploidy can arise from failed mitosis due to chromosome nondisjunction, mitotic slippage or incomplete cytokinesis.Citation30,Citation31 In these scenarios, mitotic cells decondense their chromatin and proceed to G1 with a tetraploid chromosome complement and may subsequently activate a p53-dependent G1 arrest. It has been proposed that this pathway represents a “tetraploidy checkpoint” that would be defective in cancer cells with loss of p53 function.Citation30 While loss of p53 alone does not induce tetraploidy by these mechanisms, such loss may be permissive to the survival of tetraploid cells.
As demonstrated here, tetraploidy can also result from the complete bypass of mitosis, such that cells with replicated chromosomes proceed directly from G2 to G1-S phase. The mitotic bypass that we see in our CHK1-haploinsufficient cells, while similar in effect, appears to be mechanistically distinct from the DNA damage-dependent tetraploidization described by Davoli et al. In that study, DNA damage signals that induced tetraploidization caused a ~90-fold increase in DNA damage foci and activation of the G2/M checkpoint. Prolonged G2 arrest was eventually resolved by mitotic bypass. While it is impossible to completely rule out an effect of endogenous DNA damage in our system, CHK1-haploinsufficient cells prone to tetraploidization exhibited small numbers of repair foci and no evidence of growth inhibition or checkpoint activation. Furthermore, we demonstrate that CHK1S317A/- cells, defective in the activation of the G2/M checkpoint,Citation10 had the same tendency to become tetraploid as checkpoint-proficient cells with one wild-type CHK1 allele ().
The finding that cells expressing only hypomorphic ATR were stably diploid () likewise suggests that the function of Chk1 in preventing mitotic bypass may be independent of upstream DNA damage signals. The role for Chk1 as a suppressor of tetraploidization is therefore distinct from its role in suppressing fragile site expression, another form of genetic instability that is dependent on upstream ATR.Citation32 Chk1 also differs in this regard from the structurally unrelated checkpoint kinase Chk2, which has been implicated in the maintenance of genetic stability. HCT116 cells homozygous for CHK2-knockout alleles were observed to exhibit aneuploidy after prolonged serial passaging.Citation33,Citation34 This form of instability, attributed to missegregation during mitosis, is primarily manifest as single chromosome gains and losses but does not involve changes in overall ploidy.
The role for Chk1 as a suppressor of tetraploidization in somatic cells is also consistent with recent studies that describe a role for Chk1 in the regulation of ploidy during development.Citation35,Citation36 During the growth of the mammalian trophoblast, mitotic bypass and endoreduplication are triggered in endotrophoblast mouse cells by concomitant downregulation of Chk1 and the accumulation of p21 and p57, which thereby inhibits Cdk1.Citation35,Citation36 p57 was not expressed at detectable levels our cell panel (not shown), indicating that this physiologic developmental pathway may not be active in adult somatic cells.
Studies of Chk1-haploinsufficient mouse cells have revealed mitosis-related phenotypes that are distinct from those observed here.Citation37 A reduced gene dosage of Chk1 in mouse mammary epithelial cells leads to incomplete cytokinesis and the appearance of binucleate cells,Citation38 which were not observed in our system. The frequency of mitotic entry was not directly investigated in these studies, which instead focused on mitotic progression in prometaphase-arrested cells. When comparing results obtained from different genetic systems, it is important to consider that the progressive activation of Cdk1 is required not only for mitotic entry, but also for subsequent mitotic progression.Citation19 The threshold Cdk1 levels needed to execute the early and later stages of mitosis may differ in different tissues or different species. Indeed, our previous studies revealed significant differences in tetraploidization between p53-deficient primary human fibroblasts and p53-deficient epithelial cancer cells.Citation17
Tissue-specific heterozygous disruption of murine Chk1 and p53 together promotes mammary tumor formation.Citation39 While Chk1 has not been found to be mutated in a significant number of cancers, reduced expression of CHK1 has been reported in human lymphomasCitation40 as well as chromosomally unstable T-cell acute lymphoblastic leukemias induced by the oncogene TLX1.Citation41 It remains to be determined if aberrant Chk1 expression contributes more broadly to human tumorigenesis.
Materials and Methods
Cells and gene targeting.
Human colorectal cancer cells DLD-1 and HCT116 were cultured in McCoy’s 5A medium supplemented with fetal calf serum (HyClone) and penicillin/streptomycin (Invitrogen). The endogenous CHK1 locus was disrupted using a recombinant adeno-associated virus as described in reference Citation10. Cells harboring alterations in both CHK1 and p53 were generated by first replacing the mutant p53 sequence in DLD-1 cells with wild-type sequence by homologous integration of a knockin vectorCitation12 and then disrupting the endogenous CHK1 loci. The wild-type p53 alleles of HCT116 were disrupted with a p53 locus-specific knockout vector,Citation42 and endogenous CHK1 was subsequently disrupted to obtain the double mutants.
Antibodies and immunoblotting.
Cell lysates were denatured and fractionated on NuPAGE gels (Invitrogen). Proteins were transferred to nylon membranes (Millipore) then incubated with antibodies directed against Chk1, Chk2, αTubulin, Cdc2 (Santa Cruz Biotechnology), Cdc2-Y15-P, Chk1-S317-P, Chk2-T68-P (Cell Signaling Technologies), γH2AX, Histone H3Ser10-P (Millipore), 53BP1 (Novus Biologicals) and γ-tubulin (Sigma) under conditions recommended by the manufacturers. Blots were developed using enhanced chemiluminescence (Amersham).
Karyotyping and fluorescence in situ hybridization.
Cells growing in subconfluent monolayers were incubated in 50 ng/ ml colcemid (Sigma-Aldrich) for one hour and then harvested, sequentially treated with 75 mM KCl and Carnoy’s fixative (3:1 methanol/acetic acid vol/vol) and applied to glass slides. Chromosomes were stained with DAPI. For enumeration of interphase chromosomes, fixed cells were permeabilized with pepsin and hybridized to a CEP18 probe specific for the centromeric region of chromosome 18 (Vysis), according to the manufacturer’s protocols. Nuclei were counterstained with DAPI. At least 200 interphase cells of each genotype were visualized and scored.
Immunofluorescence and cell imaging.
Subconfluent cell cultures growing on chamber slides (Nunc) were fixed in 3% paraformaldehyde, permeabilized with 0.5% triton X-100 and incubated with primary antibodies overnight. DNA damage foci were visualized using primary antibodies to γH2AX (Millipore) and 53BP1 (Novus Biologicals) and a secondary antibody conjugated to AF488 (Invitrogen). Centrosomes were visualized by direct immunofluorescence using a CY3-conjugated anti-γ tubulin antibody (Sigma-Aldrich). Mitotic spindles were visualized using an anti-α tubulin antibody (Santa Cruz Biotechnology) and a secondary antibody conjugated to AF488 (Invitrogen). Images were captured at room temperature AxioImager Z1 microscope equipped with an AxioCam HRm camera, Axiovision 4.6.3 software and a Plan Neofluar 63x/1.25NA lens (Zeiss). For time-lapse studies, transfected cells were analyzed on a TE200 inverted microscope (Nikon) with a 10x objective enclosed in a temperature and CO2-controlled incubator (Neve). Fluorescent and phase images were acquired at 10 min intervals for 72 h.
EdU incorporation assay.
EdU incorporation was assessed with the Click-iT EdU Flow Cytometry Assay Kit (Invitrogen). Cells were synchronized by treatment with media containing 2.5 mM thymidine for 18 h, washed three times with Hank’s Balanced Saline Solution (HBSS), grown for 8 h in untreated media, and then treated with media containing 2 ∝g/ml aphidicolin (Sigma) for 16 h. Cells were washed three times with HBSS and immediately labeled with 10 ∝M EdU for 2 h. Asynchronous cells were similarly labeled with 10 ∝M EdU for 2 h. Inc., EdU was detected with Alexa Fluor 488 and total DNA content was measured by 7-AAD incorporation, according to the manufacturer’s recommendations.
Histone H1 kinase assay.
Cells were synchronized by double thymidine block as previously described in reference Citation43. After release into fresh growth medium, cells were washed and harvested at the indicated times. Non-denatured extracts for in vitro kinase assays were prepared as previously described in reference Citation24, and kinase complexes were immunoprecipitated by 2 ∝g anti-Cdc2 monoclonal antibody (Santa Cruz Biotechnology) and collected with protein A-sepharose (Invitrogen). Precipitated complexes were washed with lysis buffer and incubated in 25 ∝l of a solution containing 20 mM TRIS-HCl (pH 7.5), 7.5 mM magnesium chloride, 1 mM dithiothreitol, 50 ∝M adenosine triphosphate (ATP), 20 ∝Ci [γ32P]ATP (6,000 Ci/mmol; Perkin Elmer) and 1 ∝g of histone H1 protein (Boehringer-Manheim) for 30 min at 30°C. After addition of 25 ∝l of 2x sample buffer and SDS-PAGE, 32P-labeled histone H1 was visualized by autoradiography. Excised bands were quantitatively analyzed on a scintillation counter (Beckman-Coulter).
Transfection and selection of stable clones.
The stable expression of histone H2B-GFP was achieved by transfection of cells with the plasmid pBOS H2BGFP-N1,Citation44 followed by selection of pooled clones with blasticidin. HA-tagged Cdk1 was stably expressed in DLD-1 CHK1+/- cells by co-transfection of the plasmid pCDL-Sra296-Cdc2Citation44 and plasmid conferring resistance to puromycin. A pool of puromycin-resistant cells was grown for 30 generations along with transfection control cells expressing only pPur.
Acknowledgements
The authors gratefully acknowledge the expert assistance of the staff of the Kimmel Cancer Center Cell Imaging Core facility and the Flow Cytometry Core facility. We thank David Morgan for Cdk1 expression plasmids. Support for this work was provided by the Flight Attendant Medical Research Institute and the National Cancer Institute grants R01CA157535 (F.B.) and F32CA119724 (D.W.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Ganem NJ, Pellman D. Limiting the proliferation of polyploid cells. Cell 2007; 131:437 - 40; http://dx.doi.org/10.1016/j.cell.2007.10.024; PMID: 17981108
- Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol 2009; 10:478 - 87; http://dx.doi.org/10.1038/nrm2718; PMID: 19546858
- Chung JH, Bunz F. Cdk2 is required for p53-independent G2/M checkpoint control. PLoS Genet 2010; 6:e1000863; http://dx.doi.org/10.1371/journal.pgen.1000863; PMID: 20195506
- Enders GH. Expanded roles for Chk1 in genome maintenance. J Biol Chem 2008; 283:17749 - 52; http://dx.doi.org/10.1074/jbc.R800021200; PMID: 18424430
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 2008; 9:616 - 27; http://dx.doi.org/10.1038/nrm2450; PMID: 18594563
- Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 2001; 21:4129 - 39; http://dx.doi.org/10.1128/MCB.21.13.4129-4139.2001; PMID: 11390642
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 2000; 14:1448 - 59; PMID: 10859164
- Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev 2003; 17:615 - 28; http://dx.doi.org/10.1101/gad.1067403; PMID: 12629044
- Niida H, Katsuno Y, Banerjee B, Hande MP, Nakanishi M. Specific role of Chk1 phosphorylations in cell survival and checkpoint activation. Mol Cell Biol 2007; 27:2572 - 81; http://dx.doi.org/10.1128/MCB.01611-06; PMID: 17242188
- Wilsker D, Petermann E, Helleday T, Bunz F. Essential function of Chk1 can be uncoupled from DNA damage checkpoint and replication control. Proc Natl Acad Sci U S A 2008; 105:20752 - 7; http://dx.doi.org/10.1073/pnas.0806917106; PMID: 19091954
- Rago C, Vogelstein B, Bunz F. Genetic knockouts and knockins in human somatic cells. Nat Protoc 2007; 2:2734 - 46; http://dx.doi.org/10.1038/nprot.2007.408; PMID: 18007609
- Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA Jr., Kinzler KW, et al. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci U S A 2009; 106:3964 - 9; http://dx.doi.org/10.1073/pnas.0813333106; PMID: 19225112
- Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature 1997; 386:623 - 7; http://dx.doi.org/10.1038/386623a0; PMID: 9121588
- Michel LS, Liberal V, Chatterjee A, Kirchwegger R, Pasche B, Gerald W, et al. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 2001; 409:355 - 9; http://dx.doi.org/10.1038/35053094; PMID: 11201745
- Jallepalli PV, Waizenegger IC, Bunz F, Langer S, Speicher MR, Peters JM, et al. Securin is required for chromosomal stability in human cells. Cell 2001; 105:445 - 57; http://dx.doi.org/10.1016/S0092-8674(01)00340-3; PMID: 11371342
- Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, et al. Inactivation of hCDC4 can cause chromosomal instability. Nature 2004; 428:77 - 81; http://dx.doi.org/10.1038/nature02313; PMID: 14999283
- Bunz F, Fauth C, Speicher MR, Dutriaux A, Sedivy JM, Kinzler KW, et al. Targeted inactivation of p53 in human cells does not result in aneuploidy. Cancer Res 2002; 62:1129 - 33; PMID: 11861393
- Sherwood R, Takahashi TS, Jallepalli PV. Sister acts: coordinating DNA replication and cohesion establishment. Genes Dev 2010; 24:2723 - 31; http://dx.doi.org/10.1101/gad.1976710; PMID: 21159813
- Lindqvist A, Rodríguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol 2009; 185:193 - 202; http://dx.doi.org/10.1083/jcb.200812045; PMID: 19364923
- Davoli T, Denchi EL, de Lange T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell 2010; 141:81 - 93; http://dx.doi.org/10.1016/j.cell.2010.01.031; PMID: 20371347
- Syljuåsen RG, Sørensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, et al. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol 2005; 25:3553 - 62; http://dx.doi.org/10.1128/MCB.25.9.3553-3562.2005; PMID: 15831461
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005; 434:907 - 13; http://dx.doi.org/10.1038/nature03485; PMID: 15829965
- Lam MH, Liu Q, Elledge SJ, Rosen JM. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell 2004; 6:45 - 59; http://dx.doi.org/10.1016/j.ccr.2004.06.015; PMID: 15261141
- Hurley PJ, Wilsker D, Bunz F. Human cancer cells require ATR for cell cycle progression following exposure to ionizing radiation. Oncogene 2007; 26:2535 - 42; http://dx.doi.org/10.1038/sj.onc.1210049; PMID: 17043640
- Murga M, Bunting S, Montaña MF, Soria R, Mulero F, Cañamero M, et al. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat Genet 2009; 41:891 - 8; http://dx.doi.org/10.1038/ng.420; PMID: 19620979
- Ruzankina Y, Schoppy DW, Asare A, Clark CE, Vonderheide RH, Brown EJ. Tissue regenerative delays and synthetic lethality in adult mice after combined deletion of Atr and Trp53. Nat Genet 2009; 41:1144 - 9; http://dx.doi.org/10.1038/ng.441; PMID: 19718024
- Shimada M, Niida H, Zineldeen DH, Tagami H, Tanaka M, Saito H, et al. Chk1 is a histone H3 threonine 11 kinase that regulates DNA damage-induced transcriptional repression. Cell 2008; 132:221 - 32; http://dx.doi.org/10.1016/j.cell.2007.12.013; PMID: 18243098
- Mochida S, Maslen SL, Skehel M, Hunt T. Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 2010; 330:1670 - 3; http://dx.doi.org/10.1126/science.1195689; PMID: 21164013
- Gharbi-Ayachi A, Labbé JC, Burgess A, Vigneron S, Strub JM, Brioudes E, et al. The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science 2010; 330:1673 - 7; http://dx.doi.org/10.1126/science.1197048; PMID: 21164014
- Andreassen PR, Lohez OD, Lacroix FB, Margolis RL. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell 2001; 12:1315 - 28; PMID: 11359924
- Stukenberg PT. Triggering p53 after cytokinesis failure. J Cell Biol 2004; 165:607 - 8; http://dx.doi.org/10.1083/jcb.200405089; PMID: 15184396
- Durkin SG, Arlt MF, Howlett NG, Glover TW. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene 2006; 25:4381 - 8; http://dx.doi.org/10.1038/sj.onc.1209466; PMID: 16732333
- Stolz A, Ertych N, Kienitz A, Vogel C, Schneider V, Fritz B, et al. The CHK2-BRCA1 tumour suppressor pathway ensures chromosomal stability in human somatic cells. Nat Cell Biol 2010; 12:492 - 9; http://dx.doi.org/10.1038/ncb2051; PMID: 20364141
- Stolz A, Bastians H. A novel role for Chk2 after DNA damage in mitosis?. Cell Cycle 2010; 9:25 - 6; http://dx.doi.org/10.4161/cc.9.1.10579; PMID: 20016261
- Ullah Z, Kohn MJ, Yagi R, Vassilev LT, DePamphilis ML. Differentiation of trophoblast stem cells into giant cells is triggered by p57/Kip2 inhibition of CDK1 activity. Genes Dev 2008; 22:3024 - 36; http://dx.doi.org/10.1101/gad.1718108; PMID: 18981479
- Ullah Z, de Renty C, DePamphilis ML. Checkpoint kinase 1 prevents cell cycle exit linked to terminal cell differentiation. Mol Cell Biol 2011; 31:4129 - 43; http://dx.doi.org/10.1128/MCB.05723-11; PMID: 21791608
- Peddibhotla S, Rosen JM. Chking and executing cell division to prevent genomic instability. Cell Cycle 2009; 8:2339 - 42; http://dx.doi.org/10.4161/cc.8.15.9169; PMID: 19571679
- Peddibhotla S, Lam MH, Gonzalez-Rimbau M, Rosen JM. The DNA-damage effector checkpoint kinase 1 is essential for chromosome segregation and cytokinesis. Proc Natl Acad Sci U S A 2009; 106:5159 - 64; http://dx.doi.org/10.1073/pnas.0806671106; PMID: 19289837
- Fishler T, Li YY, Wang RH, Kim HS, Sengupta K, Vassilopoulos A, et al. Genetic instability and mammary tumor formation in mice carrying mammary-specific disruption of Chk1 and p53. Oncogene 2010; 29:4007 - 17; http://dx.doi.org/10.1038/onc.2010.163; PMID: 20473325
- Tort F, Hernández S, Beà S, Camacho E, Fernández V, Esteller M, et al. Checkpoint kinase 1 (CHK1) protein and mRNA expression is downregulated in aggressive variants of human lymphoid neoplasms. Leukemia 2005; 19:112 - 7; PMID: 15526025
- De Keersmaecker K, Real PJ, Gatta GD, Palomero T, Sulis ML, Tosello V, et al. The TLX1 oncogene drives aneuploidy in T cell transformation. Nat Med 2010; 16:1321 - 7; http://dx.doi.org/10.1038/nm.2246; PMID: 20972433
- Topaloglu O, Hurley PJ, Yildirim O, Civin CI, Bunz F. Improved methods for the generation of human gene knockout and knockin cell lines. Nucleic Acids Res 2005; 33:e158; http://dx.doi.org/10.1093/nar/gni160; PMID: 16214806
- Gatei M, Young D, Cerosaletti KM, Desai-Mehta A, Spring K, Kozlov S, et al. ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nat Genet 2000; 25:115 - 9; http://dx.doi.org/10.1038/75508; PMID: 10802669
- Kanda T, Sullivan KF, Wahl GM. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol 1998; 8:377 - 85; http://dx.doi.org/10.1016/S0960-9822(98)70156-3; PMID: 9545195
- Gu Y, Rosenblatt J, Morgan DO. Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J 1992; 11:3995 - 4005; PMID: 1396589