Abstract
In this manuscript, we present experimental evidence that PKCs phosphorylate p27 at T198 in vitro and in vivo, resulting in p27 stabilization and cell cycle arrest in MCF-7 and HeLa cells. Our findings indicate that (1) recombinant PKCα, βII, δ, η and θ isoforms phosphorylate, in in vitro kinase assays, wild-type recombinant p27 protein expressed in E. coli and wild-type p27 protein immunoprecpitated from transfected HEK-293 cells but not the T198A mutant, (2) adoptive expressed PKCα and δ phosphorylate both transfected and endogenous p27 at T198 in HEK-293 cells, (3) T198 phosphorylation of transfected and endogenous p27 is increased by PKC activators [Phorbol 12-myristate 13-acetate (PMA)] and suppressed by PKC inhibitors (Rottlerin A, G06976, Calphostin C), (4) in parallel with increased T198 phosphorylation, PMA induces stabilization of p27 protein in HeLa cells, whereas PKC inhibitors induce a decrease in p27 stability and, finally, (5) PMA-induced p27 upregulation is necessary for growth arrest of HeLa and MCF-7 cells induced by PKC activation by PMA.
Overall, these results suggest that PKC-dependent upregulation of p27 induced by its phosphorylation at T198 represents a mechanism that mediates growth arrest promoted by PMA and provide novel insights on the ability of different PKC isoforms to play a role in controlling cell cycle progression.
Keywords: :
Introduction
The cyclin-dependent kinase (Cdk) inhibitor p27Kip1 (p27) is an important regulator of the mammalian cell cycle.Citation1,Citation2 p27 negatively regulates G1 progression by binding to cyclin-Cdk1, cyclin-Cdk2 and cyclin-Cdk4/6 complexes and preventing their activity.Citation3-Citation6 The activity of p27 is controlled by its concentration, its distribution among different cellular complexes and its subcellular localization.Citation7,Citation8 One key mechanism involved in the regulation of p27 abundance is proteolysis by the ubiquitin-proteasome pathway.Citation9 Accordingly, the levels of p27 protein are high in quiescent cells and decline upon mitogenic stimulation.Citation10,Citation11
Both the intracellular concentration and the localization of p27 are regulated by extensive posttranslational modifications.Citation12,Citation13 p27 protein is phosphorylated at multiple sites: serine 10, threonines 157, 187, 198 and tyrosines 74, 88, 89. Phosphorylation of p27 has been studied extensively, and for each of the phosphorylation sites, several kinases have been reported, and different functions have been ascribed.Citation14 When phosphorylated on Thr187 by cyclin E-Cdk2,Citation15-Citation18 p27 is recognized and targeted for ubiquitylation by the SCF Skp2 ubiquitin-protein ligase and its cofactor, the Cdk subunit 1.Citation19-Citation24 In addition, p27 undergoes rapid translocation from the nucleus into the cytoplasm followed by degradation through the ubiquitin-proteasome pathway by undetermined mechanisms that involve phosphorylation of Ser10, Thr157 or Thr198 by hKIS, AKT or RSK, respectively.
As to Thr198, several papers have illustrated the role of its phosphorylation in the regulation of p27 localization and stability. Fujita et al.Citation25 and Motti and coworkersCitation26 identified Thr198 as a residue phosphorylated by Akt. Subsequent work demonstrated that Thr198 is phosphorylated also by p90 ribosomal protein S6 kinases (RSKs)Citation27 and by the AMP-activated protein kinase (LKB1-AMPK).Citation28 Phosphorylation of p27 at Thr198 apparently increases p27 stabilityCitation29 and regulates cell motility.Citation30
Bioinformatic analysis of p27 phosphorylation sites has suggested that Thr198 may be the target of other kinases, including protein kinase C. Protein kinase C (PKC) is a family of serine-threonine kinases that regulate several cellular processes, including cellular growth and proliferation, transformation, differentiation and apoptosis.Citation31,Citation32 Numerous PKC isoenzymes have been described so far and are classified according to their structures and mechanisms of activation.Citation33 Accordingly, PKC isoforms can be classified into three groups based on their structure and cofactor requirement: classical PKC isoforms (α, β1, β2, γ), which are regulated by diacylglycerol (DAG) and Ca2+, novel PKC isoforms (δ, ε, η, θ) which are regulated by DAG but not by Ca2+, and atypical PKC isoforms (ζ, ι), which are not responsive to either DAG or Ca2+.Citation34,Citation35
In this manuscript, we have identified PKC as a novel bona fide Thr198 kinase and characterized the effects of PKC-dependent phoshorylation of p27 on cell proliferation.
Results
PKCs phosphorylate p27 at T198 in vitro
It has been shown that the intracellular levels of p27 protein are tightly regulated by its phosphorylation status.Citation9 A computational model based on kinase-specific phosphorylation site prediction group-based prediction system (GPS) version 2.1 software (http://gps.biocuckoo.org/)Citation36 was used to identify novel putative p27 kinases. GPS analysis identified threonine 198 as a potential phosphorylation site by the members of PKC kinase family ().
Figure 1. PKC phosphorylates p27 in vitro. (A) p27 phosphorylation site prediction by the Group-based Prediction System software. (B) In vitro kinase assays. Recombinant p27 was incubated with several recombinant PKCs in presence of 32P-labeled adenosine triphosphate (ATP), fractionated by SDS-PAGE, and transferred to nylon membrane. p27 phosphorylation was determined either by autoradiography. Histone H1 was used as control of PKCs’ activity.
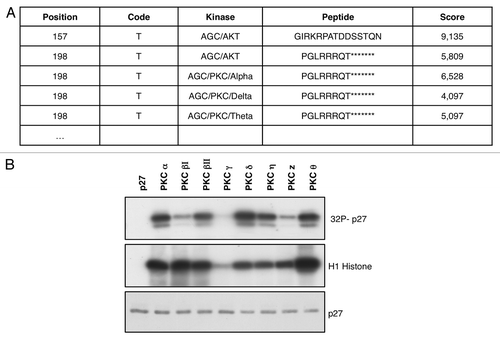
Therefore, we performed in vitro kinase assays to determine whether PKCs were able to phosphorylate p27. We set up the kinase reactions, incubating PKCs and 6-His-tagged p27 recombinant proteins in presence of 32P-labeled adenosine triphosphate (ATP). We tested different isoforms of PKC (α; βΙ; βΙΙ; γ; δ; η; ζ; θ) using recombinant histone H1 as control. We found that all the isoforms tested except PKCγ were able to phosphorylate p27, though with different efficacy ().
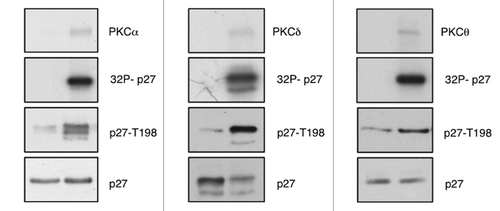
In order to investigate whether PKC was able to phosphorylate p27 at T198, we performed immunoblots using an antibody that recognizes the phosphorylated Thr198 (pT198).Citation26 Filters were incubated with total α-p27 antibody in order to normalize the results (). We found that all PKCs that phosphorylate p27 induced a marked increase in pT198 staining. Data shown in are related to kinase assays performed with PKCα, PKCδ and PKCθ.
Figure 2. PKC phosphorylates p27 at T198. In vitro kinase assays. Recombinant p27 was incubated with recombinant PKCs in presence of 32P-labeled adenosine triphosphate (ATP), fractionated by SDS-PAGE, and transferred to nylon membrane. p27 phosphorylation was determined either by autoradiography or by immunoblot using an antibody specific for phosphorylated T198 (anti-pT198).
We also performed an immunocomplex kinase assay to confirm that PKCs were able to phosphorylate p27. To this aim, HAp27-wt and HAp27-T198A were transiently transfected into HEK-293 cells, and total cell extracts were immunoprecipitated with anti-HA antibody. The immunocomplexes were incubated with recombinant PKCs proteins (PKCα and PKCδ) in the presence of [γ-32P]-ATP; T198 phosphorylation was detected by western blotting with pT198 antibody. Results were similar to those obtained with recombinant p27 and also indicated that PKCs were able to phosphorylate HAp27-wt but not the mutant HAp27-T198A (data not shown).
PKCs phosphorylate at T198 p27 in vivo
We also performed transfection experiments to determine whether PKCs were able to phosphorylate p27 in vivo. To this aim, HEK-293 cells were transiently transfected with HAp27-wt in the presence or absence of PKCα, PKCδ () or PKCθ (not shown). Total cell extracts were analyzed for the phosphorylation status at p27-T198A by immunoblot with the pT198 antibody and then with an antibody anti-p27 to normalize for protein levels.
Figure 3. PKC phosphorylates p27 at T198 in vivo. In vivo kinase assays. (A) Active PKCα or PKCδ were transfected with HAp27-wt or HAp27-T198A into HEK-293 cells. P27 phosphorylation was determined by immunoblot with anti-pT198. (B) HEK-293 cells were stimulated with or without PMA. (C) HEK-293 cells were transiently transfected with HAp27-wt and/or active PKCα and cells were cultured with or without PMA. P27 phosphorylation was determined by immunoblot using an antibody specific for phosphorylated T198.
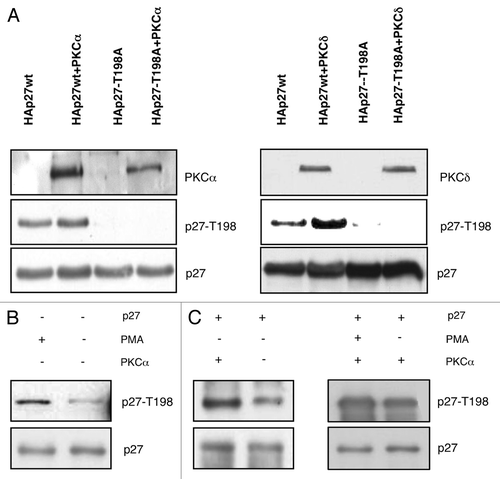
The phorbol ester PMA is a potent activator of PKC in eukaryotic cells.Citation37,Citation38 Therefore, we analyzed the effect of PMA treatment on p27 phosphorylation. We found that treatment with PMA increased T198 phosphorylation of endogenous p27 in untransfected HEK-293 cells () or of transfected HAp27-wt (, left panel). In addition, T198 phosphorylation of HAp27-wt transfected into HEK-293 cells was markedly stimulated by PMA treatment of HEK-293 cells transduced with PKCα (, right panel). As expected, PMA treatment was accompained by activation of PKCα ().
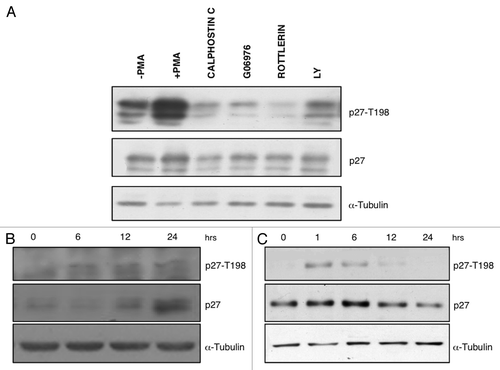
To provide complementary evidence that PKC phosphorylates p27 at T198, we transiently transfected HAp27-wt into HEK-293 that had been prior treated with PKC inhibitors (calphostin C, G06976, Rottlerin) or the PI-3 kinase inhibitor (LY294002) as control (). We found that PMA-dependent T198 phosphorylation of p27 in HEK-293 cells was markedly reduced, if PMA treatment was preceded by use of PKC inhibitors. These results support the hypothesis that p27 is a novel substrate of PKC.
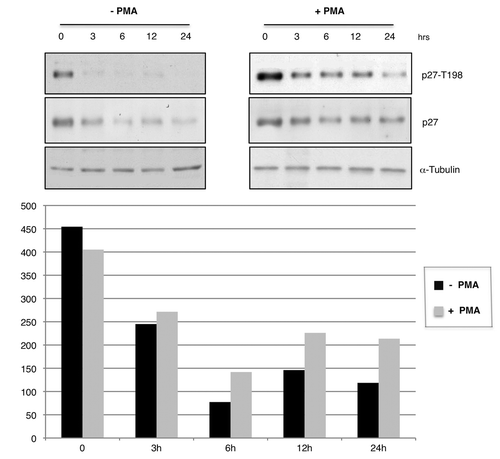
Figure 4. PMA induces T198 phosphorylation and increases p27 protein levels in HeLa and MCF7 cells. (A) Immunoblot analysis of phospho-T198 and p27 protein levels in HEK-293 cells transfected with HAp27-wt and treated with PMA with or without PKC inhibitors before adding PMA as indicated. LY294002 was used as control. (B) Kinetic analysis of p27 T198 phosphorylation in HeLa cells. Immunoblot analysis of phospho-T198 and p27 protein levels after stimulation with PMA at various time points. (C) Kinetic analysis of p27 T198 phosphorylation in MCF7 cells. Immunoblot analysis of phospho-T198 and p27 protein levels after stimulation with PMA at various time points.
PMA-dependent PKC activation increases p27 phosphorylation and stability
To further explore the effect of PMA on p27 phosphorylation, we determined the kinetic of p27 phosphorylation in MCF7 breast cancer cells and HeLa cervical cancer cells stimulated with PMA over a 24 h time course (). Total cell extracts were loaded on SDS-page gel and were immunoblotted with antibodies specific for p27 and its T198 phosphorylated form. We found that PMA increases the levels of T198 phosphorylation of endogenous p27 at 1–12 h in MCF7 cells and at 6–24 h in HeLa cells, respectively. As expected, the increased phosphorylation observed in MCF7 and HeLa cells after PMA treatment was reflected into an increased levels of total p27 levels ().
To test whether the stability of p27 was dependent on PKC-mediated phosphorylation at T198, we compared the half-life of p27 protein in the presence or absence of PMA. To this aim, HeLa cells after PMA stimulation, were treated with cycloheximide (CHX), a potent inhibitor of protein translation. The stability of p27 protein was determined by immunoblot performed on total protein extracts (). As shown in , the stability of p27 was markedly higher when HeLa cells were treated with PMA. Similar results were obtained when we compared the half-life of transfected wild-type p27-HA protein in the presence or absence of PMA in HEK-293 cells (data not shown).
Figure 5. PMA stabilizes p27 in HeLa cells. Immunoblot analysis of p27 stability in HeLa treated with solvent or PMA for 1h before cycloheximide treatment as indicated. The intensities of WB signals were quantified by NIH ImageJ software; the results are showed in the graphics. Cells were cultured with or without PMA.
PMA inhibits cell proliferation in a p27-dependent manner
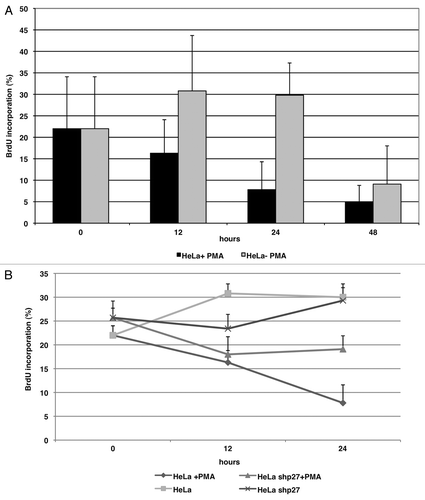
To evaluate the functional relevance of PMA-dependent p27 phosphorylation on cell cycle progression, we performed a BrdU incorporation assay in HeLa cells treated with PMA. As shown in , PMA inhibited BrdU incorporation in HeLa cells. To understand whether p27 plays a prominent role in the antiproliferative effects induced by PMA, Hela cell lines were transduced with a lentivirus expressing shp27 to knockdown the expression of p27. Cells infected with shSCR were used as control. We found that the depletion of p27 (shp27) resulted in slightly increased cell proliferation of HeLa cells compared with SCR cells. Notably, the antiproliferative effect exerted by PMA treatment of HeLa cells was suppressed by p27 knockdown (). These data indicate that p27 contributes to the cell cycle arrest induced by PKC activation. Similar results were obtained with MCF7 cell lines (data not shown).
Figure 6. p27 induction is critical for PMA-dependent inhibition of proliferation of HeLa cells. (A) BrdU incorporation of HeLa cells in presence or absence of PMA. (B) BrdU incorporation of HeLa cells infected with a control lentivirus (SCR) or a virus encoding a short hairpin RNA directed against p27 (shp27) in presence or absence of PMA.
Discussion
Phorbol 12-myristate 13-acetate (PMA) is known to affect a variety of cellular processes, including cell proliferation, differentiation and migration, through the activation of PKC kinases. Accordingly, PMA has been shown to promote antiproliferative and antimigratory effects in some types of cancer cells, including HeLa cervical cancer cells. In the present study, we report that the strong PMA-dependent antiproliferative effect is mediated at least in part through the activation of PKC and the subsequent phosphorylation and stabilization of the cell cycle inhibitor p27.
Our findings indicate that (1) the PKCα, βII, δ, η and θ isoforms preferentially phosphorylate p27 in vitro and in vivo at the T198; no such phosphorylation was observed with PKCβΙ, ζ and, to a lesser degree γ; (2) PKCα and δ physically interact in vivo with p27 in HEK-293 and HeLa cells; (3) T198 phosphorylation can be detected in immunoprecipitates of transfected and/or endogenous p27, and it is increased by PMA and abolished by PKC inhibitors, such as Calphostin C, G06976 and Rottlerin; (4) in parallel with increased p27 phosphorylation at T198, PMA induces stabilization of p27 protein in HeLa cells, whereas PKC inhibitors induce a decrease in p27 stability; (5) PMA-induced p27 upregulation is necessary for growth arrest of HeLa cells following PKC activation by PMA. Overall, these results suggest that PKC-dependent phosphorylation of p27 at T198 represents one of the mechanisms that mediates the growth arrest promoted by PMA treatment in some cells, possibly by increasing p27 stability, and provide novel insights on the ability of different PKC isoforms to play a role in controlling cell cycle progression.
Phosphorylation of key amino acid residues appears to be a critical mechanism whereby both the stability and the localization of p27 are controlled. In cells arrested in the G0 phase of the cell cycle, p27 is expressed at high levels in the nucleus and contributes to the maintenance of the quiescent state.Citation39,Citation40 Such high expression levels are due to enhanced p27 mRNA translation rate and/or stabilization of the protein as a result of phosphorylation at serine 10 (S10) and threonine198 (T198).Citation29,Citation41 Serine 10 represents the major phosphorylation site of p27 and accounts for about 80% of the total p27 phosphorylation. Myrk/Dyrk1B phosphorylates p27 during the G0 phase of the cell-cycle, stabilizing and maintaining it within the nucleus, where it can bind to CDK2 and induce cell cycle arrest.Citation42 Conversely, the Kis kinase phosphorylates p27 at ser10 in the G1 phase, enabling it to bind CRM1 and be transported into the cytoplasm for degradation.Citation43
T198 is mainly phosphorylated in G0. However, as with S10, T198 is also subject to modification early in G1. At least four kinases have been demonstrated to phosphorylate p27 at T198, including AKT, RSK1, RSK2 and AMPK.Citation25,Citation27 These kinases are activated in response to mitogenic stimuli or to nutrient deprivation and induce p27 cytoplasmic delocalization. However, phosphorylation of p27 at T198 also appears to increase p27 stability. Nutrient-deprived cells showed AMPK-dependent elevation of T198 phosphorylation and increased p27 stability.Citation28 Accordingly, cells treated with PMA or transfected with activated PKCα or δ show increased T198 phosphorylation and increased stability of p27 protein, though the subcellular localization of p27 is apparently unaltered. The findings summarized above suggest a model in which PKC-dependent phosphorylation of p27 at T198 early in G1 controls the stability of p27 but not its subcellular localization. The difference with the reported effects of AKT and RSK phosphorylation of p27 localization may be due to different kinetics of PKC phosphorylation compared with AKT and RSK, the simultaneous phosphorylation of other residues (i.e., T157 or S10) or the different cellular contest.Citation25-Citation27
The mechanism underlying the stabilization of p27 by T198 phosphorylation in G0 and G1 cells remains to be determined. Also, it is unclear whether phosphorylation at S10 and T198 cooperate in the stabilization of p27 in G0/G1 cells, or whether one or the other phosphorylation is sufficient to achieve maximal p27 stability. The finding that PKC phosphorylates T198 but not S10 or T157 suggests that the different phosphorylation events are not necessarily linked.
It is worth noting that the PKC isoforms that more efficiently phosphorylate p27 at T198 are PKCα and δ, whereas PKCβΙ and ζ and, to a lesser degree, PKCγ are relatively inefficient in their ability to phosphorylate p27 at T198. Such differential phosphorylation by PKC has often been observed; for example, only PKCα, βI, βII, γ, δ and θ, but not PKCε, ζ and η, phosphorylate S1674 of Cav1.2 α1c,Citation44 and may be associated with the different roles that PKC isoforms play in transformation.
In fact, PKCs have long been recognized to have a role in regulating different aspects of tumor growth and development,Citation45 although this role is clearly complex and highly tissue-dependent, because in some cases they act as tumor promoters, and in others, they appear to function as tumor suppressors. For example, overexpression of some PKCs has been demonstrated in tissue samples of prostate, endometrial, high-grade urinary bladder and hepatocellular cancers,Citation46-Citation49 while PKCα, βΙ, δ and ζ have been shown to be decreased at different stages of neoplastic progression.Citation50-Citation56 It is of particular interest that in rat intestinal epithelial crypt cells, PKC signaling, and in particular PKCα, mediates cell cycle exit through rapid downregulation of G1 cyclins and increased expression of Cip/Kip cyclin-dependent kinase inhibitors.Citation50,Citation57-Citation66 Similarly, PMA has been shown to be a potent inhibitor of thyroid cancer cell proliferation and migration by a mechanism involving an increase in the levels of the cell cycle regulators p21 and p27 and a decrease in the levels of cyclin D3, and the cyclin-dependent kinases Cdk4 and Cdk6 decreased.Citation67
In conclusion, we demonstrate that some PKC isoforms, including PKCα and δ, are novel bona fide p27 kinases, and that PKC-dependent p27 phosphorylation at T198 increases the stability of p27 protein, which, at least in part, mediates cell cycle arrest of MCF-7 and HeLa cells induced by PMA.
Experimental Procedures
In vitro kinase assay
The recombinant 6-His-tagged-p27 proteins (or the recombinant Histone H1 used as control), were incubated for 30 min at 30°C in kinase assay buffer containing recombinant PKC. The PKC kinase assay buffer (20 mM Hepes, pH 7.4, 10 mM MgCl2, 0.1 mM CaCl2 or EGTA, 150 μM ATP, 0,1μl [γ32-P]ATP per reaction) was completed with 10 μg of phosphatidylserine (PS) and 2 μg of diacylglycerol (DAG) (SIGMA-Aldrich). For reactions with PKC ζ, kinase buffer required only PS. After incubation, the reaction mixtures were boiled in 2x Laemmli buffer, separated by 12% SDS-PAGE, transferred to nitrocellulose membrane and exposed to X-ray film.
For the immune complex protein kinase assay, cell lysates were immunoprecipitated with anti-HA antibody as described above. The immunoprecipitates were washed three times with cold lysis buffer and once with PKC kinase assay buffer containing recombinant PKC. After incubation for 30 min at 30°C, the reaction mixtures were boiled in 2x Laemmli buffer, separated by 12% SDS-PAGE, transferred to nitrocellulose membrane, and the filters were immunoblotted with the specific antibodies.
The recombinant proteins used in this work were: histone H1 from calf thymus; human recombinant PKCα; human recombinant PKCβΙ; human recombinant PKCβΙΙ; human recombinant PKCγ; human recombinant PKCδ; human recombinant PKCη; human recombinant PKCζ; human recombinant PKCθ. All recombinant proteins were purchased from SIGMA-Aldrich. The 6-His-tagged-p27 fusion proteins used as substrates were produced as described previously.Citation26
Constructs and lentiviral production
HA-tagged wild-type p27 has already been described.Citation68 The HA-tagged p27T198A was generated with a site-specific mutagenesis kit (Stratagene). The PKC plasmids used in this work are: Myr-PKCalpha-FLAG, PKC-Δ-wt and pBS-PKCtheta, and were purchased from AddGene.
pLenti PKCδ was generated as follows: PKCδ cDNA was cloned into pEntry vector and then a recombination reaction with a pLenti-DEST vector (Gateway® Technology cloning method) was performed. shRNA-targeting p27 (shp27) and shSCR constructs were purchased from Sigma-Aldrich. For transient transfection, HEK-293T cells seeded at 60–80% confluency in 100-mm dishes were transfected with appropriate expression plasmids and harvested after 24 h.
For viral production of shRNA, HEK-293T were transfected with 12μg of VSVG, 18μg of Δ8.9 and 13μg of shRNA. Forty-eight hours after transfection, media containing viral particles were collected and filtered through a 0.45-μm cellulose acetate filter and stored at -80°C. HeLa and Mcf7 cells were infected with appropriate constructs 24 h after seeding and treated with or without PMA for the indicated times.
Cell cultures
HEK-293 (Human Embryonic Kidney), HeLa, MCF-7, wild-type and p27-T198A MEF cells were cultured in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich) containing 10% fetal calf serum and antibiotics (50 U/ml penicillin and 50 μg/ml streptomycin, and glutamine) in a humidified incubator at 37°C. PMA (Phorbol 12-myristate 13-acetate, Sigma-Aldrich) was added to the medium at a concentration of 100 nM for the indicated times. Prior to PMA stimulation, the medium was replaced with DMEM containing 10% Fetal Bovine Serum Charcoal Stripped (Sigma-Aldrich).
LY294002 and PKC inhibitors were purchased from Calbiochem.
For determination of protein half-life, exponentially growing cells were seeded at 60% confluency, grown overnight and treated with 10 μg/mL cycloheximide for the indicated times.
BrdU incorporation
The BrdU (5-bromo-2′-deoxyuridine Labeling and Detection Kit I, Roche) incorporation assay was performed as follows. Cells (5 × 105) were plated into 60-mm-diameter dishes on 12mm coverslips and allowed to attach for 24 h. Then cells were cultured in the presence or absence of PMA for the indicated times. The labeling procedure was performed for 2 h at 37°C as recommended by manufacturer; BrdU labeling reagent was used at a final concentration of 10μM. After fixation, the cultures were incubated with anti-BrdU antibody (1:10 – mouse) for 30 min at 37°C. Then cells were washed with PBS and incubated with the anti-mouse-Ig-fluorescein for 30 min at 37°C. The cells were washed four times with PBS. The nuclei were simultaneously stained with 10 μg/mL of 4V, 6-diamidino-2-phenylindole. Fluorescence was visualized with a Zeiss 140 epifluorescence microscope equipped with filters allowing discrimination between FITC and DAPI and cells were counted.
Protein extraction, western blotting and Immunoprecipitation
Total cell extracts were prepared with lysis buffer: 50 mM HEPES pH 7.5, 5 mM EDTA, 250 mM NaCl,, 1 mM dithiothreitol, 0,5% Nonidet P40, 1mM Na3VO4, 1mM NaF, 1mM PMSF and a mix of protease inhibitors (SIGMAFAST protease inhibitor tablets for general use).
Lysates were centrifuged at 13,000 rpm for 30 min at +4 ◦C and the supernatants were collected. The protein concentration was determined using the Biorad Protein Assay (BIORAD). Samples were subjected to SDS/PAGE (12% polyacrylamide). The proteins were transferred onto nitrocellulose membrane (Whatman Protran nitrocellulose transfer membrane) by western blotting. Western blot analysis was performed using specific antibodies to the indicated proteins. The secondary antibodies used were horseradish peroxidase-conjugated anti-mouse and anti-rabbit antibodies (GE Healthcare). The proteins were detected by enhanced chemiluminescence (Amersham ECL Western Blotting Detection Reagents, GE Healthcare). Densitometric analysis was performed using ImageJ program for image analysis (NIH).
For immunoprecipitation experiments, the HA-fusion proteins were immunoprecipitated from 500 μg of the cell extracts incubating samples over night at 4°C with anti-HA antibody and recovering the beads by centrifugation at 8,000 rpm for 1 min at + 4°C.
The antibodies used in this study were anti-p27 C19 (Santa Cruz Biotechnology Inc.), anti-pT198 p27 (R&D Systems), anti-HA Affinity Matrix (Roche), anti-PKCα Η7 (Santa Cruz Biotechnology Inc.), anti-PKCδ (BD Transduction LaboratoriesTM), anti-PKCθ (Cell Signaling Technology), anti-α-Tubulin Ab-2 (Neo-Markers).
Acknowledgments
This work was supported by MIUR (PRIN, 20087FSFFP_001) to GV. Marianna Scrima was supported by a fellowship from FIRC.
Disclosure of Potential Conflicts of Interest
The authors have no potential conflicts of interest to disclose.
References
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999; 13:1501 - 12; http://dx.doi.org/10.1101/gad.13.12.1501; PMID: 10385618
- Hengst L, Reed SI. Inhibitors of the Cip/Kip family. Curr Top Microbiol Immunol 1998; 227:25 - 41; http://dx.doi.org/10.1007/978-3-642-71941-7_2; PMID: 9479824
- Pines J. Cyclins and cyclin-dependent kinases: a biochemical view. Biochem J 1995; 308:697 - 711; PMID: 8948422
- Nigg EA. Cyclin-dependent protein kinases: key regulators of the eukaryotic cell cycle. Bioessays 1995; 17:471 - 80; http://dx.doi.org/10.1002/bies.950170603; PMID: 7575488
- Alessandrini A, Chiaur DS, Pagano M. Regulation of the cyclin-dependent kinase inhibitor p27 by degradation and phosphorylation. Leukemia 1997; 11:342 - 5; http://dx.doi.org/10.1038/sj.leu.2400581; PMID: 9067571
- Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 1994; 78:59 - 66; http://dx.doi.org/10.1016/0092-8674(94)90572-X; PMID: 8033212
- Ekholm SV, Reed SI. Regulation of G(1) cyclin-dependent kinases in the mammalian cell cycle. Curr Opin Cell Biol 2000; 12:676 - 84; http://dx.doi.org/10.1016/S0955-0674(00)00151-4; PMID: 11063931
- Slingerland J, Pagano M. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol 2000; 183:10 - 7; http://dx.doi.org/10.1002/(SICI)1097-4652(200004)183:1<10::AID-JCP2>3.0.CO;2-I; PMID: 10699961
- Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, et al. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science 1995; 269:682 - 5; http://dx.doi.org/10.1126/science.7624798; PMID: 7624798
- Nourse J, Firpo E, Flanagan WM, Coats S, Polyak K, Lee MH, et al. Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature 1994; 372:570 - 3; http://dx.doi.org/10.1038/372570a0; PMID: 7990932
- Agrawal D, Hauser P, McPherson F, Dong F, Garcia A, Pledger WJ. Repression of p27kip1 synthesis by platelet-derived growth factor in BALB/c 3T3 cells. Mol Cell Biol 1996; 16:4327 - 36; PMID: 8754833
- Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell 2008; 14:159 - 69; http://dx.doi.org/10.1016/j.devcel.2008.01.013; PMID: 18267085
- Sicinski P, Zacharek S, Kim C. Duality of p27Kip1 function in tumorigenesis. Genes Dev 2007; 21:1703 - 6; http://dx.doi.org/10.1101/gad.1583207; PMID: 17639075
- Borriello A, Cucciolla V, Oliva A, Zappia V, Della Ragione F. p27Kip1 metabolism: a fascinating labyrinth. Cell Cycle 2007; 6:1053 - 61; http://dx.doi.org/10.4161/cc.6.9.4142; PMID: 17426451
- Müller D, Bouchard C, Rudolph B, Steiner P, Stuckmann I, Saffrich R, et al. Cdk2-dependent phosphorylation of p27 facilitates its Myc-induced release from cyclin E/cdk2 complexes. Oncogene 1997; 15:2561 - 76; http://dx.doi.org/10.1038/sj.onc.1201440; PMID: 9399644
- Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev 1997; 11:1464 - 78; http://dx.doi.org/10.1101/gad.11.11.1464; PMID: 9192873
- Vlach J, Hennecke S, Amati B. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J 1997; 16:5334 - 44; http://dx.doi.org/10.1093/emboj/16.17.5334; PMID: 9311993
- Montagnoli A, Fiore F, Eytan E, Carrano AC, Draetta GF, Hershko A, et al. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev 1999; 13:1181 - 9; http://dx.doi.org/10.1101/gad.13.9.1181; PMID: 10323868
- Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol 1999; 1:193 - 9; http://dx.doi.org/10.1038/12013; PMID: 10559916
- Sutterlüty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Müller U, et al. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol 1999; 1:207 - 14; http://dx.doi.org/10.1038/12027; PMID: 10559918
- Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol 1999; 9:661 - 4; http://dx.doi.org/10.1016/S0960-9822(99)80290-5; PMID: 10375532
- Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J 2000; 19:2069 - 81; http://dx.doi.org/10.1093/emboj/19.9.2069; PMID: 10790373
- Ganoth D, Bornstein G, Ko TK, Larsen B, Tyers M, Pagano M, et al. The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat Cell Biol 2001; 3:321 - 4; http://dx.doi.org/10.1038/35060126; PMID: 11231585
- Spruck C, Strohmaier H, Watson M, Smith AP, Ryan A, Krek TW, et al. A CDK-independent function of mammalian Cks1: targeting of SCF(Skp2) to the CDK inhibitor p27Kip1. Mol Cell 2001; 7:639 - 50; http://dx.doi.org/10.1016/S1097-2765(01)00210-6; PMID: 11463388
- Fujita N, Sato S, Katayama K, Tsuruo T. Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J Biol Chem 2002; 277:28706 - 13; http://dx.doi.org/10.1074/jbc.M203668200; PMID: 12042314
- Motti ML, De Marco C, Califano D, Fusco A, Viglietto G. Akt-dependent T198 phosphorylation of cyclin-dependent kinase inhibitor p27kip1 in breast cancer. Cell Cycle 2004; 3:1074 - 80; http://dx.doi.org/10.4161/cc.3.8.1073; PMID: 15280662
- Fujita N, Sato S, Tsuruo T. Phosphorylation of p27Kip1 at threonine 198 by p90 ribosomal protein S6 kinases promotes its binding to 14-3-3 and cytoplasmic localization. J Biol Chem 2003; 278:49254 - 60; http://dx.doi.org/10.1074/jbc.M306614200; PMID: 14504289
- Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol 2007; 9:218 - 24; http://dx.doi.org/10.1038/ncb1537; PMID: 17237771
- Kossatz U, Vervoorts J, Nickeleit I, Sundberg HA, Arthur JS, Manns MP, et al. C-terminal phosphorylation controls the stability and function of p27kip1. EMBO J 2006; 25:5159 - 70; http://dx.doi.org/10.1038/sj.emboj.7601388; PMID: 17053782
- Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev 2004; 18:862 - 76; http://dx.doi.org/10.1101/gad.1185504; PMID: 15078817
- Nishizuka Y. Studies and perspectives of protein kinase C. Science 1986; 233:305 - 12; http://dx.doi.org/10.1126/science.3014651; PMID: 3014651
- Lévesque JT, Sirard MA. Effects of different kinases and phosphatases on nuclear and cytoplasmic maturation of bovine oocytes. Mol Reprod Dev 1995; 42:114 - 21; http://dx.doi.org/10.1002/mrd.1080420115; PMID: 8562045
- Saxon ML, Zhao X, Black JD. Activation of protein kinase C isozymes is associated with post-mitotic events in intestinal epithelial cells in situ. J Cell Biol 1994; 126:747 - 63; http://dx.doi.org/10.1083/jcb.126.3.747; PMID: 8045938
- Spitaler M, Cantrell DA. Protein kinase C and beyond. Nat Immunol 2004; 5:785 - 90; http://dx.doi.org/10.1038/ni1097; PMID: 15282562
- Shirai Y, Saito N. Activation mechanisms of protein kinase C: maturation, catalytic activation, and targeting. J Biochem 2002; 132:663 - 8; http://dx.doi.org/10.1093/oxfordjournals.jbchem.a003271; PMID: 12417013
- Zhou FF, Xue Y, Chen GL, Yao X. GPS: a novel group-based phosphorylation predicting and scoring method. Biochem Biophys Res Commun 2004; 325:1443 - 8; http://dx.doi.org/10.1016/j.bbrc.2004.11.001; PMID: 15555589
- Ballester R, Rosen OM. Fate of immunoprecipitable protein kinase C in GH3 cells treated with phorbol 12-myristate 13-acetate. J Biol Chem 1985; 260:15194 - 9; PMID: 3905792
- Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J 1995; 9:484 - 96; PMID: 7737456
- Coats S, Flanagan WM, Nourse J, Roberts JM. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science 1996; 272:877 - 80; http://dx.doi.org/10.1126/science.272.5263.877; PMID: 8629023
- Rivard N, L’Allemain G, Bartek J, Pouysségur J. Abrogation of p27Kip1 by cDNA antisense suppresses quiescence (G0 state) in fibroblasts. J Biol Chem 1996; 271:18337 - 41; http://dx.doi.org/10.1074/jbc.271.31.18337; PMID: 8702474
- Besson A, Gurian-West M, Chen X, Kelly-Spratt KS, Kemp CJ, Roberts JM. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev 2006; 20:47 - 64; http://dx.doi.org/10.1101/gad.1384406; PMID: 16391232
- Deng X, Mercer SE, Shah S, Ewton DZ, Friedman E. The cyclin-dependent kinase inhibitor p27Kip1 is stabilized in G(0) by Mirk/dyrk1B kinase. J Biol Chem 2004; 279:22498 - 504; http://dx.doi.org/10.1074/jbc.M400479200; PMID: 15010468
- Boehm M, Yoshimoto T, Crook MF, Nallamshetty S, True A, Nabel GJ, et al. A growth factor-dependent nuclear kinase phosphorylates p27(Kip1) and regulates cell cycle progression. EMBO J 2002; 21:3390 - 401; http://dx.doi.org/10.1093/emboj/cdf343; PMID: 12093740
- Batlle E, Verdú J, Domínguez D, del Mont Llosas M, Díaz V, Loukili N, et al. Protein kinase C-alpha activity inversely modulates invasion and growth of intestinal cells. J Biol Chem 1998; 273:15091 - 8; http://dx.doi.org/10.1074/jbc.273.24.15091; PMID: 9614119
- Abraham C, Scaglione-Sewell B, Skarosi SF, Qin W, Bissonnette M, Brasitus TA. Protein kinase C alpha modulates growth and differentiation in Caco-2 cells. Gastroenterology 1998; 114:503 - 9; http://dx.doi.org/10.1016/S0016-5085(98)70533-5; PMID: 9496940
- Kharait S, Dhir R, Lauffenburger D, Wells A. Protein kinase Cdelta signaling downstream of the EGF receptor mediates migration and invasiveness of prostate cancer cells. Biochem Biophys Res Commun 2006; 343:848 - 56; http://dx.doi.org/10.1016/j.bbrc.2006.03.044; PMID: 16564022
- Haughian JM, Reno EM, Thorne AM, Bradford AP. Protein kinase C alpha-dependent signaling mediates endometrial cancer cell growth and tumorigenesis. Int J Cancer 2009; 125:2556 - 64; http://dx.doi.org/10.1002/ijc.24633; PMID: 19672862
- Margolis EJ, Choi JC, Shu WP, Liu BC. Specific sequences of fibronectin activate the protein kinase C signal transduction pathway in invasive bladder cancer. Cancer Lett 1996; 100:163 - 8; http://dx.doi.org/10.1016/0304-3835(95)04096-X; PMID: 8620437
- Wu TT, Hsieh YH, Wu CC, Hsieh YS, Huang CY, Liu JY. Overexpression of protein kinase C alpha mRNA in human hepatocellular carcinoma: a potential marker of disease prognosis. Clin Chim Acta 2007; 382:54 - 8; http://dx.doi.org/10.1016/j.cca.2007.03.018; PMID: 17459358
- Weinstein IB. Nonmutagenic mechanisms in carcinogenesis: role of protein kinase C in signal transduction and growth control. Environ Health Perspect 1991; 93:175 - 9; http://dx.doi.org/10.1289/ehp.9193175; PMID: 1773790
- McGarrity TJ, Peiffer LP. Protein kinase C activity as a potential marker for colorectal neoplasia. Dig Dis Sci 1994; 39:458 - 63; http://dx.doi.org/10.1007/BF02088328; PMID: 8131680
- Kahl-Rainer P, Karner-Hanusch J, Weiss W, Marian B. Five of six protein kinase C isoenzymes present in normal mucosa show reduced protein levels during tumor development in the human colon. Carcinogenesis 1994; 15:779 - 82; http://dx.doi.org/10.1093/carcin/15.4.779; PMID: 8149496
- Kahl-Rainer P, Sedivy R, Marian B. Protein kinase C tissue localization in human colonic tumors suggests a role for adenoma growth control. Gastroenterology 1996; 110:1753 - 9; http://dx.doi.org/10.1053/gast.1996.v110.pm8964400; PMID: 8964400
- Brasitus TA, Bissonnette M. PKC isoforms: villains in colon cancer?. Gastroenterology 1998; 115:225 - 7; http://dx.doi.org/10.1016/S0016-5085(98)70387-7; PMID: 9649481
- Verstovsek G, Byrd A, Frey MR, Petrelli NJ, Black JD. Colonocyte differentiation is associated with increased expression and altered distribution of protein kinase C isozymes. Gastroenterology 1998; 115:75 - 85; http://dx.doi.org/10.1016/S0016-5085(98)70367-1; PMID: 9649461
- Black JD. Protein kinase C isozymes in colon carcinogenesis: guilt by omission. Gastroenterology 2001; 120:1868 - 72; http://dx.doi.org/10.1053/gast.2001.25287; PMID: 11375968
- Frey MR, Saxon ML, Zhao X, Rollins A, Evans SS, Black JD. Protein kinase C isozyme-mediated cell cycle arrest involves induction of p21(waf1/cip1) and p27(kip1) and hypophosphorylation of the retinoblastoma protein in intestinal epithelial cells. J Biol Chem 1997; 272:9424 - 35; http://dx.doi.org/10.1074/jbc.272.14.9424; PMID: 9083081
- Frey MR, Clark JA, Leontieva O, Uronis JM, Black AR, Black JD. Protein kinase C signaling mediates a program of cell cycle withdrawal in the intestinal epithelium. J Cell Biol 2000; 151:763 - 78; http://dx.doi.org/10.1083/jcb.151.4.763; PMID: 11076962
- Clark JA, Black AR, Leontieva OV, Frey MR, Pysz MA, Kunneva L, et al. Involvement of the ERK signaling cascade in protein kinase C-mediated cell cycle arrest in intestinal epithelial cells. J Biol Chem 2004; 279:9233 - 47; http://dx.doi.org/10.1074/jbc.M312268200; PMID: 14670956
- Fukumoto S, Nishizawa Y, Hosoi M, Koyama H, Yamakawa K, Ohno S, et al. Protein kinase C delta inhibits the proliferation of vascular smooth muscle cells by suppressing G1 cyclin expression. J Biol Chem 1997; 272:13816 - 22; http://dx.doi.org/10.1074/jbc.272.21.13816; PMID: 9153238
- Toyoda M, Gotoh N, Handa H, Shibuya M. Involvement of MAP kinase-independent protein kinase C signaling pathway in the EGF-induced p21(WAF1/Cip1) expression and growth inhibition of A431 cells. Biochem Biophys Res Commun 1998; 250:430 - 5; http://dx.doi.org/10.1006/bbrc.1998.9332; PMID: 9753647
- Ashton AW, Watanabe G, Albanese C, Harrington EO, Ware JA, Pestell RG. Protein kinase Cdelta inhibition of S-phase transition in capillary endothelial cells involves the cyclin-dependent kinase inhibitor p27(Kip1). J Biol Chem 1999; 274:20805 - 11; http://dx.doi.org/10.1074/jbc.274.30.20805; PMID: 10409620
- Shanmugam M, Krett NL, Maizels ET, Murad FM, Rosen ST, Hunzicker-Dunn M. A role for protein kinase C delta in the differential sensitivity of MCF-7 and MDA-MB 231 human breast cancer cells to phorbol ester-induced growth arrest and p21(WAFI/CIP1) induction. Cancer Lett 2001; 172:43 - 53; http://dx.doi.org/10.1016/S0304-3835(01)00602-4; PMID: 11595128
- Page K, Li J, Corbit KC, Rumilla KM, Soh JW, Weinstein IB, et al. Regulation of airway smooth muscle cyclin D1 transcription by protein kinase C-delta. Am J Respir Cell Mol Biol 2002; 27:204 - 13; PMID: 12151312
- Nakagawa M, Oliva JL, Kothapalli D, Fournier A, Assoian RK, Kazanietz MG. Phorbol ester-induced G1 phase arrest selectively mediated by protein kinase Cdelta-dependent induction of p21. J Biol Chem 2005; 280:33926 - 34; http://dx.doi.org/10.1074/jbc.M505748200; PMID: 16055435
- Pysz MA, Leontieva OV, Bateman NW, Uronis JM, Curry KJ, Threadgill DW, et al. PKCalpha tumor suppression in the intestine is associated with transcriptional and translational inhibition of cyclin D1. Exp Cell Res 2009; 315:1415 - 28; http://dx.doi.org/10.1016/j.yexcr.2009.02.002; PMID: 19232344
- Afrasiabi E, Ahlgren J, Bergelin N, Törnquist K. Phorbol 12-myristate 13-acetate inhibits FRO anaplastic human thyroid cancer cell proliferation by inducing cell cycle arrest in G1/S phase: evidence for an effect mediated by PKCdelta. Mol Cell Endocrinol 2008; 292:26 - 35; http://dx.doi.org/10.1016/j.mce.2008.04.018; PMID: 18541361
- Baldassarre G, Barone MV, Belletti B, Sandomenico C, Bruni P, Spiezia S, et al. Key role of the cyclin-dependent kinase inhibitor p27kip1 for embryonal carcinoma cell survival and differentiation. Oncogene 1999; 18:6241 - 51; http://dx.doi.org/10.1038/sj.onc.1203031; PMID: 10597222