Abstract
Histone deacetylases (HDACs) control fundamental physiological processes such as proliferation and differentiation. HDAC inhibitors (HDACi) induce cell cycle arrest and apoptosis of tumor cells. Therefore, they represent promising cancer therapeutics that appear particularly useful in combination therapies. Although HDACi are tested in current clinical trials, the molecular mechanisms modulating the cellular responses toward HDACi are incompletely understood. To gain insight into pathways that limit HDACi efficacy in gastric cancer, we treated a panel of gastric cancer cells with the clinically relevant HDACi suberoylanilide hydroxamic acid (SAHA). We report that higher expression levels of the anti-apoptotic BCL2 family members MCL1 and BCLXL were detectable in cells with high inhibitory concentration 50 (IC50) values for SAHA. Using RNAi, we show that MCL1 and BCLXL lower the efficacy of SAHA. To find strategies to interfere with MCL1 and BCLXL expression, we investigated molecular regulation of both proteins. We show that specific siRNAs against c-MYC as well as pharmacological inhibition of this cancer-relevant transcription factor reduced MCL1 and BCLXL expression. Subsequently, we observed an increase in SAHA efficacy. Our data furthermore demonstrate that two different molecular mechanisms are responsible for the modulation of these factors. Whereas c-MYC controls transcription of MCL1 directly, regulation of BCLXL was due to c-MYC’s capability to regulate the eIF4E gene, which encodes a rate-limiting factor of eukaryotic translation. Our data reveal a new molecular mechanism for how c-MYC controls cell autonomous apoptosis and provide a rationale for a concerted inhibition of HDACs and c-MYC in gastric cancer.
Introduction
Histone deacetylases (HDACs) are divided into four distinct classes according to phylogenetic analysis, sequence homology and function. The Zn2+-dependent enzymes are grouped into class I (HDAC1, 2, 3 and 8), class II (HDAC4, 5, 6, 7, 9 and 10) and class IV (HDAC11) enzymes, while the NAD+-dependent sirtuins (SIRT 1–7) represent class III.Citation1 HDACs are involved in the control of proliferation, apoptosis, differentiation, migration and angiogenesis of cancer cells.Citation2 Thus, they are promising therapeutic targets.Citation3-Citation5 Furthermore, a recent study demonstrates the sensitivity of tumor-initiating cells toward HDAC inhibitors (HDACi), pointing to a good therapeutic potential in solid tumors.Citation6 The pan-HDACi suberoylanilide hydroxamic acid (SAHA; Vorinostat) and the cyclic peptide depsipeptide (Romidepsin; FK-228), which preferably inhibits class I HDACs, have been approved by the FDA for the treatment of cutaneous T-cell lymphoma (CTCL).Citation7 Although HDACi are currently being tested in diverse clinical trials for solid cancers and leukemias, mechanisms controlling the efficacy of HDACi and predictive markers for these agents are still not fully identified.
Class I HDACs are often overexpressed in solid tumors, and high expression of these enzymes is also found in gastric cancer.Citation8-Citation12 Of note, the concomitant nuclear expression of HDAC 1, 2 and 3 correlates strongly with reduced 3-y survival rates.Citation11 Furthermore, high HDAC1 expression is a predictor of reduced survival in a group of gastric cancer patients responding to a 5-Fluorouracil (5-FU)/platinum-based neoadjuvant chemotherapy.Citation12 It has been postulated that the levels of class I HDACs predicts the responsiveness of gastric cancer toward HDACi.Citation11 However, recent observations suggest that neither HDAC1 nor HDAC2 expression are convincing markers for HDACi sensitivity of gastric cancer cell lines.Citation12
Results
BCLXL and MCL1 expression levels inversely correlate with the responsiveness of gastric cancer cells toward HDACi.
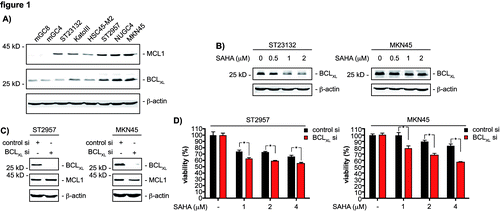
To elucidate mechanisms controlling the sensitivity of gastric cancer cells exposed to HDACi, we used a panel of murine (mGC4 and mGC8) and human (ST23132, KatoIII, HSC45-M2, ST2957, NUGC4 and MKN45) gastric cancer cell lines. These were treated with increasing concentrations of SAHA, and IC50 values were determined for each cell line ( and Fig. S1). SAHA IC50 values ranged between 1 ∝M and 3.2 ∝M. This finding is consistent with recent observations.Citation12,Citation13 One important response of tumor cells toward current cancer therapies is the induction of programmed cell death, which is regulated by the fine-tuned interplay of BCL2 family members.Citation14 In this family, pro-survival factors, such as BCL-2, BCLXL or MCL1, counteract the action of death inducers, which are the multidomain pro-death proteins BAX and BAK or the BH3-only proteins, such as BIM, PUMA, NOXA or BID.Citation14 As shown in , BCLXL and MCL1 protein levels correlated with SAHA IC50 values. This finding provided us a first hint that both proteins modulate the response of gastric cancer cells to HDACi.
Table 1. Response of gastric cancer cell lines to SAHA
Figure 1. BCLXL restricts SAHA efficacy of gastric cancer cells. (A) western blot analysis of MCL1 and BCLXL expression of the indicated gastric cancer cell lines. β-actin served as loading control. (B) ST23132 and MKN45 cells were treated with increasing doses SAHA for 24 h as indicated or were left as vehicle treated controls. Western blots detected expression of BCLXL and β-actin (loading control). (C) ST2957 and MKN45 cells were transfected with the indicated siRNAs. 48 h after the transfection whole-cell lysates were prepared and western blots detected expression of BCLXL, MCL1 and β-actin (loading control). (D) ST2957 and MKN45 were transfected with the indicated siRNAs. 48 h after the transfection cells were treated with SAHA as indicated for additional 24 h. Viability of cells was measured in MTT assays and viability vehicle treated controls were arbitrary set to 100% in order to compare the SAHA responses within each siRNA condition. Data are presented as mean and standard error of the mean (S.E.M). (Student’s t-test: * p < 0.05 vs. controls).
BCLXL and MCL1 counteract the efficacy of HDACi.
To investigate regulation of BCLXL by HDACi, we performed western blot analysis. As shown in , SAHA treatment downregulated BCLXL protein expression in the SAHA-sensitive ST23132 cells (IC50 1.4 ∝M). No regulation of BCLXL was detected in the more resistant MKN45 cells (IC50 3.2 ∝M), when SAHA concentrations below the IC50 value were used (). SAHA concentrations over the IC50 value decreased BCLXL expression also in the more resistant MKN45 and ST2957 (IC50 2.8 ∝M) cells (). We used RNAi to analyze the contribution of BCLXL for cellular responses toward SAHA. Knockdown of this protein in ST2957 cells and MKN45 cells was specific and did not influence MCL1 expression (). In both cell lines, a reduction of BCLXL decreased the viability of both cell lines upon treatment with SAHA (). Thus, BCLXL limits the pro-apoptotic potency of HDACi against gastric cancer cells.
Figure 2. MCL1 restricts SAHA efficacy of gastric cancer cells. (A) ST23132 and MKN45 cells were treated for 24 h with increasing doses SAHA as indicated or left as vehicle treated controls. Western blots detected expression of MCL1 and β-actin (loading control). (B) ST2957 and MKN45 cells were transfected with the indicated siRNAs. Forty-eight hours after transfection whole-cell lysates were prepared and western blots detected expression of MCL1, BCLXL and β-actin (loading control). (C) ST2957 and MKN45 cells were transfected with the indicated siRNAs. 48 h after the transfection cells were treated with SAHA as indicated for additional 24 h. Viability of cells was measured in MTT assays and viability of vehicle treated controls were arbitrary set to 100% in order to compare the SAHA responses within each siRNA condition. Data are presented as mean and standard error of the mean (S.E.M). (D) ST2957 and MKN45 cells were transfected with the indicated siRNAs. For MCL1, the MCL1–1 siRNA was used. siRNA amount was kept constant using control siRNA. Forty-eight hours after transfection cells were treated with SAHA for additional 24 h as indicated or were left as untreated controls. Western blots detected expression of MCL1, BCLXL and β-actin (loading control). (E) ST2957 and MKN45 cells were transfected with the indicated siRNAs. For MCL1, the MCL1–1 siRNA was used. siRNA amount was kept constant using control siRNA. Forty-eight hours after transfection cells were treated with SAHA for additional 24 h as indicated or were left as untreated controls. Cells were stained with Propidium iodide (PI) and FITC-labeled anti-Annexin V. Depicted is the Annexin V positive fraction (early apoptosis = Annexin V+/PI- and late apoptosis = Annexin V+/PI+). (Student’s t-test: * p < 0.05 vs. controls).
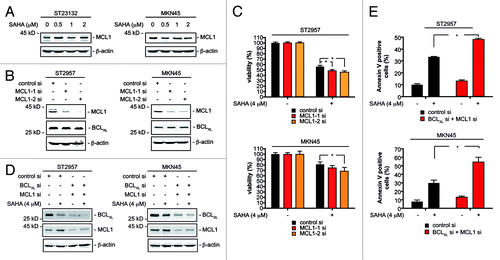
HDACi have been reported to induce apoptosis of leukemic cells without decreasing MCL1 protein expression.Citation15 We observed that SAHA treatment neither altered MCL1 expression in ST23132 nor in MKN45 gastric cancer cells (). Also, SAHA concentration over the IC50 value failed to downregulate MCL1 protein expression (). Nevertheless, attenuation of MCL1 levels was reported to enhance HDACi-induced cytotoxicity in leukemic cells.Citation15 This notion prompted us to test how MCL1 affects the HDACi responses of our cellular systems. Knockdown of MCL1 with RNAi in ST2957 and MKN45 cells was specific and did not influence BCLXL expression (). The reduction of MCL1 protein expression slightly increased the SAHA-induced therapeutic response of ST2957 and MKN45 cells (). The fact that this observation reached statistical significance argues that MCL1 counteracts HDACi-induced death programs in gastric cancer cells. To corroborate our notion that BCLXL and MCL1 limit SAHA efficacy, we simultaneously depleted both BCL2 family members in ST2957 and MKN45 cells (). Simultaneous knockdown of BCLXL and MCL1 distinctly increased SAHA-induced apoptosis compared with the knockdown of each anti-apoptotic protein alone ().
MYC controls BCLXL, MCL1 and the HDACi response.
Having found that MCL1 and BCLXL counteract the HDACi response, we deciphered pathways that control their expression in gastric cancer cells. After excluding NFκB and STAT3 as main MCL1 and BCLXL regulators in ST2957 and MKN45 gastric cancer cells (Fig. S2), we investigated c-MYC, since gene expression patterns regulated by c-MYC are associated with therapeutic resistance.Citation16-Citation19 Knockdown of c-MYC distinctly decreased MCL1 protein and mRNA levels in ST2957 and MKN45 gastric cancer cells (). While BCLXL protein expression was distinctly reduced in c-MYC siRNA transfected cells (), BCLXL mRNA expression was not altered significantly (). These observations argue for a non-genomic function of c-MYC on BCLXL.
Figure 3. c-MYC controls MCL1 and BCLXL expression of gastric cancer cells. (A) ST2957 and MKN45 cells were transfected with the indicated siRNAs. 48 h after the transfection whole-cell lysates were prepared and western blots detected expression of c-MYC, BCLXL, MCL1 and β-actin (loading control). (B) ST2957 and MKN45 cells were transfected with the indicated siRNAs. 48 h after the expression of c-MYC, MCL1 and BCLXL mRNAs were determined by quantitative RT-PCR using cyclophilinA mRNA as reference. (C) ST2957 and MKN45 cells were transfected with the indicated siRNAs. 48 h after the transfection cells were treated with SAHA as indicated for additional 24 h. Viability of cells was measured in MTT assays and viability of vehicle treated controls were arbitrary set to 100% in order to compare the SAHA responses within each siRNA condition. Data are presented as mean and standard error of the mean (S.E.M). (Student’s t-test: * p < 0.05 vs. controls).
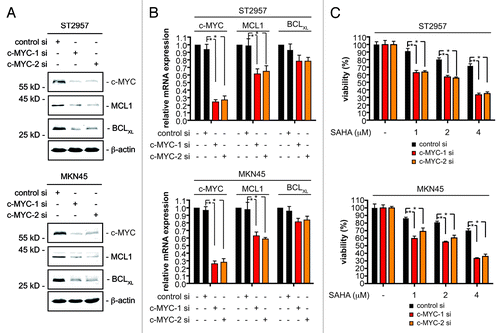
Knockdown of c-MYC in ST2957 and MKN45 cells significantly reduces viability of cells (Fig. S3), confirming a role for c-MYC in tumor maintenance of gastric cancer cells.Citation20,Citation21 Remarkably, the cellular responses toward SAHA in the rather HDACi-resistant cells (ST2957 and MKN45) was markedly increased upon attenuation of c-MYC (). Hence, this factor appears to counteract the effects of HDACi on gastric cancer cells. To validate the RNAi results, we treated ST2957 and MKN45 cells with the c-MYC inhibitor (myc-I) 10058-F4, which interferes with c-MYC/MAX heterodimerization.Citation22 Dose-dependent downregulation of MCL1 and BCLXL was observed in both cell lines (). Myc-I as well as SAHA treatment induced apoptosis of ST2957 and MKN45 cells, which was further increased by the myc-I and SAHA co-treatment ().
Figure 4. MCL1 is a direct c-MYC target in gastric cancer cells. (A) ST2957 and MKN45 cells were treated with the MYC inhibitor (myc-I) for 24 h. Western blots detected the expression of BCLXL, MCL1 and β-actin (loading control). (B) ST2957 and MKN45 cells were treated with the MYC inhibitor (myc-I), SAHA or the combination of both for 24 h as indicated or were left as an untreated control. Cells were stained with Propidium iodide (PI) and FITC-labeled anti-Annexin V. Depicted is the Annexin V positive fraction (early apoptosis = Annexin V+/PI- and late apoptosis = Annexin V+/PI+). (Student’s t-test: * p < 0.05). (C) and (D) ST2957 and MKN45 cells were treated with the MYC inhibitor for 24 h or were left as an vehicle treated control. ChIP analysis revealing the binding of c-MYC (C) or the RNA Polymerase II (D) to the E-box of the MCL1 promoter or an 13 kb 3′ control.
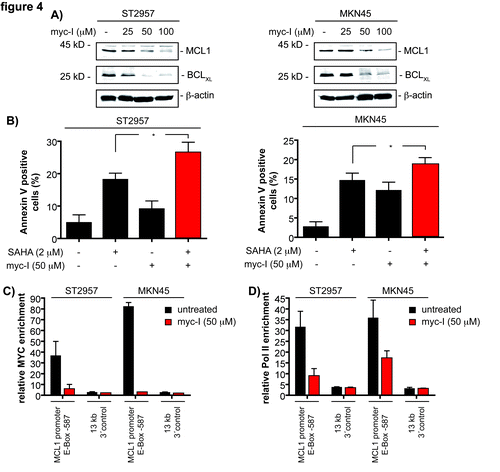
To test the direct binding of c-MYC to the MCL1 promoter, we used quantitative chromatin immunoprecipitation (qChIP). In MKN45 as well as in ST2957 cells, c-MYC was recruited to the E-box in the promoter of the MCL1 gene (). As expected, binding of c-MYC was reduced after treatment of both cell lines with the myc-I (). Furthermore, c-MYC binding correlated with the binding of RNA polymerase II to the MCL1 promoter, which was reduced by the myc-I treatment in both cell lines (). Together, the ChIP data determine the MCL1 gene as a c-MYC target gene in gastric cancer cells.
c-MYC-induced transcription of eIF4E regulates BCLXL expression in gastric cancer cells.
c-MYC can regulate protein translation, e.g., via control of genes coding for translation initiation factors, such as eIF4E.Citation23-Citation25 Accordingly, we observed downregulation of eIF4E expression in c-MYC siRNA-transfected ST2957 and MKN45 cells at the protein () and mRNA levels (). Furthermore, reduced eIF4E expression was also detected in myc-I treated gastric cancer cells (). To prove direct transcriptional regulation of eIF4E by c-MYC, we again used qChIP. Analogously to the MCL1 promoter, we detected c-MYC recruitment to the E-box in the proximal eIF4E promoter. As expected, recruitment of this transcription factor was impaired after myc-I treatment (). Moreover, reduced binding of c-MYC to the eIF4E promoter was accompanied by a decline in RNA polymerase II recruitment to this promoter in ST2957 and MKN45 cells (). These data show transcriptional control of eIF4E by c-MYC in gastric cancer cells.
Figure 5. MYC controls eIF4E transcription to regulate BCLXL expression in gastric cancer cells. (A) ST2957 and MKN45 cells were transfected with the indicated siRNAs. 48 h after the transfection whole-cell lysates were prepared and western blots detected expression of c-MYC, eIF4E and β-actin (loading control). (B) ST2957 and MKN45 cells were transfected with the indicated siRNAs. 48 h after the expression of the eIF4E mRNA were determined by quantitative RT-PCR using cyclophilinA mRNA as reference. (C) ST2957 and MKN45 cells were treated with the MYC inhibitor for 24 h. Western blots detected the expression of eIF4E and β-actin (loading control). (D) and (E) ST2957 and MKN45 cells were treated with the MYC inhibitor for 24 h or were left as an vehicle treated control. ChIP analysis revealing the binding of c-MYC (D) or the RNA Polymerase II (E) to the E-box of the eIF4E promoter or an cyclophilin A 3′ control. (F) ST2957 and MKN45 cells were transfected with the indicated siRNAs. 48 h after the transfection whole-cell lysates were prepared and western blots detected expression of eIF4E, BCLXL and β-actin (loading control).
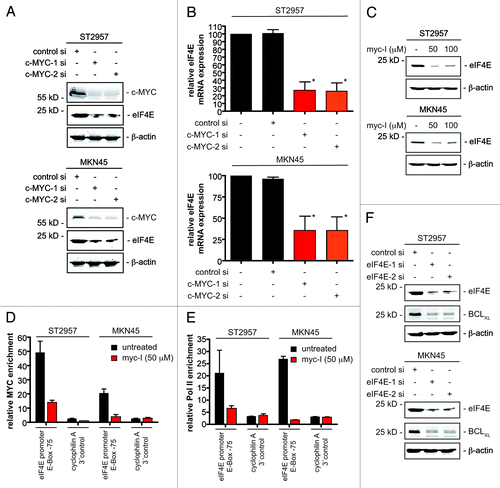
To corroborate regulation of BCLXL expression by eIF4E, we used two specific eIF4E siRNAs. As shown in , transfection of ST2957 and MKN45 cells with such siRNAs indeed blocked BCLXL expression. From these findings, we conclude that a c-MYC-eIF4E axis controls BCLXL translation in gastric cancer cells.
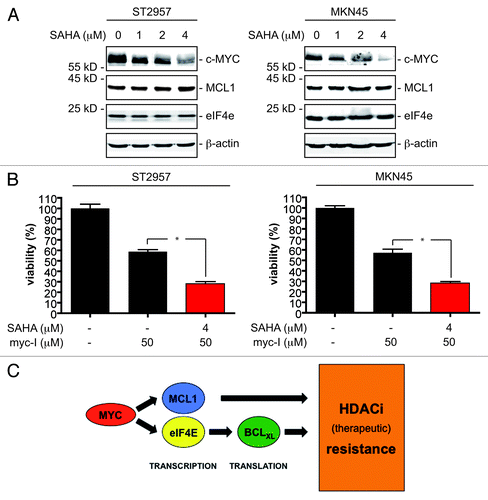
Since expression of c-MYC is downregulated by HDACi in various cell types,Citation26-Citation28 we tested c-MYC expression upon SAHA treatment. Interestingly, although the c-MYC protein expression was downregulated in ST2957 and MKN45 cells upon SAHA treatment, we observed no regulation of the herein described c-MYC target genes MCL1 and eIF4E (), which may argue that both genes are actively repressed upon inhibition of c-MYC by a HDAC-dependent repressor complex. This observation might also explain the rather small effect of the simultaneous treatment of both cell lines with the myc-I and SAHA toward apoptosis induction () compared with the distinct increase in the therapeutic efficacy of SAHA after c-MYC depletion via RNAi (). Consequently, we concluded that drug scheduling composed out of a 24 h myc-I pretreatment, to assure downregulation of MCL1 and eIF4E, followed by a combination of myc-I and SAHA might be more potent. Indeed, we observed a distinct increase of the therapeutic SAHA efficacy in ST2957 and MKN45 cells ().
Figure 6. MYC expression is regulated by HDACi in gastric cancer cells. (A) ST2957 and MKN45 cells were treated with increasing doses SAHA for 24 h as indicated or were left as vehicle treated controls. Western blots detected expression of c-MYC, MCL1, eIF4E and β-actin (loading control). (B) ST2957 and MKN45 cells were pre-treated with myc-I as indicated or were left as an untreated control. After 24 h cells were again treated with myc-I or with the combination of the myc-I and SAHA for additional 24 h. Untreated cells remained untreated. Viability of cells was measured in MTT assays and viability of vehicle treated controls were arbitrary set to 100%. Data are presented as mean and standard error of the mean (S.E.M). (Student’s t-test: * p < 0.05 vs. controls). (C) Schematic presentation of how gene expression signatures induced by c-MYC restrain the efficacy of HDACi against gastric cancer cells.
Discussion
Most patients with gastric cancer are diagnosed with an advanced tumor stage, in which systemic chemotherapy remains the key treatment option. Compared with best supportive care, chemotherapy in advanced gastric cancer improves median survival.Citation29 Nonetheless, chemotherapy remains palliative, and 5-y survival rates are below 10%. HDACi are currently tested in advanced gastric cancer (www.clinicaltrials.gov), but molecular mechanisms and markers relevant for the efficacy of HDACi in gastric cancer cells are largely unknown. Here we provide evidence that c-MYC counteracts HDACi efficacy in gastric cancer cells by regulating the expression of the anti-apoptotic BCL2 family members BCLXL and MCL1. Our data illustrate two independent mechanisms by which c-MYC protects gastric cancer cells from HDACi. First, c-MYC directly controls transcription of MCL1 and, second, regulation of BCLXL protein expression was due to c-MYC’s capability to control the eIF4E gene and thereby translation of the BCLXL mRNA ().
Apoptosis is one major cellular response of cancer cells treated with HDACi and was also observed in SAHA-treated gastric cancer cells.Citation30 Induction of this type of cell death predicts a beneficial therapeutic success of these agents.Citation31 Consistently, we observed that the pro-survival BCL2 family members BCLXL and MCL1 limit the efficacy of these compounds in a cell-based model of gastric cancer. Furthermore, our results are in line with in vivo data demonstrating that pro-survival BCL2 family members limit the efficacy of HDACi.Citation31 High MCL1 expression has been detected in about 70% of patients with gastric cancer, and expression of MCL1 is an independent prognostic factor.Citation32 Furthermore, lowering MCL1 expression by an antisense oligonucleotide in NCI-N87 gastric cancer cells increased sensitivity toward docetaxel and cisplatin.Citation33 MCL1 hence appears relevant for gastric tumorigenesis and appears as a valid chemotherapeutic marker protein. In addition to MCL1, BCLXL is overexpressed in gastric cancers, and depletion of BCLXL leads to an increased sensitivity of such cells toward extrinsic apoptosis.Citation34-Citation36 Therefore, our observation that c-MYC controls expression of important anti-apoptotic BCL2 family members might argue that c-MYC is a common mediator of therapeutic resistance of gastric cancer cells. This conclusion is supported by recent microarray data, which point to a role of c-MYC in primary and secondary resistance of gastric cancer patients toward conventional chemotherapy.Citation37,Citation38
The oncogene and basic helix-loop-helix leucine-zipper transcription factor c-MYC heterodimerizes with MAX. These bind to cis-acting elements, the so called E-boxes (CAC GTG) of cancer-promoting genes.Citation39 Inhibition of c-MYC has recently been demonstrated to be well tolerated in vivo, and inhibition of c-MYC eradicated tumors in a murine Kras-dependent lung cancer model.Citation40 Therefore, several approaches to inhibit c-MYC are currently under development.Citation41 c-MYC overexpression has frequently been observed in gastric cancer and is associated with therapeutic resistance of certain tumor entities.Citation16-Citation19,Citation42 A high-affinity c-MYC binding site in the MCL1 promoter has also been found in U937 histiocytoma cells.Citation43 Consistent with the downregulation of MCL1 protein and mRNA after the knockdown of c-MYC, we detected binding of c-MYC to the MCL1 promoter in gastric cancer cell lines, demonstrating direct transcriptional regulation.
In addition to c-MYC’s direct transcriptional mode of action, this factor can regulate protein expression at the level of translation. c-MYC can control genes coding for translation initiation factors, such as eIF4E.Citation23-Citation25 Dysregulated translational control can contribute to diverse human diseases, including cancer.Citation44 Translational initiation is regulated by the eIF4F complex, which is composed of the RNA helicase eIF4A, the scaffolding protein eIF4G and the cap-binding protein eIF4E, which is believed to be the rate-limiting protein in the eIF4F complex.Citation44,Citation45 eIF4E harbors transforming characteristics in vivo and in vitro and was found to be overexpressed in various tumors, contributing to proliferation, survival, angiogenesis and metastasis of cancer cells.Citation44-Citation46 Also in gastric cancer patients, high expression of eIF4E was correlated with vascular invasion and a worse outcome for patients.Citation47 Our data demonstrate that c-MYC controls the transcription of eIF4E in gastric cancer cells by direct binding to its proximal high affinity E-box. Since eIF4E knockdown results in a distinct downregulation of BCLXL, our data argue that BCLXL is regulated at the translational level in gastric cancer cells. These observations are consistent with observations from breast cancer models in which the knockdown of eIF4E reduced BCLXL expression.Citation48 The observation that SAHA doses above the IC50 value regulate BCLXL expression in the cellular models investigated, without changing eIF4E expression, argues that HDACi control BCLXL beyond the c-MYC-eIF4E axis.
The c-MYC antagonist of the MAD family of transcription factors are alternative binding partners of MAX, primarily acting as repressors.Citation49 For instance, MAD/MAX heterodimers repress transcription by the recruitment of an mSin3/HDAC containing co-repressor complex.Citation50 We detected downregulation of the c-MYC protein expression in the investigated gastric cancer cells upon HDACi treatment. In contrast, the c-MYC target genes MCL1 and eIF4E were not altered by HDACi treatment, which might argue for the involvement of an HDAC containing co-repressor complex. Such observations are important, since they argue for a specific drug regimes to assure optimal therapeutic efficacy. Consistently, inhibition of c-MYC before HDACi treatment revealed an increased efficacy. The observation that HDACi synergize with DNA damaging agents only in case of simultaneous applicationCitation51 argues that each HDACi combination therapy posses its own application schedule. Although attractive, these possibilities clearly need more experimental evaluations.
In contrast to our data obtained with HDACi in gastric cancer cells, there is evidence that c-MYC can promote the efficacy of diverse chemotherapeutics and γ-irradiation in other tumor entities.Citation52 While the molecular determinants that define the pro-therapeutic or therapeutic-resistance role of c-MYC are still unclear, these data clearly argue for the need to determine the molecular actions of novel therapeutic strategies in pre-clinical settings, especially considering that even novel targeted therapies are used in combinations with conventional therapeutics.
Here we have demonstrated that the anti-apoptotic BCL2 family members MCL1 and BCLXL limit the biological effects HDACi exert against gastric cancer cells. Since MCL1 and BCLXL are regulated by c-MYC in gastric cancer cells, c-MYC inhibition applied in combination with HDACi might be a rationally based therapeutic option for this type of cancer. Since c-MYC is included in a three gene predictor indicating poor prognosis for cisplatin/5-FU-treated gastric cancer patients and a c-MYC-controlled gene set is enriched in gastric cancer patients with acquired resistance toward cisplatin/5-FU treatment, c-MYC might limit the therapeutic response of gastric cancers beyond HDACi.Citation37,Citation38
Material and Methods
Cell culture, reagents, siRNAs, transfection, viability assay and annexin V FACS analysis.
Suberoylanilide hydroxamic acid (SAHA; Vorinostat) was purchased from LC Laboratories, and the MYC inhibitor (myc-I, 10058-F4; (Z,E)-5-(4-Ethylbenzylidine)-2-thioxothiazolidin-4-one) was purchased from EMD Biosciences. Cell lines were cultivated in DMEM or RPMI medium supplemented with 10% fetal calf serum and 1% (w/v) penicillin/streptomycin (Invitrogen) as recently described in references Citation53–Citation55. KatoIII were purchased from ATCC and MKN45 cells from DSMZ. The murine MGC4 and MGC8 cells were a kind gift from Dr. W. Zimmermann, HSC45-M2 cells a kind gift from Dr. Senekowitsch-Schmidtke and ST23123, ST2957 and NUGC4 cells were a kind gift from Dr. R. Mejías-Luque.Citation56 Double-stranded siRNAs were transfected with polyethylenimine (Sigma-Aldrich) at a final concentration of 50 nM as recently described in reference Citation54. siRNAs were purchased from Eurofins. Target sequences of the siRNAs used were: control siRNA 5'-CAG TCG CGT TTG CGA CTG G-3'; MCL1-1 si 5'-GAA ACG CGG TAA TCG GAC T 3'; MCL1–2 si 5'-CGC CGA ATT CAT TAA TTT A-3'; BCLXL si 5'-GGA GAT GCA GGT ATT GGT G-3'; RelA si 5'-GAT CAA TGG CTA CAC AGG A-3'; STAT3 5'-CAT CTG CCT AGA TCG GCT A-3'; c-MYC-1 si 5'-CTT CTA CCA GCA GCA GCA G-3'; c-MYC-2 si 5'-GAA CAC ACA ACG TCT TGG A-3'; eIF4E-1 5'-si GGA CGA TGG CTA ATT ACA T-3'; eIF4E-2 si 5'-GGA TGG TAT TGT GCC TAT G-3'. Viability of the cells was measured using MTT-assays performed according to the manufacturer’s protocol (Roche Applied Science) and described recently in reference Citation53 and Citation54. To determine Annexin V, cells were stained with Propidium iodide (PI) and FITC-labeled anti-Annexin V using the FITC Annexin V Apoptosis Detection Kit I (BD Biosciences) and analyzed by FACS.Citation54
Total cell lysates and western blot.
Whole-cell lysates were prepared and western blots were performed as recently described in reference Citation53. The following antibodies were used: anti-MCL1 (Santa Cruz Biotechnology, sc-819), anti-BCLXL (Cell signaling technology, #2762S), STAT3 (Cell signaling technology, #9132), RelA (Santa Cruz Biotechnology, sc-372), c-MYC (Cell signaling technology, #9402S), eIF4E (BD Biosciences, #610269) and β-actin (Sigma-Aldrich). Western blots were performed using Odyssey Infrared Imaging System (LI-COR Biosciences) as described in reference Citation53.
Quantitative reverse-transcriptase PCR.
Total RNA was isolated from pancreatic carcinoma cell lines using the RNeasy kit (Qiagen) following the manufacturer’s instructions. Quantitative mRNA analyses were performed as previously described using real-time PCR analysis (TaqMan, PE Applied Biosystems).Citation53 The following primers were used: cyclophilinA-fw 5'-ATG GTC AAC CCC ACC GTG T-3'; cyclophilinA-rv 5'-TCT GCT GTC TTT GGG ACC TTG TC-3'; c-MYC-fw 5'-AGC GAC TCT GAG GAG GAA CA-3'; c-MYC-rv 5'-CTC TGA CCT TTT GCC AGG AG 3'; MCL1-fw 5'-GCA TCG AAC CAT TAG CAG AAA G-3'; MCL1-rv 5'-AAA GCC AGC AGC ACA TTC-3'; BCLXL-fw 5'-CCA CTT ACC TGA ATG ACC ACC TAG A-3'; BCLXL-rv 5'-GCT GCA TTG TTC CCA TAG AGT TC-3'; eIF4E-fw 5'-ACA AGT CAG TCT GAA ACC ATC GAA C-3'; eIF4E-rv 5'-CTT CAT CCT CTT CGG CCA CTC CTC C-3'.
Quantitative chromatin immunoprecipitation.
Chromatin Immunoprecipitations were performed using magnetic SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling technology) according to the manufacturer’s protocol with the following antibodies: c-MYC (Cell signaling technology, #9402S), RNA Polymerase II (Santa Cruz Biotechnology, N-20) and rabbit IgG (Cell signaling technology) as control. c-MYC and RNA Polymerase II binding was determined by quantitative PCR.57 ΔCt values for each sample were calculated using ΔCt = [Ct (sam ple) - Ct (input)]. ΔΔCt was calculated using ΔΔCt = [ΔCt (experimental sample) -ΔCt (negative control)]. Shown is the fold difference between the experimental sample and the negative control (IgG), which was determined as 2-ΔΔCt. The following primers with similar amplification efficiencies were used: MCL1–587-fw 5'-TTA GGG TAG CAC GTG GAG CA-3'; MCL1–587-rv 5'-ACG GGG AAG GTT CAG TGA TGG-3'; eIF4E-75-fw 5'-TAC TCA CGC AGC CGC AGT C-3'; eIF4E-75-rv 5'-TCG CAC AAC CGC TCC AG-3'; MCL1–13 kb 3'-fw 5'-GCT GTG CTG AGA GGC CTG GG-3'; MCL1–13 kb 3'-rv AGC ACA CAA ACA TGC CGA CCC-3'; cyclophilin A 3' control-fw 5'-GCG ACC TTG AGG CCT GCG TT-3'; cyclophilin A 3'-control-rv 5'-CGG CTC TTC GGC CGT TGT CA-3'.
Statistical methods.
A two-tailed Student t-test was used to test statistical significance. p-values are indicated, and * denotes a p-value of at least < 0.05. Inhibitor concentration 50 (IC50) values were calculated with GraphPad Prism4 software using a nonlinear regression model with classic equations as described in reference Citation53.
Additional material
Download Zip (570.7 KB)Acknowledgements
We thank Dr. R. Mejías-Luque, Dr. W. Zimmermann and Dr. R. Senekowitsch-Schmidtke for providing cell lines. The work was supported by KAAD, Bundesministerium für Bildung und Forschung, MoBiMed Programm (01 EZ 0802), Deutsche Krebshilfe (Grant 109265) and Deutsche Forschungsgemeinschaft (Grant SCHN 959/1–2).
Disclosure of potential Conflicts of interest
No potential conflicts of interest were disclosed.
Note
Supplemental material can be found at: www.landesbioscience.com/journals/cc/article/20008
References
- Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol 2008; 9:206 - 18; http://dx.doi.org/10.1038/nrm2346; PMID: 18292778
- Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene 2007; 26:5420 - 32; http://dx.doi.org/10.1038/sj.onc.1210610; PMID: 17694083
- Müller S, Krämer OH. Inhibitors of HDACs--effective drugs against cancer?. Curr Cancer Drug Targets 2010; 10:210 - 28; http://dx.doi.org/10.2174/156800910791054149; PMID: 20201785
- Marks PA. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin Investig Drugs 2010; 19:1049 - 66; http://dx.doi.org/10.1517/13543784.2010.510514; PMID: 20687783
- Thurn KT, Thomas S, Moore A, Münster PN. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future Oncol 2011; 7:263 - 83; http://dx.doi.org/10.2217/fon.11.2; PMID: 21345145
- Sikandar S, Dizon D, Shen X, Li Z, Besterman J, Lipkin SM. The class I HDAC inhibitor MGCD0103 induces cell cycle arrest and apoptosis in colon cancer initiating cells by upregulating Dickkopf-1 and non-canonical Wnt signaling. Oncotarget 2010; 1:596 - 605; PMID: 21317455
- Wagner JM, Hackanson B, Lübbert M, Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics 2010; 1:117 - 36; http://dx.doi.org/10.1007/s13148-010-0012-4; PMID: 21258646
- Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett 2009; 280:168 - 76; http://dx.doi.org/10.1016/j.canlet.2008.10.047; PMID: 19103471
- Choi JH, Kwon HJ, Yoon BI, Kim JH, Han SU, Joo HJ, et al. Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn J Cancer Res 2001; 92:1300 - 4; http://dx.doi.org/10.1111/j.1349-7006.2001.tb02153.x; PMID: 11749695
- Song J, Noh JH, Lee JH, Eun JW, Ahn YM, Kim SY, et al. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS 2005; 113:264 - 8; http://dx.doi.org/10.1111/j.1600-0463.2005.apm_04.x; PMID: 15865607
- Weichert W, Röske A, Gekeler V, Beckers T, Ebert MP, Pross M, et al. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: a retrospective analysis. Lancet Oncol 2008; 9:139 - 48; http://dx.doi.org/10.1016/S1470-2045(08)70004-4; PMID: 18207460
- Mutze K, Langer R, Becker K, Ott K, Novotny A, Luber B, et al. Histone deacetylase (HDAC) 1 and 2 expression and chemotherapy in gastric cancer. Ann Surg Oncol 2010; 17:3336 - 43; http://dx.doi.org/10.1245/s10434-010-1182-1; PMID: 20585871
- Huang C, Ida H, Ito K, Zhang H, Ito Y. Contribution of reactivated RUNX3 to inhibition of gastric cancer cell growth following suberoylanilide hydroxamic acid (vorinostat) treatment. Biochem Pharmacol 2007; 73:990 - 1000; http://dx.doi.org/10.1016/j.bcp.2006.12.013; PMID: 17276407
- Kelly PN, Strasser A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ 2011; 18:1414 - 24; http://dx.doi.org/10.1038/cdd.2011.17; PMID: 21415859
- Inoue S, Walewska R, Dyer MJ, Cohen GM. Downregulation of Mcl-1 potentiates HDACi-mediated apoptosis in leukemic cells. Leukemia 2008; 22:819 - 25; http://dx.doi.org/10.1038/leu.2008.1; PMID: 18239621
- Sklar MD, Prochownik EV. Modulation of cis-platinum resistance in Friend erythroleukemia cells by c-myc. Cancer Res 1991; 51:2118 - 23; PMID: 2009531
- Mizutani Y, Fukumoto M, Bonavida B, Yoshida O. Enhancement of sensitivity of urinary bladder tumor cells to cisplatin by c-myc antisense oligonucleotide. Cancer 1994; 74:2546 - 54; http://dx.doi.org/10.1002/1097-0142(19941101)74:9<2546::AID-CNCR2820740924>3.0.CO;2-Y; PMID: 7923012
- Leonetti C, Biroccio A, Candiloro A, Citro G, Fornari C, Mottolese M, et al. Increase of cisplatin sensitivity by c-myc antisense oligodeoxynucleotides in a human metastatic melanoma inherently resistant to cisplatin. Clin Cancer Res 1999; 5:2588 - 95; PMID: 10499637
- Pyndiah S, Tanida S, Ahmed KM, Cassimere EK, Choe C, Sakamuro D. c-MYC suppresses BIN1 to release poly(ADP-ribose) polymerase 1: a mechanism by which cancer cells acquire cisplatin resistance. Sci Signal 2011; 4:ra19; http://dx.doi.org/10.1126/scisignal.2001556; PMID: 21447800
- Khanna A, Böckelman C, Hemmes A, Junttila MR, Wiksten JP, Lundin M, et al. MYC-dependent regulation and prognostic role of CIP2A in gastric cancer. J Natl Cancer Inst 2009; 101:793 - 805; http://dx.doi.org/10.1093/jnci/djp103; PMID: 19470954
- Chen JP, Lin C, Xu CP, Zhang XY, Fu M, Deng YP, et al. Molecular therapy with recombinant antisense c-myc adenovirus for human gastric carcinoma cells in vitro and in vivo. J Gastroenterol Hepatol 2001; 16:22 - 8; http://dx.doi.org/10.1046/j.1440-1746.2001.02361.x; PMID: 11206311
- Yin X, Giap C, Lazo JS, Prochownik EV. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 2003; 22:6151 - 9; http://dx.doi.org/10.1038/sj.onc.1206641; PMID: 13679853
- Rosenwald IB, Rhoads DB, Callanan LD, Isselbacher KJ, Schmidt EV. Increased expression of eukaryotic translation initiation factors eIF-4E and eIF-2 alpha in response to growth induction by c-myc. Proc Natl Acad Sci USA 1993; 90:6175 - 8; http://dx.doi.org/10.1073/pnas.90.13.6175; PMID: 8327497
- Jones RM, Branda J, Johnston KA, Polymenis M, Gadd M, Rustgi A, et al. An essential E box in the promoter of the gene encoding the mRNA cap-binding protein (eukaryotic initiation factor 4E) is a target for activation by c-myc. Mol Cell Biol 1996; 16:4754 - 64; PMID: 8756633
- Lin CJ, Cencic R, Mills JR, Robert F, Pelletier J. c-Myc and eIF4F are components of a feedforward loop that links transcription and translation. Cancer Res 2008; 68:5326 - 34; http://dx.doi.org/10.1158/0008-5472.CAN-07-5876; PMID: 18593934
- Skov S, Rieneck K, Bovin LF, Skak K, Tomra S, Michelsen BK, et al. Histone deacetylase inhibitors: a new class of immunosuppressors targeting a novel signal pathway essential for CD154 expression. Blood 2003; 101:1430 - 8; http://dx.doi.org/10.1182/blood-2002-07-2073; PMID: 12393479
- Bommi PV, Dimri M, Sahasrabuddhe AA, Khandekar J, Dimri GP. The polycomb group protein BMI1 is a transcriptional target of HDAC inhibitors. Cell Cycle 2010; 9:2663 - 73; http://dx.doi.org/10.4161/cc.9.13.12147; PMID: 20543557
- Kretzner L, Scuto A, Dino PM, Kowolik CM, Wu J, Ventura P, et al. Combining histone deacetylase inhibitor vorinostat with aurora kinase inhibitors enhances lymphoma cell killing with repression of c-Myc, hTERT, and microRNA levels. Cancer Res 2011; 71:3912 - 20; http://dx.doi.org/10.1158/0008-5472.CAN-10-2259; PMID: 21502403
- Wagner AD, Unverzagt S, Grothe W, Kleber G, Grothey A, Haerting J, et al. Chemotherapy for advanced gastric cancer. Cochrane Database Syst Rev CD004064
- Frew AJ, Johnstone RW, Bolden JE. Enhancing the apoptotic and therapeutic effects of HDAC inhibitors. Cancer Lett 2009; 280:125 - 33; http://dx.doi.org/10.1016/j.canlet.2009.02.042; PMID: 19359091
- Lindemann RK, Newbold A, Whitecross KF, Cluse LA, Frew AJ, Ellis L, et al. Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc Natl Acad Sci USA 2007; 104:8071 - 6; http://dx.doi.org/10.1073/pnas.0702294104; PMID: 17470784
- Maeta Y, Tsujitani S, Matsumoto S, Yamaguchi K, Tatebe S, Kondo A, et al. Expression of Mcl-1 and p53 proteins predicts the survival of patients with T3 gastric carcinoma. Gastric Cancer 2004; 7:78 - 84; http://dx.doi.org/10.1007/s10120-004-0272-9; PMID: 15224193
- Wacheck V, Cejka D, Sieghart W, Losert D, Strommer S, Crevenna R, et al. Mcl-1 is a relevant molecular target for antisense oligonucleotide strategies in gastric cancer cells. Cancer Biol Ther 2006; 5:1348 - 54; http://dx.doi.org/10.4161/cbt.5.10.3224; PMID: 16969094
- Kondo S, Shinomura Y, Kanayama S, Higashimoto Y, Kiyohara T, Zushi S, et al. Modulation of apoptosis by endogenous Bcl-xL expression in MKN-45 human gastric cancer cells. Oncogene 1998; 17:2585 - 91; http://dx.doi.org/10.1038/sj.onc.1202194; PMID: 9840921
- Smith L, Berrieman HK, O’Kane SL, Campbell A, Maraveyas A, Cawkwell L. Immunohistochemical detection of apoptotic markers in gastric cancer. Oncol Res 2006; 15:441 - 4; PMID: 16555550
- Kondo S, Shinomura Y, Kanayama S, Higashimoto Y, Miyagawa JI, Minami T, et al. Over-expression of bcl-xL gene in human gastric adenomas and carcinomas. Int J Cancer 1996; 68:727 - 30; http://dx.doi.org/10.1002/(SICI)1097-0215(19961211)68:6<727::AID-IJC6>3.0.CO;2-5; PMID: 8980174
- Kim HK, Choi IJ, Kim CG, Kim HS, Oshima A, Michalowski A, et al. A gene expression signature of acquired chemoresistance to cisplatin and fluorouracil combination chemotherapy in gastric cancer patients. PLoS ONE 2011; 6:e16694; http://dx.doi.org/10.1371/journal.pone.0016694; PMID: 21364753
- Kim HK, Choi IJ, Kim CG, Kim HS, Oshima A, Yamada Y, et al. Three-gene predictor of clinical outcome for gastric cancer patients treated with chemotherapy. Pharmacogenomics J 2010; In press http://dx.doi.org/10.1038/tpj.2010.87; PMID: 21173787
- Eilers M, Eisenman RN. Myc’s broad reach. Genes Dev 2008; 22:2755 - 66; http://dx.doi.org/10.1101/gad.1712408; PMID: 18923074
- Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, et al. Modelling Myc inhibition as a cancer therapy. Nature 2008; 455:679 - 83; http://dx.doi.org/10.1038/nature07260; PMID: 18716624
- Prochownik EV, Vogt PK. Therapeutic Targeting of Myc. Genes Cancer 2010; 1:650 - 9; http://dx.doi.org/10.1177/1947601910377494; PMID: 21132100
- Calcagno DQ, Leal MF, Assumpcao PP, Smith MA, Burbano RR. MYC and gastric adenocarcinoma carcinogenesis. World J Gastroenterol 2008; 14:5962 - 8; http://dx.doi.org/10.3748/wjg.14.5962; PMID: 18932273
- Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, et al. Genomic targets of the human c-Myc protein. Genes Dev 2003; 17:1115 - 29; http://dx.doi.org/10.1101/gad.1067003; PMID: 12695333
- Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 2009; 136:731 - 45; http://dx.doi.org/10.1016/j.cell.2009.01.042; PMID: 19239892
- Fischer PM. Cap in hand: targeting eIF4E. Cell Cycle 2009; 8:2535 - 41; http://dx.doi.org/10.4161/cc.8.16.9301; PMID: 19597330
- Graff JR, Konicek BW, Carter JH, Marcusson EG. Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer Res 2008; 68:631 - 4; http://dx.doi.org/10.1158/0008-5472.CAN-07-5635; PMID: 18245460
- Chen CN, Hsieh FJ, Cheng YM, Lee PH, Chang KJ. Expression of eukaryotic initiation factor 4E in gastric adenocarcinoma and its association with clinical outcome. J Surg Oncol 2004; 86:22 - 7; http://dx.doi.org/10.1002/jso.20037; PMID: 15048676
- Soni A, Akcakanat A, Singh G, Luyimbazi D, Zheng Y, Kim D, et al. eIF4E knockdown decreases breast cancer cell growth without activating Akt signaling. Mol Cancer Ther 2008; 7:1782 - 8; http://dx.doi.org/10.1158/1535-7163.MCT-07-2357; PMID: 18644990
- Rottmann S, Lüscher B. The Mad side of the Max network: antagonizing the function of Myc and more. Curr Top Microbiol Immunol 2006; 302:63 - 122; http://dx.doi.org/10.1007/3-540-32952-8_4; PMID: 16620026
- Laherty CD, Yang WM, Sun JM, Davie JR, Seto E, Eisenman RN. Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell 1997; 89:349 - 56; http://dx.doi.org/10.1016/S0092-8674(00)80215-9; PMID: 9150134
- Luchenko VL, Salcido CD, Zhang Y, Agama K, Komlodi-Pasztor E, Murphy RF, et al. Schedule-dependent synergy of histone deacetylase inhibitors with DNA damaging agents in small cell lung cancer. Cell Cycle 2011; 10:3119 - 28; http://dx.doi.org/10.4161/cc.10.18.17190; PMID: 21900747
- Larsson LG, Henriksson MA. The Yin and Yang functions of the Myc oncoprotein in cancer development and as targets for therapy. Exp Cell Res 2010; 316:1429 - 37; http://dx.doi.org/10.1016/j.yexcr.2010.03.025; PMID: 20382143
- Fritsche P, Seidler B, Schüler S, Schnieke A, Göttlicher M, Schmid RM, et al. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut 2009; 58:1399 - 409; http://dx.doi.org/10.1136/gut.2009.180711; PMID: 19528037
- Wirth M, Fritsche P, Stojanovic N, Brandl M, Jaeckel S, Schmid RM, et al. A simple and cost-effective method to transfect small interfering RNAs into pancreatic cancer cell lines using polyethylenimine. Pancreas 2011; 40:144 - 50; http://dx.doi.org/10.1097/MPA.0b013e3181f7e41c; PMID: 20938367
- Stauber RH, Knauer SK, Habtemichael N, Bier C, Unruhe B, Weisheit S, et al. Drug-induced head and neck cancer cell killing by downregulating EGFR and triggering BIM-induced apoptosis. Oncotarget 2012; 3:31 - 43; PMID: 22289787
- Thompson J, Epting T, Schwarzkopf G, Singhofen A, Eades-Perner AM, van Der Putten H, et al. A transgenic mouse line that develops early-onset invasive gastric carcinoma provides a model for carcinoembryonic antigen-targeted tumor therapy. Int J Cancer 2000; 86:863 - 9; http://dx.doi.org/10.1002/(SICI)1097-0215(20000615)86:6<863::AID-IJC16>3.0.CO;2-4; PMID: 10842202
- Mukhopadhyay A, Deplancke B, Walhout AJ, Tissenbaum HA. Chromatin immunoprecipitation (ChIP) coupled to detection by quantitative real-time PCR to study transcription factor binding to DNA in Caenorhabditis elegans. Nat Protoc 2008; 3:698 - 709; http://dx.doi.org/10.1038/nprot.2008.38; PMID: 18388953