Abstract
The capacity of β cells to expand in response to insulin resistance is a critical factor in the development of type 2 diabetes. Proliferation of β cells is a major component for these adaptive responses in animal models. The extracellular signals responsible for β-cell expansion include growth factors, such as insulin, and nutrients, such as glucose and amino acids. AKT activation is one of the important components linking growth signals to the regulation of β-cell expansion. Downstream of AKT, tuberous sclerosis complex 1 and 2 (TSC1/2) and mechanistic target of rapamycin complex 1 (mTORC1) signaling have emerged as prime candidates in this process, because they integrate signals from growth factors and nutrients. Recent studies demonstrate the importance of mTORC1 signaling in β cells. This review will discuss recent advances in the understanding of how this pathway regulates β-cell mass and present data on the role of TSC1 in modulation of β-cell mass. Herein, we also demonstrate that deletion of Tsc1 in pancreatic β cells results in improved glucose tolerance, hyperinsulinemia and expansion of β-cell mass that persists with aging.
Introduction
Type 2 diabetes is characterized by defects in β-cell expansion and function in conditions of insulin resistance. The extracellular signals responsible for β-cell expansion include growth factors, insulin and nutrients, such as glucose and amino acids. AKT (also known as protein kinase B) signaling links growth signals to the regulation of β-cell expansion. Downstream of AKT, TSC1/2 (tuberous sclerosis complex) and mTORC1 (mechanistic target of rapamycin, complex 1) are key players in this process, because they integrate signals from growth factors and nutrients. mTORC1 controls growth (cell size), proliferation (cell number) and metabolism directly, by modulating eukaryotic initiation factor 4E-binding proteins (4E-BP 1, 2 and 3) and ribosomal protein S6 kinases (S6K 1 and 2), and indirectly, by attenuating AKT signaling via an mTORC1/S6K-mediated negative feedback loop.1‑10 How TSC/mTOR signaling regulates β-cell mass expansion is not completely understood. A better understanding of how mTORC1 regulates β-cell mass and the role of interactions between mTORC1 and other signaling pathways in this process are critical to discovering how β-cell mass adapts to insulin resistance and autoimmune injury and, ultimately, how it impacts diabetes management. In this review, we will discuss our current knowledge of the importance of mTORC1 signaling in modulation of β-cell mass and function, and we will also present data demonstrating the importance of TSC1 signaling in β cells.
mTOR: The Signaling Network
Upstream of mTORC1.
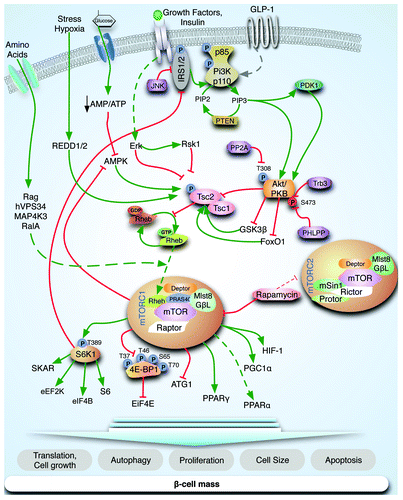
mTOR is a serine/threonine kinase that is conserved through evolution and is essential for cell growth and proliferation.11 mTOR constitutes the catalytic unit of two distinct complexes, mTORC1 and 2,12,13 which both contain mTOR, mLST8 (mammalian lethal with SEC13 protein)/GβL (G-protein β-protein subunit-like) and Deptor (DEP domain-containing mTOR-interacting protein) (). mTORC1 constitutes the rapamycin-sensitive arm of mTOR signaling and contains Raptor (regulatory associated protein of mTOR) and PRAS40 (proline-rich AKT substrate of 40 kDa).14,15 mTORC2 is insensitive to rapamycin and also contains Rictor (rapamycin in-sensitive companion of mTOR), mSIN1 (mammalian stress-activated MAPK-interacting protein 1) and Protor 1/2 (protein observed with rictor 1/2).12,13 mTORC2 phosphorylates AKT on serine 473, suggesting that this pathway could be indirectly linked to proliferation.16 mTORC1 activity is modulated by multiple upstream signals, which include growth factors, amino acids, cytokines, energy and cellular stressors. mTORC1 is negatively regulated by TSC1 and TSC2 and the small G protein RHEB (Ras homolog enriched in brain) (). The major function of TSC1 is to stabilize TSC2 and prevent its ubiquitin-mediated degradation.17,18 TSC2 phosphorylation and inactivation by AKT and ERK, among others, releases the inhibition of RHEB, leading to activation of mTOR.19-24 In contrast, phosphorylation and activation of TSC2 by AMPK and GSK3β inhibits mTOR signaling.23,25 Although amino acids are important regulators of mTORC1 activity, the mechanisms responsible for this activation are not as well-characterized as the regulation by insulin or growth factors. RAG-GTPases have been identified as important regulators of this process by transporting mTORC1 to specific cellular compartments and facilitating the ability of RHEB-GTP to activate mTORC1.26,27 Ste20-related kinase, MAP4K3 and the class III PI3K, mVPS34, have also been shown to link amino acid sensing to mTORC1.28-30 Stress and hypoxia act to limit growth and protein synthesis by mechanisms that are incompletely understood but involve HIF-1 and REDD1.31,32 Finally, reduction in ATP levels in conditions of energy stress suppresses mTORC1 signaling via several mechanisms, including both AMPK-dependent and independent pathways.23,33,34
Figure 1. mTOR signaling pathway. mTOR is part of two major complexes, mTORC1 and mTORC2. Insulin and growth factor receptors induce PI3K, which results in phosphoinositide-dependent kinase-1 (PDK1)-dependent phosphorylation of T308 of AKT. mTORC1 is negatively regulated by TSC1 and TSC2 and positively regulated by the small G protein RHEB. Akt phosphorylates TSC2 and disrupts the complex of mTORC1, which inhibits the GTPase activating function (GAP activity) of the TSC1-TSC2 complex toward RHEB and favors the GTP bound form of RHEB (active). S6K regulates cell size, ribosomal biogenesis and protein synthesis. In addition, S6K signaling inhibits insulin signaling by phosphorylation of IRS1 and possibly IRS2. mTORC1 phosphorylates 4E-BP at multiple sites, leading to dissociation of eIF4E and resulting in cap-dependent translation. Additional mTORC1 targets include ATG1, PPARγ, PGC1α and HIF-1. Energy depletion and glucose starvation increase the AMP/ATP ratio resulting in augmented AMPK activity. Amino acid activation of mTORC1 is more complex and not completely understood but several pathways have been proposed (Rag GTPases, hVPS34, MAP4K3 and RalA). Stress condition such as hypoxia can regulate TSC complex through REDD1/2.
Downstream of mTORC1.
Activation of mTORC1 signaling leads to translation initiation, which includes the synthesis of secreted proteins. mTORC1 controls growth (cell size) and proliferation (cell number) by modulating mRNA translation through phosphorylation of 4E-BPs and the S6 kinases ().35-38 The mTORC1-S6K arm regulates multiple biological processes, including cell growth/size, gene transcription, cell survival, synaptic plasticity and adipocyte differentiation.39,40 S6K promotes mRNA translation initiation and other steps of protein synthesis by phosphorylating an array of substrates, including ribosomal S6 protein, a component of the 40S ribosome, and eIF4B,41,42 PDCD4 (programmed cell death 4), eEF2K,43 SKAR (S6K1 Aly/REF-like substrate),44 CCTβ,45 and CBP80.45 However, chronic activation of S6K negatively regulates IRS1, therefore inactivating the PI3K/AKT signaling pathway.1-4,47 Phosphorylation of the 4E-BPs triggers the release of eIF4E, initiating cap-dependent translation.48,49 This arm of mTORC1 signaling has been linked to induction of cell proliferation independent of the mTORC1-S6K1 arm.50 Together these substrates downstream of mTORC1 play an integral role in mRNA translation initiation and progression, thus controlling the rate of protein synthesis.
TSC/mTOR Plays a Critical Role in Whole-Body Metabolism
mTORC1 is overactive in many organs in animal models of obesity and insulin resistance.1,4,5 Additionally, chronic mTORC1 activation plays an important role in the pathogenesis of insulin resistance in states of nutrient excess.1,4,8 This effect is mediated by an S6K1-dependent negative feedback loop resulting in phosphorylation and degradation of IRS1.1-4 Until recently, most of the evidence supporting the role of mTORC1 in insulin-sensitive tissues was derived from the use of rapamycin and alterations in insulin sensitivity in global S6K and 4E-bp-deficient mice.1,9,51 However, abnormalities in multiple tissues observed in global knockouts and systemic effects of rapamycin make it difficult to identify the functions of different mTORC1 signaling components in various tissues. Recent experiments using conditional knockout mice have begun to unravel the tissue-specific functions of different mTOR signaling components (e.g., Raptor and Rictor) and the contribution of these pathways to whole-body metabolism.52,53 Adipocyte-specific deficient mice for Raptor exhibit improved glucose tolerance, insulin sensitivity and resistance to high-fat feeding.52 In contrast, mice with a muscle-specific knockout of Raptor are glucose-intolerant and display downregulation of proteins involved in mitochondrial biogenesis (such as Pgc1α) and hyperactivation of Akt (due to loss of negative feedback).53 Most recently, conditional deletion of Raptor in the liver uncovered an important role of mTorc1 in regulation of adaptation to the fasting state due to a defect in ketogenesis,54 caused by loss of mTORC1-dependent Pparα signaling activation.54 While there are many studies in Raptor, the role of Rictor has been less well-characterized. Conditional deletion of Rictor in the muscle has no apparent phenotype.53 In contrast, deletion of Rictor in β cells results in glucose intolerance caused by a reduction in β-cell mass, β-cell proliferation, pancreatic insulin content and glucose-stimulated insulin secretion.55 Currently, little is known about the importance of Raptor in β cells. However, the above observations illustrate that the functions of Raptor are tissue-specific, and the next challenge will be to determine the contribution of this complex to glucose homeostasis.
mTORC1 Regulates b-Cell Proliferation, Cell Size and Mass
Studies with rapamycin.
Rapamycin treatment has been shown to have mixed effects on insulin secretion, depending on the experimental conditions.56-59 The role of mTORC1 in β-cell proliferation using rapamycin has been explored in multiple studies, which have resulted in five important observations. (1) Rapamycin treatment blocks β-cell expansion, cell size and proliferation induced by an activation of AKT in β cells.60 (2) Inhibition of β-cell proliferation by rapamycin results from alterations in cyclin D2 and D3 levels and Cdk4 activity. (3) Rapamycin modulates cyclin D2 synthesis and stability, suggesting that mTORC1 regulates post-transcriptional modifications of cell cycle components in β cells. (4) Rapamycin also ameliorates β-cell expansion in a model of insulin resistance, suggesting that mTORC1 coordinates β-cell adaptation to hyperglycemia and type 2 diabetes.9 (5) Rapamycin treatment results in reduced proliferation in β cells in pregnant mice and causes anti-proliferative effects in transplanted rat β cells in vivo.61,62 Together, these studies suggest a role of mTORC1 in the regulation of β-cell mass and cell cycle and imply that mTORC1 can mediate adaptation of β cells to insulin resistance. However, it is important to note that in vivo studies designed to reveal the importance of mTORC1 using rapamycin are less than ideal because of modulation of insulin sensitivity.63,64 In addition, it is now accepted that a major limitation of rapamycin for in vivo and in vitro studies is the inhibition of mTORC1 activity toward only a subset of mTORC1 substrates.8,9,65-67 Therefore, inhibiting mTORC1 signaling in vivo by genetic deletion of Raptor in β cells will provide new insights into the role of this pathway in nutrient- and growth factor-dependent regulation of β-cell proliferation.
Evidence from Genetic Models
TSC2.
The role of mTORC1 in β cells has been explored using animal models with gain of mTORC1 function. Mice with conditional deletion of Tsc2 in β cells (βTsc2-/-) exhibit lower glucose levels, hyperinsulinemia and improved glucose tolerance. These changes are explained by increases in β-cell mass, proliferation and cell size.7 A separate study demonstrated that conditional Tsc2 deletion in β cells exhibited a similar phenotype, but these mice developed diabetes and β-cell failure after 40 weeks.8 The differences in the phenotype between these reports are most likely explained by different genetic backgrounds and the RIP-Cre line (hypothalamic expression) used. Further confirmation of the effect of mTORC1 activation in increased β-cell mass comes from mice overexpressing Rheb in β cells.68 While the effect on cell size has been regularly described in these models, it is important to note that the effect of mTORC1 on β-cell proliferation has not been consistently observed in these studies. It is possible that different experimental conditions could, in part, explain these differences. However, one important conclusion derived from these studies is that more research is needed to determine the mechanisms and signaling components downstream of mTORC1 that control β-cell mass, proliferation and function.
TSC1.
The TSC1/TSC2 complex acts as a functional inhibitor of mTOR. Studies in human, mice, flies and yeast strongly suggest that their gene products are interdependent, and that these proteins function primarily as a complex.18,69-71 The role of Tsc1 in β cells was evaluated by deletion of Tsc1 specifically in β cells using RIP2-Cre mice.72 These mice presented a complex phenotype, with older mice displaying hyperphagia, obesity and diabetes due to the activation of mTorc1 in RIP2-Cre-expressing hypothalamic neurons.73 However, evaluation of the β-cell phenotype prior to the onset of obesity and insulin resistance in 4-week-old mice showed that Tsc1-deficient mice exhibit augmented β-cell hypertrophy, increased insulin synthesis and release and consequent hyperinsulinemia. Interestingly, this biphasic phenotype observed is similar to that observed in mice with conditional deletion of Tsc2 in β cells using the same RIP2-Cre line.8 Therefore, it remains unclear if the disparate β-cell phenotypes observed following conditional deletion of Tsc1 vs. to Tsc2 are consequences of β-cell specific functions of these proteins or alterations resulting from the Cre line used.
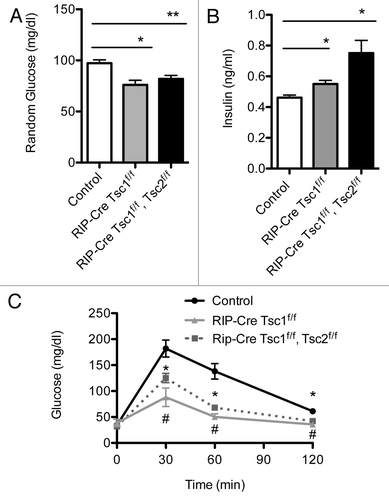
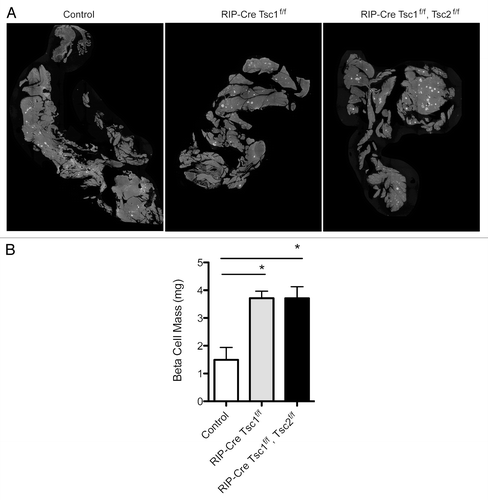
To further examine the consequences of conditional deletion of Tsc1 in β cells, we crossed Tsc1 floxed mice with a RIP-Cre line that displays little hypothalamic expression (βTsc1-/-).74 Absence of Tsc1 expression in islets from βTsc1-/- mice was demonstrated by immunostaining and immunoblotting (data not shown). Evaluation of glucose and insulin levels in 5- to 6-month-old βTsc1-/- mice revealed lower random glucose and hyperinsulinemia (). βTsc1-/- mice also displayed improved glucose tolerance when compared with control (), and this phenotype was also observed in older mice (10–12 mo, data not shown). In a parallel experiment, we set out to determine if concomitant deletion of Tsc2 in a Tsc1-deleted background has additional effects in β cells. βTsc1-/-/Tsc2‑/- (RIP-Cre Tsc1f/f Tsc2f/f) mice showed lower basal glucose levels and hyperinsulinemia compared with control mice, but the levels were not significantly different than those observed in βTsc1-/- mice (). Interestingly, glucose tolerance was also improved in βTsc1-/-/Tsc2-/- mice but not significantly different from βTsc1-/- mice (). Evaluation of β-cell morphometry demonstrated that βTsc1-/- mice had increased in β-cell mass that was not significantly different than that observed in βTsc1-/-/Tsc2-/- mice (). The results of these experiments demonstrate that deletion of Tsc1 in β cells has a positive impact on β-cell mass expansion, insulin levels and glucose tolerance that persists with aging. Moreover, these data suggest that deletion of both Tsc1 and Tsc2 does not have an additive effect on glucose metabolism and β-cell mass.
Figure 2. Metabolic assessment of Rip-Cre Tsc1 f/f and Rip-Cre Tsc1f/f, Tsc2 f/f. Random glucose (A) and insulin (B) levels obtained in the same group and age of mice. Intraperitoneal glucose tolerance tests (IPGTT) (C) were performed on the same group of mice. Measurements were performed on overnight -fasted 5–7 mo-old male control mice or Tsc 1f/f or Tsc1 f/f, Tsc2 f/f mice. Data are presented as mean SEM (n > 3). *p< 0.05 when compared with Rip-Cre Tsc1 f/f (solid gray line), Rip-Cre Tsc1 f/f Tsc2 f/f (dotted line) and control (solid black line).
How Downstream mTORC1 Targets Regulate β-Cell Mass Expansion
mTORC1 signaling activation has been implicated in regulation of β-cell mass, although the role of different direct downstream targets is unknown. An emerging view is that the 4E-BP and S6K signaling arms have distinct roles in regulation of proliferation, growth and possibly function.75 However, it is unclear whether the effects of activation of the 4E-BP/eIF pathway in β cells are different than those observed following induction of S6K signaling.76 Further studies will need to be conducted in order to elucidate these differences.
4E-BP proteins.
The members of the 4E-BP family include three paralogs (4E-BP 1, 2 and 3) with different tissue expression profiles. 4E-BP proteins are repressors of the translation initiation factor 4E (eIF4E) and therefore inhibit protein translation. Phosphorylation of 4E-BPs at multiple sites by mTORC1 leads to dissociation of eIF4E from 4E-BP, allowing elF4E binding to eIF4F and eIF4G. This complex formation promotes the translational machinery of mRNAs with high 5'-UTR secondary structures, such as those that encode ribosomal proteins, elongation factors and other proteins involved in the assembly and function of the translational machinery.38,77,78 This pathway regulates translation of mRNAs that encode important proteins for many biological processes, including proliferation. Our most current knowledge on the role of these proteins has been based on experiments using 4E-BP1-deficient cells or mice. The combined disruption of 4e-bp1 and 4e-bp2 in mice increased their sensitivity to diet-induced obesity by accelerated adipogenesis. These animals displayed increased insulin resistance associated with increased S6K activity and impaired Irs2/Akt signaling in peripheral tissues. Unfortunately, the specific β-cell phenotype of these animals was not analyzed.51 Recent studies have also revealed an important role for 4E-BP in protection against endoplasmic reticulum (ER) stress in β cells. Expression of 4e-bp1 was increased by direct transcriptional activation of the Eif4e-bp1 gene in islets under ER stress in several mouse models of diabetes.79 Most importantly, islets from 4e-bp1-null mutant mice were more susceptible to ER stress-induced apoptosis, suggesting that 4E-BP1 could be a survival factor for β cells. In vitro studies have demonstrated that growth factors, such as insulin, and nutrients, such as amino acids and glucose, induce phosphorylation of 4E-BP1 in islets and insulinoma cells.80-83 However, little is known about the importance of different 4E-BPs and the function of these proteins on regulation of β-cell proliferation, size, survival, mass and function. In summary, studies to determine the importance of 4E-BP1 in β cells have been limited by the alteration on insulin sensitivity observed in the global knockout model. Our understanding of the role of 4E-BP2 in different tissues is limited, but this protein is abundant in the brain and is required for learning and memory, indicating that in tissues it has functions independent of 4E-BP1.84
S6K.
The ribosomal protein S6 kinase (S6K) is described as a regulator of cell growth, protein translation and proliferation.85 The activity of S6K1 is regulated by both mTORC1 and PDK1, and several of its downstream targets have been identified. S6K1 and S6K2 regulate ribosomal protein S6, the eukaryotic initiation factor 4B (eIF4B), SKAR and eukaryotic elongation factor 2 kinase (eEF2K).86 S6K1- and S6K2-mediated phosphorylation is required for full activation and subsequent induction of the 40S ribosomal protein S6, which can then induce cell growth and proliferation. The importance of S6K signaling in β cells has been assessed in genetically modified mouse models. S6K1-deficient mice are viable and fertile and only present mild phenotypes during development because of a compensatory increase in levels of S6K2.87,88 Additionally, these mice display glucose intolerance and hypoinsulinemia with impaired insulin secretion.87 The β cells display a reduced size and decreased transcription of insulin, demonstrating the importance of S6K1 in regulating glucose homeostasis as well as cell growth. Interestingly, S6K1-deficient mice are resistant to age and high-fat diet-induced obesity and remain insulin-sensitive due to the loss of the negative feedback loop from S6K on Irs1 and Irs2. Together, these results suggest an in vivo role for S6K in desensitizing tissues to insulin,1,3,47 although the significance of this feedback regulation on IRS1 has recently been questioned.89 In contrast, S6K2-deficient mice are phenotypically similar to the control mice, suggesting that S6K1 might be more important than S6K2 for glucose homeostasis.90 Recent findings also demonstrated that S6K is important for insulinoma formation induced by activation of AKT signaling, implicating this kinase in regulation of β-cell cell cycle progression.91 Little is known about the signaling events downstream of S6K. The importance of ribosomal S6 protein was assessed using knock-in mice containing alanine substitutions at all five phosphorylatable serine residues of their S6 protein.92 These mice exhibit impaired glucose tolerance, lower insulin levels and increased insulin sensitivity. The pancreatic insulin content was reduced in these mice but was not associated with alterations in β-cell mass, implying an effect of S6 protein on insulin synthesis. The similarity of this phenotype with that of S6K-deficient mice suggests that ribosomal S6 protein is a critical substrate in relating metabolic signals from S6K.
To study the role of S6K activation in pancreatic β cells, we developed transgenic mice overexpressing a constitutively active form of S6K in β cells (S6KCARIP).76 Activation of S6K signaling in these mice improves insulin secretion in the absence of changes in β-cell mass. These findings revealed for the first time that activation of S6K induces insulin resistance in β cells by feedback inhibition of IRS signaling. Importantly, these studies also demonstrated that S6K regulates IRS2 levels, a major determinant of β-cell proliferation and survival. The β-cell insulin resistance induced by this mechanism had major negative effects on cell cycle progression by modulating levels of Cdk2, p27 and p16. Another major effect of insulin resistance in this model was increased apoptosis due to decreased survival signals from AKT. The balance between increased cell cycle entry with slow progression and enhanced apoptosis resulted in lack of β-cell mass expansion. Furthermore, these observations underscore the importance of the S6K-mediated negative feedback on IRS1/2 signaling in this phenotype and suggest that one of the major consequences of chronic exposure of β cells to nutrient overload is the development of impaired IRS signaling. In addition, these studies show that S6K activation recapitulates the cell size but not the proliferative phenotype of models with activation of mTORC1 signaling. These findings serve to elucidate some of the abnormalities observed in adaptive responses of β cells to nutrient excess and tentatively explain some of the mechanisms involved in glucose toxicity. Together, these data suggest that the negative feedback of S6K on IRS signaling can be a major modulator of β-cell mass and function in vivo.
How Decreased AKT Signaling by mTORC1-Mediated Negative Feedback Modulates β-cell Mass
Obesity induced by excess nutrient intake leads to the upregulation of mTORC1/S6K1 signaling in insulin-sensitive tissues, including β cells.1,4,8 Current evidence suggests that chronic mTORC1 activation by nutrient excess contributes to obesity by controlling appetite as well as fat deposition in white adipose tissue, liver and muscle cells.52-54,93 One consequence of chronic mTORC1 hyperactivation is the induction of an S6K1-dependent negative feedback loop, leading to attenuation of AKT signaling in multiple tissues and insulin resistance.1-4 It has been proposed that in contrast to classic feedback loops, where a threshold is reached before inhibition occurs, mTORC1 seems to suppress growth factor signaling in a more gradual and continual fashion. Therefore, it is conceivable that mTORC1 activation could play an initial physiological role in adaptation of nutrient excess and obesity, but chronic and persistent hyperactivation could lead to development of insulin resistance by a negative feedback loop on IRS signaling. Thus, it is also possible that S6K activation plays a beneficial compensatory role by increasing insulin secretion during early adaptation of β-cell cells to insulin resistance. However, chronic and persistent hyperactivation of S6K during later stages of adaptation to conditions of nutrient excess, such obesity, could have detrimental effects by inhibiting IRS/AKT signaling and β-cell expansion. How decreased IRS/AKT signaling by this feedback loop modulates β-cell mass expansion is unclear, but islets from S6KCARIP mice showed reduced phosphorylation of Bad, Gsk3β and FoxO1, suggesting that these proteins could be involved.76 Uncertainties still remain as to (1) what are the underlying mechanisms and key downstream effector(s) of mTORC1 that mediate growth and proliferative functions; (2) how inhibition on IRS/AKT signaling by the negative feedback loop regulates β-cell expansion; and (3) what is the physiological role of gain of mTORC1 function in adaptation of β cells to insulin resistance. Answers to these questions would further our understanding of the role of AKT signaling in conjunction with mTORC1 signaling mechanisms and how β cells adapt to conditions of nutrient excess.
Methods
Islet morphometry, β-cell mass and metabolic analysis.
Paraformaldehyde-fixed pancreatic tissues were embedded in paraffin using standard techniques. Sections were deparaffinized, rehydrated and incubated with blocking solution. Sections were incubated overnight at 4°C with antibodies against insulin (Linco Research), followed by secondary antibodies conjugated to FITC (Jackson Immunoresearch). DAPI-containing mounting media (Vector Laboratories) was added to the coverslips. β-cell mass assessment was performed by point counting morphometry from five insulin-stained sections (5 μm) separated by 200 μm using NIH Image Pro software (Media Cybernetics, Inc.).
Metabolic studies glucose, intraperitoneal glucose tolerance test, ELISA.
Fasting glucose levels were measured after an overnight fasting using an AccuChek II glucometer (Roche Diagnostics). Fed glucose levels were obtained at the same time in the morning of each day. Glucose tolerance test was performed by intraperitoneal delivery of 2 g/kg glucose to mice after 12 h fasting. Blood glucose was monitored for 2 h after glucose delivery. Plasma insulin levels were measured using mouse insulin ELISA kit (ALPCO).
Statistical analysis.
All data are represented as the mean ± SEM from at least three independent experiments unless otherwise indicated. Data were analyzed by Student t-test or ANOVA followed by post hoc analysis where appropriate. Results were considered statistically significant when the p-value was less than 0.05 ().
Figure 3. Representative pancreatic morphology (A) from mice with different genotypes stained for insulin (green) and DAPI (blue). Quantification of β-cell mass (B) in 5–7 mo old mice (n >3). Data are mean SE ± SEM *p< 0.05.
Summary and Further Perspective
Failure of β cells to expand or adapt to insulin resistance results in type 2 diabetes. β cells adapt to insulin resistance mainly by augmenting proliferation, although cell size and apoptosis may also play a role in this process.94 Therefore, finding therapies that increase or replenish β cells through enhanced proliferation will delay, prevent or possibly even cure diabetes. The IRS2/PI3K/AKT pathway plays a critical role in the regulation of β-cell proliferation and mass in vivo and in vitro.6,95-99 A major limitation of this kinase for therapeutic benefit is its potential to induce oncogenic transformation. Downstream of AKT, the TSC/mTORC1 signaling pathway emerges as an important candidate to modulate β-cell expansion, because it integrates signals from growth factors and nutrients,69,100-105 induces controlled growth and is rarely associated with malignancy.106,107 The current evidence supports the concept that mTORC1 is active in states of increased insulin demand and plays a major role in β-cell expansion and proliferation induced by AKT and insulin resistance.1,4,5,9,60,68 Most of our knowledge about the role of mTORC1 in β cells comes from in vitro experiments using insulinoma cells or isolated islets treated with rapamycin. However, the different doses and duration of incubation makes it difficult to draw consistent conclusions. In addition, limiting the use of rapamycin to draw conclusions about the role of mTORC1 is problematic, and future experiments should be complemented by genetic approaches. Several lines of evidence support this concept: (1) The effects of acute treatment with rapamycin are different than those observed with chronic exposure. Chronic exposure to this agent disrupts the mTORC2 complex altering other signaling pathways, such as Akt signaling, a major pathway for regulation of β-cell proliferation and survival. (2) In contrast to the potent inhibition of rapamycin on S6K phosphorylation by mTORC1, the inhibitory effect on 4E-BP signaling is partial and transient. All these limitations also apply to in vivo observations in transplant patients receiving chronic rapamycin treatment. Chronic administration of rapamycin has been associated with higher incidence of diabetes, although the relative contribution of β-cell dysfunction and insulin resistance to this effect is difficult to dissect.63,64 It is possible that the alterations in insulin sensitivity could result from the effect of chronic rapamycin treatment on mTORC2/Akt signaling. Therefore, it is conceivable that rapamycin therapy could impair the adaptation of β cells to insulin resistance, resulting in diabetes. In contrast, acute treatment with rapamycin appears to improve insulin sensitivity in humans and rodents.4,108-110 How the effects of rapamycin could be used for therapeutic purposes of metabolic disease is unclear, and more research should be done to unravel the tissue-specific effects after acute vs. chronic treatment with rapamycin in different metabolically active tissues.
Future research should be aimed at determining the mechanisms by which activation of mTORC1 regulates controlled β-cell proliferation and mass. This knowledge will provide new platforms to develop novel pharmacologic strategies to induce controlled β-cell mass by selectively inducing β-cell proliferation without altering the risk of oncogenic transformation. Discovery of agents modulating this pathway could be used in translational experiments to treat diabetes by expanding β-cell mass in vivo. At the same time, the pharmacologic manipulation of this pathway can also be used to increase the pool of transplantable islets and enhance the success of islet transplantation. Another major impact of these studies is obtaining a better understanding of the effects of rapamycin in β cells’ mass and function to develop better immunosuppressive therapies for islet transplantation.
Acknowledgements
This work was supported by National Institutes of Health Grant DK-073716-(to E.B.M.), Research Grant from The Juvenile Diabetes Research Foundation and a Career Development Award from the American Diabetes Association (to E.B.M.). E.U.A. was supported by an NIH training grant (2T32DK071212-06). We would also acknowledge the MDRTC Cell and Molecular Biology Core (P60DK020572) and the Morphology Core at the University of Michigan Cancer Center for their services. We want to thank the innumerable authors who have contributed to the field of mTOR signaling and β cell signaling and apologize for not being able to cite all of their important contributions because of editorial limitations on the numbers of references allowed.
Author Contributions
M.B.R., A.Y.C., E.U.A. performed experiments, analyzed data and wrote the manuscript. A.P.G. and T.T. performed experiments. J.O.S. edited the manuscript. L.E. edited the bibliography and corrected part of the manuscript. C.C.M. designed Figure 1 and corrected part of the manuscript. E.B.M. conceived the study and wrote/edited the manuscript.
References
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004; 431:200 - 5; http://dx.doi.org/10.1038/nature02866; PMID: 15306821
- Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol 2004; 14:1650 - 6; http://dx.doi.org/10.1016/j.cub.2004.08.026; PMID: 15380067
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol 2004; 166:213 - 23; http://dx.doi.org/10.1083/jcb.200403069; PMID: 15249583
- Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology 2005; 146:1473 - 81; http://dx.doi.org/10.1210/en.2004-0921; PMID: 15604215
- Asahara S, Matsuda T, Kido Y, Kasuga M. Increased ribosomal biogenesis induces pancreatic beta cell failure in mice model of type 2 diabetes. Biochem Biophys Res Commun 2009; 381:367 - 71; http://dx.doi.org/10.1016/j.bbrc.2009.02.047; PMID: 19309774
- Briaud I, Dickson LM, Lingohr MK, McCuaig JF, Lawrence JC, Rhodes CJ. Insulin receptor substrate-2 proteasomal degradation mediated by a mammalian target of rapamycin (mTOR)-induced negative feedback down-regulates protein kinase B-mediated signaling pathway in beta-cells. J Biol Chem 2005; 280:2282 - 93; http://dx.doi.org/10.1074/jbc.M412179200; PMID: 15537654
- Rachdi L, Balcazar N, Osorio-Duque F, Elghazi L, Weiss AJ, Gould AP, et al. Disruption of Tsc2 in pancreatic beta cells induces beta cell mass expansion and improved glucose tolerance in a TORC1-dependent manner. Proc Natl Acad Sci USA 2008; 105:9250 - 5; http://dx.doi.org/10.1073/pnas.0803047105; PMID: 18587048
- Shigeyama Y, Kobayashi T, Kido Y, Hashimoto N, Asahara S, Matsuda T, et al. Biphasic response of pancreatic beta-cell mass to ablation of tuberous sclerosis complex 2 in mice. Mol Cell Biol 2008; 28:2971 - 9; http://dx.doi.org/10.1128/MCB.01695-07; PMID: 18316403
- Fraenkel M, Ketzinel-Gilad M, Ariav Y, Pappo O, Karaca M, Castel J, et al. mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes 2008; 57:945 - 57; http://dx.doi.org/10.2337/db07-0922; PMID: 18174523
- Liu H, Remedi MS, Pappan KL, Kwon G, Rohatgi N, Marshall CA, et al. Both Glycogen Synthase Kinase-3 (GSK-3) and Mammalian Target of Rapamycin (mTOR) Pathways Contribute to DNA Synthesis, Cell Cycle Progression and Proliferation in Human Islets. Diabetes 2008; http://dx.doi.org/10.2337/db07-1208; PMID: 19073772
- Murakami M, Ichisaka T, Maeda M, Oshiro N, Hara K, Edenhofer F, et al. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol Cell Biol 2004; 24:6710 - 8; http://dx.doi.org/10.1128/MCB.24.15.6710-6718.2004; PMID: 15254238
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 2004; 14:1296 - 302; http://dx.doi.org/10.1016/j.cub.2004.06.054; PMID: 15268862
- Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 2004; 6:1122 - 8; http://dx.doi.org/10.1038/ncb1183; PMID: 15467718
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002; 110:163 - 75; http://dx.doi.org/10.1016/S0092-8674(02)00808-5; PMID: 12150925
- Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 2007; 9:316 - 23; http://dx.doi.org/10.1038/ncb1547; PMID: 17277771
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 307:1098 - 101; http://dx.doi.org/10.1126/science.1106148; PMID: 15718470
- Benvenuto G, Li S, Brown SJ, Braverman R, Vass WC, Cheadle JP, et al. The tuberous sclerosis-1 (TSC1) gene product hamartin suppresses cell growth and augments the expression of the TSC2 product tuberin by inhibiting its ubiquitination. Oncogene 2000; 19:6306 - 16; http://dx.doi.org/10.1038/sj.onc.1204009; PMID: 11175345
- Chong-Kopera H, Inoki K, Li Y, Zhu T, Garcia-Gonzalo FR, Rosa JL, et al. TSC1 stabilizes TSC2 by inhibiting the interaction between TSC2 and the HERC1 ubiquitin ligase. J Biol Chem 2006; 281:8313 - 6; http://dx.doi.org/10.1074/jbc.C500451200; PMID: 16464865
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 2002; 10:151 - 62; http://dx.doi.org/10.1016/S1097-2765(02)00568-3; PMID: 12150915
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 2002; 4:648 - 57; http://dx.doi.org/10.1038/ncb839; PMID: 12172553
- Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, et al. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest 2003; 112:1223 - 33; PMID: 14561707
- Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell 2003; 11:1457 - 66; http://dx.doi.org/10.1016/S1097-2765(03)00220-X; PMID: 12820960
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003; 115:577 - 90; http://dx.doi.org/10.1016/S0092-8674(03)00929-2; PMID: 14651849
- Dan HC, Sun M, Yang L, Feldman RI, Sui XM, Ou CC, et al. Phosphatidylinositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor complex by phosphorylation of tuberin. J Biol Chem 2002; 277:35364 - 70; http://dx.doi.org/10.1074/jbc.M205838200; PMID: 12167664
- Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006; 126:955 - 68; http://dx.doi.org/10.1016/j.cell.2006.06.055; PMID: 16959574
- Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 2008; 10:935 - 45; http://dx.doi.org/10.1038/ncb1753; PMID: 18604198
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008; 320:1496 - 501; http://dx.doi.org/10.1126/science.1157535; PMID: 18497260
- Findlay GM, Yan L, Procter J, Mieulet V, Lamb RFA. A MAP4 kinase related to Ste20 is a nutrient-sensitive regulator of mTOR signalling. Biochem J 2007; 403:13 - 20; http://dx.doi.org/10.1042/BJ20061881; PMID: 17253963
- Byfield MP, Murray JT, Backer JM. hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J Biol Chem 2005; 280:33076 - 82; http://dx.doi.org/10.1074/jbc.M507201200; PMID: 16049009
- Nobukuni T, Joaquin M, Roccio M, Dann SG, Kim SY, Gulati P, et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci USA 2005; 102:14238 - 43; http://dx.doi.org/10.1073/pnas.0506925102; PMID: 16176982
- Reiling JH, Hafen E. The hypoxia-induced paralogs Scylla and Charybdis inhibit growth by down-regulating S6K activity upstream of TSC in Drosophila. Genes Dev 2004; 18:2879 - 92; http://dx.doi.org/10.1101/gad.322704; PMID: 15545626
- Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 2004; 18:2893 - 904; http://dx.doi.org/10.1101/gad.1256804; PMID: 15545625
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008; 30:214 - 26; http://dx.doi.org/10.1016/j.molcel.2008.03.003; PMID: 18439900
- Zheng M, Wang YH, Wu XN, Wu SQ, Lu BJ, Dong MQ, et al. Inactivation of Rheb by PRAK-mediated phosphorylation is essential for energy-depletion-induced suppression of mTORC1. Nat Cell Biol 2011; 13:263 - 72; http://dx.doi.org/10.1038/ncb2168; PMID: 21336308
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell 2007; 12:9 - 22; http://dx.doi.org/10.1016/j.ccr.2007.05.008; PMID: 17613433
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev 2004; 18:1926 - 45; http://dx.doi.org/10.1101/gad.1212704; PMID: 15314020
- Harris TE, Lawrence JC Jr.. TOR signaling. Sci STKE 2003; 2003:re15; http://dx.doi.org/10.1126/stke.2122003re15; PMID: 14668532
- Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev 2002; 16:1472 - 87; http://dx.doi.org/10.1101/gad.995802; PMID: 12080086
- Weber JD, Gutmann DH. Deconvoluting mTOR biology. Cell Cycle 2012; 11:236 - 48; http://dx.doi.org/10.4161/cc.11.2.19022; PMID: 22214661
- Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J 2012; 441:1 - 21; http://dx.doi.org/10.1042/BJ20110892; PMID: 22168436
- Raught B, Peiretti F, Gingras AC, Livingstone M, Shahbazian D, Mayeur GL, et al. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J 2004; 23:1761 - 9; http://dx.doi.org/10.1038/sj.emboj.7600193; PMID: 15071500
- Shahbazian D, Roux PP, Mieulet V, Cohen MS, Raught B, Taunton J, et al. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J 2006; 25:2781 - 91; http://dx.doi.org/10.1038/sj.emboj.7601166; PMID: 16763566
- Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J 2001; 20:4370 - 9; http://dx.doi.org/10.1093/emboj/20.16.4370; PMID: 11500364
- Richardson CJ, Bröenstrup M, Fingar DC, Jülich K, Ballif BA, Gygi S, et al. SKAR is a specific target of S6 kinase 1 in cell growth control. Curr Biol 2004; 14:1540 - 9; http://dx.doi.org/10.1016/j.cub.2004.08.061; PMID: 15341740
- Wilson KF, Wu WJ, Cerione RA. Cdc42 stimulates RNA splicing via the S6 kinase and a novel S6 kinase target, the nuclear cap-binding complex. J Biol Chem 2000; 275:37307 - 10; http://dx.doi.org/10.1074/jbc.C000482200; PMID: 10973943
- Tremblay F, Marette A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem 2001; 276:38052 - 60; PMID: 11498541
- Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol 2004; 167:399 - 403; http://dx.doi.org/10.1083/jcb.200408161; PMID: 15533996
- Lin TA, Kong X, Haystead TA, Pause A, Belsham G, Sonenberg N, et al. PHAS-I as a link between mitogen-activated protein kinase and translation initiation. Science 1994; 266:653 - 6; http://dx.doi.org/10.1126/science.7939721; PMID: 7939721
- Pause A, Belsham GJ, Gingras AC, Donzé O, Lin TA, Lawrence JC Jr., et al. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature 1994; 371:762 - 7; http://dx.doi.org/10.1038/371762a0; PMID: 7935836
- Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol Cell Biol 2004; 24:200 - 16; http://dx.doi.org/10.1128/MCB.24.1.200-216.2004; PMID: 14673156
- Le Bacquer O, Petroulakis E, Paglialunga S, Poulin F, Richard D, Cianflone K, et al. Elevated sensitivity to diet-induced obesity and insulin resistance in mice lacking 4E-BP1 and 4E-BP2. J Clin Invest 2007; 117:387 - 96; http://dx.doi.org/10.1172/JCI29528; PMID: 17273556
- Polak P, Cybulski N, Feige JN, Auwerx J, Rüegg MA, Hall MN. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab 2008; 8:399 - 410; http://dx.doi.org/10.1016/j.cmet.2008.09.003; PMID: 19046571
- Bentzinger CF, Romanino K, Cloëtta D, Lin S, Mascarenhas JB, Oliveri F, et al. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab 2008; 8:411 - 24; http://dx.doi.org/10.1016/j.cmet.2008.10.002; PMID: 19046572
- Sengupta S, Peterson TR, Laplante M, Oh S, Sabatini DM. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 2010; 468:1100 - 4; http://dx.doi.org/10.1038/nature09584; PMID: 21179166
- Gu Y, Lindner J, Kumar A, Yuan W, Magnuson MA. Rictor/mTORC2 is essential for maintaining a balance between beta-cell proliferation and cell size. Diabetes 2011; 60:827 - 37; http://dx.doi.org/10.2337/db10-1194; PMID: 21266327
- Zhang N, Su D, Qu S, Tse T, Bottino R, Balamurugan AN, et al. Sirolimus is associated with reduced islet engraftment and impaired beta-cell function. Diabetes 2006; 55:2429 - 36; http://dx.doi.org/10.2337/db06-0173; PMID: 16936190
- Bell E, Cao X, Moibi JA, Greene SR, Young R, Trucco M, et al. Rapamycin has a deleterious effect on MIN-6 cells and rat and human islets. Diabetes 2003; 52:2731 - 9; http://dx.doi.org/10.2337/diabetes.52.11.2731; PMID: 14578291
- Shimodahira M, Fujimoto S, Mukai E, Nakamura Y, Nishi Y, Sasaki M, et al. Rapamycin impairs metabolism-secretion coupling in rat pancreatic islets by suppressing carbohydrate metabolism. J Endocrinol 2010; 204:37 - 46; http://dx.doi.org/10.1677/JOE-09-0216; PMID: 19812126
- Johnson JD, Ao Z, Ao P, Li H, Dai LJ, He Z, et al. Different effects of FK506, rapamycin, and mycophenolate mofetil on glucose-stimulated insulin release and apoptosis in human islets. Cell Transplant 2009; 18:833 - 45; http://dx.doi.org/10.3727/096368909X471198; PMID: 19500470
- Balcazar N, Sathyamurthy A, Elghazi L, Gould A, Weiss A, Shiojima I, et al. mTORC1 activation regulates beta-cell mass and proliferation by modulation of cyclin D2 synthesis and stability. J Biol Chem 2009; 284:7832 - 42; http://dx.doi.org/10.1074/jbc.M807458200; PMID: 19144649
- Zahr E, Molano RD, Pileggi A, Ichii H, Jose SS, Bocca N, et al. Rapamycin impairs in vivo proliferation of islet beta-cells. Transplantation 2007; 84:1576 - 83; http://dx.doi.org/10.1097/01.tp.0000296035.48728.28; PMID: 18165767
- Niclauss N, Bosco D, Morel P, Giovannoni L, Berney T, Parnaud G. Rapamycin impairs proliferation of transplanted islet β cells. Transplantation 2011; 91:714 - 22; PMID: 21297554
- Teutonico A, Schena PF, Di Paolo S. Glucose metabolism in renal transplant recipients: effect of calcineurin inhibitor withdrawal and conversion to sirolimus. J Am Soc Nephrol 2005; 16:3128 - 35; http://dx.doi.org/10.1681/ASN.2005050487; PMID: 16107580
- Di Paolo S, Teutonico A, Leogrande D, Capobianco C, Schena PF. Chronic inhibition of mammalian target of rapamycin signaling downregulates insulin receptor substrates 1 and 2 and AKT activation: A crossroad between cancer and diabetes?. J Am Soc Nephrol 2006; 17:2236 - 44; http://dx.doi.org/10.1681/ASN.2006030196; PMID: 16807405
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009; 460:392 - 5; PMID: 19587680
- Veilleux A, Houde VP, Bellmann K, Marette A. Chronic inhibition of the mTORC1/S6K1 pathway increases insulin-induced PI3K activity but inhibits Akt2 and glucose transport stimulation in 3T3-L1 adipocytes. Mol Endocrinol 2010; 24:766 - 78; http://dx.doi.org/10.1210/me.2009-0328; PMID: 20203102
- Choo AY, Blenis J. Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle 2009; 8:567 - 72; http://dx.doi.org/10.4161/cc.8.4.7659; PMID: 19197153
- Hamada S, Hara K, Hamada T, Yasuda H, Moriyama H, Nakayama R, et al. Upregulation of the mammalian target of rapamycin complex 1 pathway by Ras homolog enriched in brain in pancreatic beta-cells leads to increased beta-cell mass and prevention of hyperglycemia. Diabetes 2009; 58:1321 - 32; http://dx.doi.org/10.2337/db08-0519; PMID: 19258434
- Gao X, Pan D. TSC1 and TSC2 tumor suppressors antagonize insulin signaling in cell growth. Genes Dev 2001; 15:1383 - 92; http://dx.doi.org/10.1101/gad.901101; PMID: 11390358
- Potter CJ, Pedraza LG, Huang H, Xu T. The tuberous sclerosis complex (TSC) pathway and mechanism of size control. Biochem Soc Trans 2003; 31:584 - 6; http://dx.doi.org/10.1042/BST0310584; PMID: 12773160
- Matsumoto S, Bandyopadhyay A, Kwiatkowski DJ, Maitra U, Matsumoto T. Role of the Tsc1-Tsc2 complex in signaling and transport across the cell membrane in the fission yeast Schizosaccharomyces pombe. Genetics 2002; 161:1053 - 63; PMID: 12136010
- Mori H, Inoki K, Opland D, Münzberg H, Villanueva EC, Faouzi M, et al. Critical roles for the TSC-mTOR pathway in β-cell function. Am J Physiol Endocrinol Metab 2009; 297:E1013 - 22; http://dx.doi.org/10.1152/ajpendo.00262.2009; PMID: 19690069
- Mori H, Inoki K, Münzberg H, Opland D, Faouzi M, Villanueva EC, et al. Critical role for hypothalamic mTOR activity in energy balance. Cell Metab 2009; 9:362 - 74; http://dx.doi.org/10.1016/j.cmet.2009.03.005; PMID: 19356717
- Wicksteed B, Brissova M, Yan W, Opland DM, Plank JL, Reinert RB, et al. Conditional gene targeting in mouse pancreatic ß-Cells: analysis of ectopic Cre transgene expression in the brain. Diabetes 2010; 59:3090 - 8; http://dx.doi.org/10.2337/db10-0624; PMID: 20802254
- Dowling RJO, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 2010; 328:1172 - 6; http://dx.doi.org/10.1126/science.1187532; PMID: 20508131
- Elghazi L, Balcazar N, Blandino-Rosano M, Cras-Méneur C, Fatrai S, Gould AP, et al. Decreased IRS signaling impairs beta-cell cycle progression and survival in transgenic mice overexpressing S6K in beta-cells. Diabetes 2010; 59:2390 - 9; http://dx.doi.org/10.2337/db09-0851; PMID: 20622167
- Beretta L, Gingras AC, Svitkin YV, Hall MN, Sonenberg N. Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J 1996; 15:658 - 64; PMID: 8599949
- Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem 1998; 273:14484 - 94; http://dx.doi.org/10.1074/jbc.273.23.14484; PMID: 9603962
- Yamaguchi S, Ishihara H, Yamada T, Tamura A, Usui M, Tominaga R, et al. ATF4-mediated induction of 4E-BP1 contributes to pancreatic beta cell survival under endoplasmic reticulum stress. Cell Metab 2008; 7:269 - 76; http://dx.doi.org/10.1016/j.cmet.2008.01.008; PMID: 18316032
- Xu G, Kwon G, Marshall CA, Lin TA, Lawrence JC Jr., McDaniel ML. Branched-chain amino acids are essential in the regulation of PHAS-I and p70 S6 kinase by pancreatic beta-cells. A possible role in protein translation and mitogenic signaling. J Biol Chem 1998; 273:28178 - 84; http://dx.doi.org/10.1074/jbc.273.43.28178; PMID: 9774438
- Xu G, Marshall CA, Lin TA, Kwon G, Munivenkatappa RB, Hill JR, et al. Insulin mediates glucose-stimulated phosphorylation of PHAS-I by pancreatic beta cells. An insulin-receptor mechanism for autoregulation of protein synthesis by translation. J Biol Chem 1998; 273:4485 - 91; http://dx.doi.org/10.1074/jbc.273.8.4485; PMID: 9468502
- Xu G, Kwon G, Cruz WS, Marshall CA, McDaniel ML. Metabolic regulation by leucine of translation initiation through the mTOR-signaling pathway by pancreatic beta-cells. Diabetes 2001; 50:353 - 60; http://dx.doi.org/10.2337/diabetes.50.2.353; PMID: 11272147
- Kwon G, Marshall CA, Pappan KL, Remedi MS, McDaniel ML. Signaling elements involved in the metabolic regulation of mTOR by nutrients, incretins, and growth factors in islets. Diabetes 2004; 53:Suppl 3 S225 - 32; http://dx.doi.org/10.2337/diabetes.53.suppl_3.S225; PMID: 15561916
- Bidinosti M, Ran I, Sanchez-Carbente MR, Martineau Y, Gingras A-C, Gkogkas C, et al. Postnatal deamidation of 4E-BP2 in brain enhances its association with raptor and alters kinetics of excitatory synaptic transmission. Mol Cell 2010; 37:797 - 808; http://dx.doi.org/10.1016/j.molcel.2010.02.022; PMID: 20347422
- Kimball SR, Jefferson LS. Molecular mechanisms through which amino acids mediate signaling through the mammalian target of rapamycin. Curr Opin Clin Nutr Metab Care 2004; 7:39 - 44; http://dx.doi.org/10.1097/00075197-200401000-00008; PMID: 15090902
- Um SH, D’Alessio D, Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab 2006; 3:393 - 402; http://dx.doi.org/10.1016/j.cmet.2006.05.003; PMID: 16753575
- Pende M, Kozma SC, Jaquet M, Oorschot V, Burcelin R, Le Marchand-Brustel Y, et al. Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature 2000; 408:994 - 7; http://dx.doi.org/10.1038/35050135; PMID: 11140689
- Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J 1998; 17:6649 - 59; http://dx.doi.org/10.1093/emboj/17.22.6649; PMID: 9822608
- Copps KD, Hancer NJ, Opare-Ado L, Qiu W, Walsh C, White MF. Irs1 serine 307 promotes insulin sensitivity in mice. Cell Metab 2010; 11:84 - 92; http://dx.doi.org/10.1016/j.cmet.2009.11.003; PMID: 20074531
- Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, et al. S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol 2004; 24:3112 - 24; http://dx.doi.org/10.1128/MCB.24.8.3112-3124.2004; PMID: 15060135
- Alliouachene S, Tuttle RL, Boumard S, Lapointe T, Berissi S, Germain S, et al. Constitutively active Akt1 expression in mouse pancreas requires S6 kinase 1 for insulinoma formation. J Clin Invest 2008; 118:3629 - 38; http://dx.doi.org/10.1172/JCI35237; PMID: 18846252
- Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich-Rain M, Nir T, et al. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev 2005; 19:2199 - 211; http://dx.doi.org/10.1101/gad.351605; PMID: 16166381
- Cota D, Matter EK, Woods SC, Seeley RJ. The role of hypothalamic mammalian target of rapamycin complex 1 signaling in diet-induced obesity. J Neurosci 2008; 28:7202 - 8; http://dx.doi.org/10.1523/JNEUROSCI.1389-08.2008; PMID: 18614690
- Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004; 429:41 - 6; http://dx.doi.org/10.1038/nature02520; PMID: 15129273
- Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren J-M, Previs S, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 1998; 391:900 - 4; http://dx.doi.org/10.1038/36116; PMID: 9495343
- Withers DJ, Burks DJ, Towery HH, Altamuro SL, Flint CL, White MF. Irs-2 coordinates Igf-1 receptor-mediated beta-cell development and peripheral insulin signalling. Nat Genet 1999; 23:32 - 40; http://dx.doi.org/10.1038/12631; PMID: 10471495
- Hennige AM, Burks DJ, Ozcan U, Kulkarni RN, Ye J, Park S, et al. Upregulation of insulin receptor substrate-2 in pancreatic beta cells prevents diabetes. J Clin Invest 2003; 112:1521 - 32; PMID: 14617753
- Lingohr MK, Dickson LM, McCuaig JF, Hugl SR, Twardzik DR, Rhodes CJ. Activation of IRS-2-mediated signal transduction by IGF-1, but not TGF-alpha or EGF, augments pancreatic beta-cell proliferation. Diabetes 2002; 51:966 - 76; http://dx.doi.org/10.2337/diabetes.51.4.966; PMID: 11916914
- Lingohr MK, Dickson LM, Wrede CE, Briaud I, McCuaig JF, Myers MG Jr., et al. Decreasing IRS-2 expression in pancreatic beta-cells (INS-1) promotes apoptosis, which can be compensated for by introduction of IRS-4 expression. Mol Cell Endocrinol 2003; 209:17 - 31; http://dx.doi.org/10.1016/j.mce.2003.08.003; PMID: 14604813
- Tokunaga C, Yoshino K, Yonezawa K. mTOR integrates amino acid- and energy-sensing pathways. Biochem Biophys Res Commun 2004; 313:443 - 6; http://dx.doi.org/10.1016/j.bbrc.2003.07.019; PMID: 14684182
- Meijer AJ, Dubbelhuis PF. Amino acid signalling and the integration of metabolism. Biochem Biophys Res Commun 2004; 313:397 - 403; http://dx.doi.org/10.1016/j.bbrc.2003.07.012; PMID: 14684175
- Miloloza A, Kubista M, Rosner M, Hengstschläger M. Evidence for separable functions of tuberous sclerosis gene products in mammalian cell cycle regulation. J Neuropathol Exp Neurol 2002; 61:154 - 63; PMID: 11853018
- Soucek T, Rosner M, Miloloza A, Kubista M, Cheadle JP, Sampson JR, et al. Tuberous sclerosis causing mutants of the TSC2 gene product affect proliferation and p27 expression. Oncogene 2001; 20:4904 - 9; http://dx.doi.org/10.1038/sj.onc.1204627; PMID: 11521203
- Hengstschläger M, Rodman DM, Miloloza A, Hengstschläger-Ottnad E, Rosner M, Kubista M. Tuberous sclerosis gene products in proliferation control. Mutat Res 2001; 488:233 - 9; http://dx.doi.org/10.1016/S1383-5742(01)00058-8; PMID: 11397651
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell 2006; 124:471 - 84; http://dx.doi.org/10.1016/j.cell.2006.01.016; PMID: 16469695
- Manning BD, Logsdon MN, Lipovsky AI, Abbott D, Kwiatkowski DJ, Cantley LC. Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev 2005; 19:1773 - 8; http://dx.doi.org/10.1101/gad.1314605; PMID: 16027169
- Paik J-H, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 2007; 128:309 - 23; http://dx.doi.org/10.1016/j.cell.2006.12.029; PMID: 17254969
- Tremblay F, Marette A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem 2001; 276:38052 - 60; PMID: 11498541
- Krebs M, Brunmair B, Brehm A, Artwohl M, Szendroedi J, Nowotny P, et al. The Mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes 2007; 56:1600 - 7; http://dx.doi.org/10.2337/db06-1016; PMID: 17329620
- Ueno M, Carvalheira JBC, Tambascia RC, Bezerra RMN, Amaral ME, Carneiro EM, et al. Regulation of insulin signalling by hyperinsulinaemia: role of IRS-1/2 serine phosphorylation and the mTOR/p70 S6K pathway. Diabetologia 2005; 48:506 - 18; http://dx.doi.org/10.1007/s00125-004-1662-6; PMID: 15692808
- Abe Y, Yoon SO, Kubota K, Mendoza MC, Gygi SP, Blenis J. p90 ribosomal S6 kinase and p70 ribosomal S6 kinase link phosphorylation of the eukaryotic chaperonin containing TCP-1 to growth factor, insulin, and nutrient signaling. J Biol Chem 2009; 284:14939 - 48; http://dx.doi.org/10.1074/jbc.M900097200; PMID: 19332537