Abstract
FNDC3B was recently identified in an oncogenomic screen for amplified oncogenes in hepatocellular carcinoma. It is located at 3q26 and is amplified in over 20% of cancers, usually as part of a broad amplified region encompassing the entire 3q arm. Consistent with an oncogenic role in multiple cancer types, we show here that overexpression of FNDC3B is capable of malignantly transforming mammary and kidney epithelial cells in addition to hepatocytes. To explore how FNDC3B transforms cells, we determined the cellular localization of its gene product and the cancer pathways that it activates. We found that the FNDC3B oncoprotein localizes to the Golgi network, and that its correct localization is essential for its transforming function. We found that overexpression of FNDC3B induces the epithelial-to-mesenchymal transition (EMT) and activates several cancer pathways, including PI3-kinase/Akt, Rb1 and TGFβ signaling. For TGFβ signaling, we analyzed the point in the pathway at which FNDC3B operates and obtained evidence that it induces expression of all three TGFβ ligands and also promotes TGFBR1 cell-surface localization. We found that RNAi-mediated knockdown of FNDC3B in cancer cells with 3q amplification suppressed their clonogenicity and tumorigenicity, but that the same RNAi knockdown had no effect on single-copy 3q cancer cells. These results indicate that FNDC3B is an important oncogenic driver gene of the 3q amplicon, adding to the growing list of oncogenic drivers within this commonly amplified region.
Keywords: :
Introduction
Systematic genome-wide efforts have greatly expanded the list of genes that are somatically altered in cancer and that could potentially affect tumorigenicity.Citation1 These genome-wide efforts have been driven by technological developments in DNA sequencing, copy-number analysis and gene expression analysis.Citation2 One of the next steps in studying these somatic alterations will be to determine which of these hundreds of altered genes are functional cancer genes, i.e., oncogenes or tumor suppressors. Toward this end, we recently we screened 124 genes that are focally amplified in human liver cancer for their ability, when overexpressed, to promote tumor formation in a transplantable mouse liver cancer model.Citation3 Eighteen of these amplified genes were found to be functional oncogenes, including ten that had never been implicated before in oncogenesis. The same mouse liver cancer model was used to identify 12 deleted tumor suppressor genes.Citation4 Similarly, a recent functional screen of 199 genes amplified in human melanoma led to the discovery of six new oncogenes.Citation5 As these types of screens generate increasingly larger lists of functionally validated cancer genes, one question that emerges is whether it will be possible to develop systematic approaches to understand their oncogenic mechanism. In the past, such studies have invariably focused on single genes. However, it would be advantageous to develop more systematic approaches to deciphering oncogenic mechanisms of newly validated cancer genes.
Here, we approached this problem by using generally applicable tools to investigate the oncogenic mechanisms of FNDC3B, which we recently identified as an amplified oncogene in hepatocellular carcinoma. There is very little known about the biological function of FNDC3B. It was initially discovered as a gene (FAD104) that is upregulated in the early stages of adipocyte differentiation.Citation6 Homozygous disruption of FNDC3B is lethal and has pronounced effects on the adhesion, spreading and migration of mouse embryo fibroblasts.Citation7 Although these cellular phenotypes are similar to processes that are aberrantly regulated in cancer,Citation8 and could be used as the starting point for an investigation into oncogenic function, we instead adopted a generalizable approach that could be taken with any novel cancer gene.
Results
Amplification and overexpression of FNDC3B in diverse cancers.
We picked FNDC3B for further study from the set of 10 novel HCC amplified oncogenes that we had previously discovered in reference Citation3, based on its frequent amplification and overexpression in diverse cancer types, as revealed by inspection of public databases such as Oncomine (www.oncomine.com). FNDC3B is frequently amplified (> 30%) in esophageal, lung, ovarian and breast cancers; many of these amplification events are high-level (). Several key features of FNDC3B amplification can be readily visualized by examining a heatmap of copy number status of 261 lung cancer samples for positions along chromosomal arm 3q, where FNDC3B is located at 3q26.31 (). It can be seen that FNDC3B is amplified to varying degrees in ~30% of these samples, with most of the lower level amplifications encompassing most of the 3q arm and the majority of the higher level amplifications also embedded within gain of the entire 3q arm (). Thus FNDC3B is amplified with other genes and, in most cases, is co-amplified with one or more nearby oncogenes located within the 22 Mb region of 3q26, for example, PIK3CA at 3q26.32,Citation10 SEC62 and SKIL at 3q26.2,Citation11,Citation12 and SOX2 as 3q26.33.Citation13 Although in this study we focus exclusively on FNDC3B, it is clear that deeper understanding of its role in cancer could emerge from studying in within the context of co-amplified genes. At the present time, this would be extremely challenging for both resource and technical reasons; this point is addressed in more detail in the discussion.
Figure 1. Amplification and overexpression of FNDC3B in diverse tumor types. (A) The frequency of amplification is shown for 9 different tumor types along with the overall frequency in > 3,000 diverse tumors using data from Tumorscape (www.broadinstitute.org/tumorscape). High-level amplifications are > 2-fold increase in copy number, focal amplifications are those smaller than one half of the chromosome arm (3q is 104 Mb). (B) Heatmap of copy number alterations along 3q for a large data set of 261 lung tumors.Citation9 The heatmap was created using the Integrated Genomics Viewer (www.broadinstitute.org/igv). (C) Relationship between FNDC3B expression (log2 scale) and DNA copy number in over 400 high-grade serous ovarian cancers in the TCGA data set (http://cbio.mskcc.org/gdac-portal). (D) Detection of FNDC3B protein in amplified and single-copy human liver cells. Immunoblotting with anti-peptide antibodies were used to detect endogenous FNDC3B in HCC cell lines with different copy numbers of FNDC3B as indicated. NexHep is an immortalized non-malignant human hepatocyte.
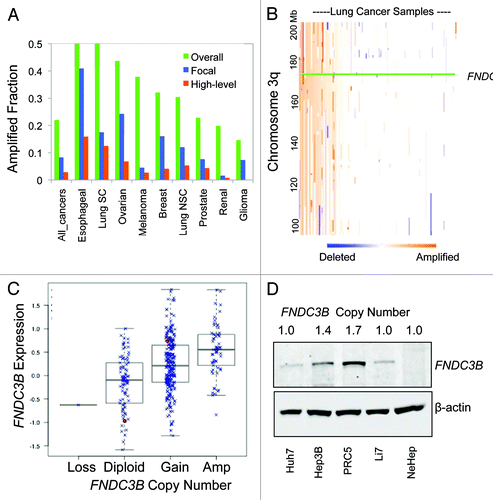
On a genome-wide scale, it has been estimated that close to 50% of DNA copy number alterations lead directly to corresponding changes in gene expression, with a 1.5-fold change in mRNA on average relative to a 2-fold change in copy number.Citation14 Examination of the relationship between FNDC3B copy number and expression in the large TCGA ovarian cancer data set clearly establishes that increased gene dosage results in greater expression, with an approximately 1.3-fold elevation in mRNA relative to the 1.5- to 2-fold change in copy number in tumors with low-level gain and a greater than 2-fold elevation in mRNA in tumors with higher-level amplification (). We observed quantitatively stronger effects on expression when we examined protein levels. FNDC3B protein expression was correlated with copy number in a series of human HCC cell lines with approximately 5-fold increase in protein seen in the PRC5 cell line which harbors a low level gain of FNDC3B ().
Malignant transformation by FNDC3B overexpression and induction of EMT.
Previously we showed that FNDC3B overexpression induced tumorigenicity in a nonmalignant murine hepatocyte.Citation3 Based on its amplification and overexpression in additional epithelial tumor types, we suspected that FNDC3B overexpression would transform other epithelial cell types as well. To test this, we overexpressed FNDC3B in two nonmalignant immortalized epithelial cells, NMuMG and RK3E, isolated from murine mammary epithelium and rat kidney epithelium, respectively.Citation15,Citation16 In both cases, FNDC3B overexpression induced malignant transformation based on induction of anchorage-independent growth in suspension culture as well as the formation of morphologically altered foci in monolayer culture (Fig. S1). In the course of performing these experiments, we noted that FNDC3B overexpression destroyed the normal compact cuboidal structures of epithelial cell colonies and instead induced more dispersed colonies with migratory cells at the periphery (Fig. S2). These structural alterations and changes in behavior are indicative of the epithelial-to-mesenchymal transition (EMT) and led us to test whether molecular alterations that have been clearly linked to EMT are found in FNDC3B transformed cells. Loss of E-cadherin expression and induction of vimentin expression have been extensively linked to EMT,Citation17 and we examined expression of these EMT-associated proteins in three different FNDC3B-transformed epithelial cells. In hepatocytes, mammary epithelial and kidney epithelial cells, FNDC3B transformation caused induction of the mesenchymal marker vimentin and suppression of the epithelial marker E-cadherin (Fig. S2). This result places FNDC3B in the category of oncogenes that induce EMT (e.g., RAS) as opposed to those that do not (e.g., CCND1).Citation18,Citation19
Golgi localization of FNDC3B protein.
Determining the subcellular localization of FNDC3B protein could provide clues to its oncogenic function. Based on its primary structure, FNDC3B is predicted to be a membrane-associated protein. Although this large 1,024 amino acid protein lacks a signal sequence, it has a hydrophobic C terminus that is similar to those found in tail-anchored proteins such as Bcl-2 family members.Citation20 Tail-anchored proteins localize to a variety of intracellular membranes including the endoplasmic reticulum, the mitochondrial outer membrane and the Golgi. In many cases, the specific localization is dependent upon targeting sequences in the cytoplasmic portion of the proteins.Citation20 The majority of the presumptive cytoplasmic portion of FNDC3B is comprised of nine fibronectin type-III domains of approximately 100 amino acids each. At its N terminus, it contains a proline-rich motif that is predicted to bind WW-domains.
We investigated its localization with two different approaches, one using GFP tagging and the other by developing specific antibodies that could be used to detect endogenous protein using immunofluorescence. GFP-protein fused to just the C-terminal tail of FNDC3B appeared to localize to the endoplasmic reticulum when expressed in hepatocytes (), as has been observed with other C-terminal tail proteins.Citation20 However, GFP fused to full-length FNDC3B appeared instead to localize to the Golgi (). Mutational and deletion analysis revealed that several different regions of FNDC3B were required for Golgi localization, including the extreme C-terminal lysine () and the first 62 N-terminal amino acids (Fig. S3). Additionally, the localization function of the transmembrane region of the C-terminal tail could not simply be replaced by another membrane localization sequence (CAAX box) (Fig. S3).
Figure 2. FNDC3B protein localizes to the Golgi network. (A) Fluorescent images of subcellular localization of FNDC3B-GFP fusion protein in C-terminal tail (GFP-C-terminal tail), full length protein (GFP-FNDC3B) and deleted C-terminal lysine (GFP-FNDC3B-ΔK) transfected PHM1 cells. See also Figure S2. (B) Co-staining of FNDC3B (red) and Golgi marker 58K (green) in HCC cell line HepG2. Double positive FNDC3B+ 58K+ cells are shown in a merged view (yellow). (C) Confocal microscopy imaging of endogenous FNDC3B (red) localization in HCC cell line HepG2. (D) AKT, pAKTS473, E-cadherin and Vimentin protein levels in untransfected (U.T), control vector (Vector), FNDC3B overexpressing (FNDC3B) and deleted C-terminal lysine overexpressing (FNDC3B-ΔK) detected by immunoblotting. β-actin was used as a loading control. See also Fig S4.
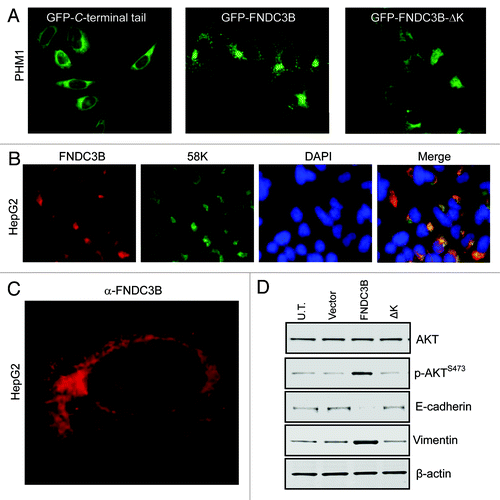
The proposed Golgi localization of FNDC3B was confirmed using specific antibodies that we developed to determine the subcellular location of endogenous FNDC3B in human HCC cells. FNDC3B was found to co-localize in two different HCC cell lines with the 58K Golgi marker formiminotransferase cyclodeaminaseCitation21 (; Fig. S4). The 58K Golgi marker is known to also associate with earlier compartments of the secretory pathway, and indeed, confocal microscopy imaging of endogenous FNDC3B localization does suggest that the protein can localize to both Golgi and cis-Golgi networks with a perinuclear localization ().
As discussed in the section below, overexpression of FNDC3B induces activation of Akt. We also observed that GFP fused to full-length FNDC3B activates, but that the mislocalized mutant missing the C-terminal lysine does not (). Similarly, full-length FNDC3B GFP-fusion protein can induce vimentin expression and cause loss of E-cadherin expression, while the mislocalized C-terminal lysine mutant cannot (). In fact, all of the mislocalized FNDC3B mutants failed to induce these molecular alterations (Fig. S5), indicating that the correct localization of FNDC3B is critical to its oncogenic function.
FNDC3B overexpression activates several cancer pathways.
The phosphorylation status of oncogenes, tumor suppressors and key signaling proteins within a pathway is often a strong indicator of pathway activity. We examined the effect of FNDC3B overexpression on 54 different phosphosites covering 40 different proteins using the KPSS 1.3 screen (www.kinexus.ca). Six proteins displayed consistent increases in protein phosphorylation, including PKCα, Rb1, p85 S6 kinase α, Raf1, Mek1/2 and Akt1 (Fig. S6). There were no decreases in protein phosphorylation of similar magnitude (Fig. S6 and Table S1).
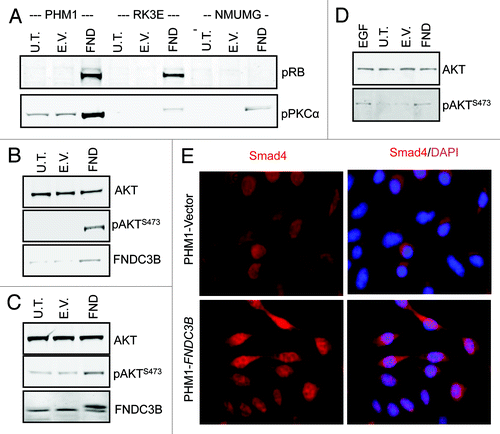
These results indicated that four of the most critical pathways involved in cancer were activated by overexpression of FNDC3B, including the RB-pathway (Rb1), the PI3-kinase/Akt pathway (Akt1, its downstream target p85 S6 kinase α and PKCα which is phosphorylated by pathway member PDK1), the Raf/Mek axis (Raf1 and Mek1/2). To confirm these findings, we analyzed the phosphorylation status of Rb1, PKC and Akt in three different epithelial cells transformed by FNDC3B overexpression. In all three cases, FNDC3B overexpression induced increased phosphorylation of these proteins, confirming that activation of the Rb and PI3-kinase/Akt pathways was a general feature of FNDC3B transformation (). Additionally, even though Smad1/2/8 were only activated in one of the two biological replicates, we thought that this data plus the strong induction of EMT suggested that FNDC3B overexpression was activating the TGF pathway. Indeed, we found that FNDC3B overexpression activated the TGFβ pathway, as it induces the nuclear translocation of the key pathway transcription factor Smad4 ().
Figure 3.FNDC3B overexpression activates multiple signaling pathways. (A) pRB and PKC-α protein levels in untransfected (U.T.), control vector (E.V.) and FNDC3B overexpressing (FND) PHM1 (left), RK3E (middle) and NMuMG (right) cells were detected by immunoblotting. (B) AKT, pAKTS473 and FNDC3B protein levels in untransfected (U.T.), control vector (E.V.) and FNDC3B overexpressing (FND) NMuMG cells were detected by immunoblotting. β-actin was used as a loading control. (C) AKT, pAKTS473 and FNDC3B protein levels in untransfected (U.T.), control vector (E.V.) and FNDC3B overexpressing (FND) RK3E cells were detected by immunoblotting. β-actin was used as a loading control. (D) AKT and pAKTS473 protein levels EGF treated (treatment with 10ng/ml recombinant EGF for 10 min) in untransfected (U.T.), control vector (E.V.) and FNDC3B overexpressing (FND) PHM1 cells were detected by immunoblotting. (E) Immunofluorescence analysis of Smad4 (red) in control vector (PHM1-Vector) or FNDC3B overexpressing (PHM1-FNDC3B). Smad4/DAPI is shown in a merged view (pink).
FNDC3B overexpression’s effects on the TGF® pathway.
We wanted to determine for one of the three cancer pathways the point at which FNDC3B overexpression induced activation. Based on its relative simplicity in terms of components, we chose the TGFβ pathway. Both the nuclear translocation of Smad4 and the increased phosphorylation of Smad1/5/9 suggested that the TGFβ receptor TGFBR1, which is the upstream kinase of Smad1/5/9, was activated in FNDC3B-transformed cells. To test this directly, we immunoprecipitated TGFBR1 from both control and FNDC3B-transformed cells and incubated it with Smad3 peptide substrate, which was then immunoblotted with anti-phospho Smad3 antibodies to determine whether it had become phosphoryated. As predicted, we found that two independent FNDC3B transformants showed clear evidence of activation of TGFBR1, whereas neither control groups showed appreciable activation (Fig. S7).
We then explored how FNDC3B overexpression could activate the cell-surface receptor TGFBR1. Clearly, one possible mechanism would be increased production of ligands for TGFBR1, for which there are three, TGFβ1–3. We examined the effect of FNDC3B overexpression on mRNA expression of these three ligands as well as 30 other ligands in the related GDF family of ligands as well as the EGF and FGF families. All three TGFBR1 ligands were clearly upregulated in cells overexpressing FNDC3B (Fig. S7).
Based on the Golgi localization of FNDC3B, we also wanted to explore whether its overexpression might affect the localization of TGFBR1, which, like many other receptors, is partitioned between the plasma membrane and endosomes.Citation22 Additionally, another Golgi protein with established oncogenic function, MGAT5, is a b1,6N-acetylglucosaminyltransferase that produces branched complex N-glycans on cell surface glycoproteins (including TGFBR1), thereby promoting their retention on the plasma membrane.Citation23 We investigated the amount of TGBR1 on the cell-surface of live cells using flow cytometry and found that FNDC3B overexpressing cells displayed a marked increase in cell-surface TGFBR1 (Fig. S7). Using immunofluorescence microscopy, we confirmed this result and, in addition, found that when cells were permeabilized, the total amount of detectable TGFBR1 was the same in controls cells and FNDC3B overexpressing cells (Fig. S8).
Selective tumor maintenance function for FNDC3B in amplified cancer cells.
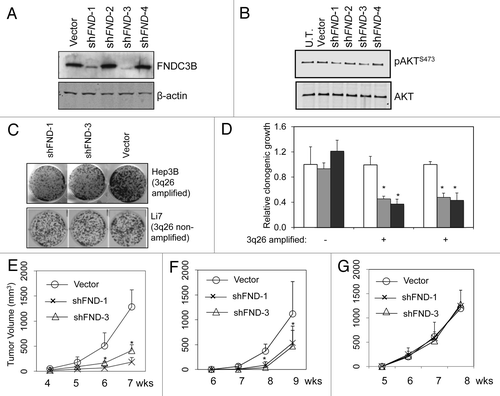
Oncogenic drivers need not be required for ongoing tumor maintenance. For example, MAD2 overexpression can initiate a wide variety of tumors in transgenic mice, but its continued expression is not required for continued tumor growth and progression.Citation24 To determine if overexpression of FNDC3B plays a tumor maintenance role in cancer cell lines in which it is amplified and overexpressed, we first designed a set of shRNAs directed against FNDC3B using the BiopredSi algorithm.Citation25 Two out of four of these shRNAs were effective at reducing FNDC3B protein levels in the PRC5 cell line, which is amplified for FNDC3B (). These two effective shRNAs were capable of reducing the level of activated Akt in PRC5, which was the first indication that FNDC3B overexpression plays a role in maintenance of the transformed state (). We then used these shRNAs to test whether FNDC3B overexpression could influence the clonogenicity of two HCC cell lines that were amplified for FNDC3B (PRC5, Hep3B) compared with a control single-copy FNDC3B HCC cell line (Li7). Clonogenicity is one of the in vitro cell culture properties that is correlated with tumorigenicityCitation26 and is particularly relevant to predicting tumor maintenance function in human HCC cell lines.Citation3 We found that the two independent shRNAs suppressing FNDC3B expression were both capable of suppressing clonogenic growth to approximately 40–50% of control levels in the two cell lines amplified for FNDC3B, but they had no effect on the clonogenic growth of the control single-copy FNDC3B HCC cell line (). Similarly, RNAi knockdown of FNDC3B expression suppressed the tumor forming ability of the two cell lines amplified for FNDC3B but had no effect on the tumorgenicity of the single-copy FNDC3B HCC cell line (). These results establish that FNDC3B amplification and overexpression is not only an oncogenic driver, but that it is required for full maintenance of the tumorigenic phenotype. Interestingly, as has been observed several times with other amplified oncogenes, the tumor maintenance function provided by expression of FNDC3B is required selectively in cancer cells that harbor the 3q or 3q26 amplicon.
Figure 4.FNDC3B overexpression contributes to tumor maintenance in 3q amplified cell lines. Panel (A) FNDC3B protein levels in PRC5 cell line (3q-amplified) following stable transfection with control vector or shRNAs targeting FNDC3B (shFND1,2,3 and 4) were detected by immunoblotting. shFND1 and shFND3 are effective in silencing FNDC3B protein expression. (B) AKT and pAKTS473 protein levels in PRC5 cell line before (U.T.) or following stable transfection with control vector or shRNAs targeting FNDC3B (shFND1,2,3 and 4) were detected by immunoblotting. (C) Clonogenicity of HepG2 cells (3q-amplified) and Li7 cells (single copy 3q) transfected with shRNAs targeting FNDC3B (shFND-1 and shFND-3) or control vector. (D) Quantification of clonogenicity in three liver cancer cell lines transfected with shRNAs targeting FNDC3B (shFND-1 and shFND-3) or control vector. Results with cells transfected with control vector are shown in white bars, shFND-1 results are shown in light gray bars and shFND-3 results are shown in dark gray bars. Error bars denote ± SD (E) Subcutaneous growth of 5x106 PRC5 cells transfected with control vector or shRNAs targeting FNDC3B (shFND-1 and shFND-3) over a period of seven weeks. n = 10 injections per group. Error bars denote ± SD (F) Subcutaneous growth of 5x106 HepG2 cells transfected with control vector or shRNAs targeting FNDC3B (shFND-1 and shFND-3) over a period of nine weeks. n = 10 injections per group. Error bars denote ± SD (G) Subcutaneous growth of 5x106 Li7 cells transfected with control vector or shRNAs targeting FNDC3B (shFND-1 and shFND-3) over a period of eight weeks. n = 10 injections per group. Error bars denote ± SD.
Discussion
In this work we’ve shown that FNDC3B is an amplified oncogene that is part of a larger amplicon encompassing several genes and often the entire chromosomal arm of 3q. FNDC3B is genetically altered by amplification and resultant overexpression in a substantial portion of human cancers, over or close to 50% of esophageal, lung squamous and ovarian cancers. We determined that the FNDC3B protein product, which contains a hydrophobic C-terminal tail similar to those found in Bcl-2 type proteins, localizes to the Golgi, and this localization is essential for its transforming function. We also determined that FNDC3B amplification is involved in tumor maintenance in addition to its ability promote cancer, and that tumor cells harboring FNDC3B amplification selectively require its function for clonogenicity and tumor formation.
We initiated this study in part to explore the potential of specific approaches to understand the oncogenic mechanism of newly validated cancer genes, with the long-term goal of developing scalable informative approaches to this problem. Although not part of the original plan, we noticed that overexpression of FNDC3B induced EMT. Given the central role that EMT plays in cancer biology and based on the relative ease with which EMT detection could be scaled up for higher throughput analysis, we believe that EMT characterization should be a standard part of novel cancer gene characterization.
One of the approaches we set out to evaluate was determining the subcellular localization of FNDC3B protein. By both GFP tagging and by use of specific antibodies we developed to FNDC3B, we determined that it localized to the Golgi. Although developing and validating specific antibodies is not readily scalable to large numbers of proteins, GFP or other epitope tagging methods are scalable, particularly since large ORFeome collections have been developed that could be used for this purpose.Citation27 However, determining the cellular localization of FNDC3B did not help decipher its oncogenic function, which involves the activation of multiple cancer pathways, none of which were mechanistically tied into its Golgi localization. Indeed, it is not clear what impact previous genome-scale efforts at subcellular localization have had in general,Citation28 and it could be that subcellular localization, while certainly important to fully understand the function of a protein, does not immediately contribute in a significant way to the understanding of newly discovered and functionally validated cancer genes.
The second specific approach we took to understand the oncogenic mechanism of FNDC3B overexpression was to profile the pathway activation status of key cancer pathways using kinase profiling by multiplex immunoblotting with phosphosite specific antibodies.Citation29 This analysis led directly to the realization that FNDC3B overexpression activates critical cancer pathways, including the PI3-kinase/Akt pathway and the Rb1 pathway. The benefits of this approach are that it directly ties the oncogenic function of new cancer genes to previously established cancer pathways and builds upon the vast knowledge of the molecular biology of cancer pathways that has accumulated over the past 30 years.Citation30 Additionally, the phosphorylation status of specific proteins is often plays a key role in the mechanism by which the activity of the pathway is controlled. Phosphorylation of Rb1 promotes its dissociation from E2F-DP heterodimers, allowing E2F to be free to transcriptionally activate cell cycle progression factors such as cyclins E and A and proliferating cell nuclear antigen.Citation31 Phosphorylation of the C terminus of Smads1/5/8 by TGFBR1 allows them to form a transcriptionally active complex with Smad4 and induce transcription of target genes.Citation32 One weakness with kinase profiling with phosphospecific antibodies is that it cannot directly readout the activity of certain cancer pathways, e.g., the p53 pathway, or the activity of Myc. In these cases, well-validated transcriptional reporter genes might be used to accurately reflect pathway activity, for example, DNA damage induction of expression of CIP1/WAF1, GADD45 and MDM2 mRNAs for p53 activity.Citation33 Another approach to measuring pathway activation that is scalable and could be useful is genome-wide expression profiing and the use of gene expression signatures or gene-set enrichment analysis as surrogates for pathway status.Citation34,Citation35 However, genes activated by a given cancer pathway can vary substantially from cell type to cell type, and additional work will be required to develop robust transcriptional biomarkers for each cancer pathway.Citation36 Regardless, determining the effect of previously uncharacterized cancer genes on established cancer pathways will clearly be beneficial as a first step in characterization, and both phosphosite profiling and transcriptional readouts can be scaled up for higher throughput analysis.
We also investigated the phenotypic effects of reversing the genomic activation of FNDC3B by suppressing its overexpression with RNAi. This addresses whether the driver gene is suitable as a therapeutic target, but it can also be used to provide complimentary analyses of the influence of a cancer gene on known cancer pathways. With FNDC3B, RNAi-mediated suppression reduced the level of activation of Akt, consistent with the forward genetic experiments showing that its overexpression activated Akt. This loss of Akt activation correlated with a > 50% reduction in clonogenicity and tumor formation in a xenograft assay. Interestingly, not all cancer cells displayed this dependency upon FNDC3B expression: Li7, which is single-copy for FNDC3B and expresses relatively low levels, showed no change in clonogenicity or tumor formation when FNDC3B expressed was further reduced by shRNAs. As has been observed often with amplified oncogenes, including ERBB2, MET, FGFR1, FGFR2, FGF19 and CCND1, only cancer cells that are amplified for the oncogene require its continued expression for a fully tumorigenic phenotype.Citation3,Citation37-Citation40
However, the previous examples of selective amplified oncogene dependency have all concerned high-level, focal amplification events. FNDC3B is rarely focally amplified and instead is more commonly amplified as a broad chromosome arm gain (3q) of a large chromosomal region (e.g., all of 3q26). Since every focal amplicon that has been closely examined has turned out to have two or more linked driver genes,Citation3,Citation9,Citation38,Citation41 it is almost a certainty that larger amplicons contain several driver genes. In terms of either therapeutic targeting or understanding the role of FNDC3B amplification in human cancer, the real question then becomes what are all the amplified driver genes on 3q, and, together, what is their impact on cancer biology, and what tumor cell dependencies do they create? Being able to address this important question will require the development of different approaches to functional genomics and cancer biology.
Materials and Methods
Cell lines, tissue culture and transformation assays.
Hepatocellular carcinoma human cell lines (Huh7, Hep3B, PRC5 and Li7), telomerase-immortalized human liver epithelial cells (NeHepLxHT), rat kidney epithelial cells (RK3E) and murine mammary epithelial cells (NMuMG) were obtained from ATCC or the Japanese Collection of Research Bioresources (JCRB) and grown as recommended by the supplier. PHM1 cells are mouse liver cells obtained from Scott Lowe’s laboratory and were cultured as previously described in reference Citation4. Cell lines overexpressing FNDC3B were generated by retroviral transfection, and shRNAs targeting FNDC3B were cloned into the MLP retroviral vector using previously described methods.Citation3 Tumorigenicity assays and transformations assays were performed as previously described in reference Citation3.
Immunoblotting, immunoprecipitation, immunofluorescence and FACS analysis.
FNDC3B peptide (DQIPLELPPLLNGE) antibody was generated by Open Biosystems (Thermo Fischer). Vendors for other antibodies were Abcam for E-cadherin, vimentin, Smad3, pSmad3 and β-actin; Cell Signaling for AKT and pAKT and Santacruz for TGFBR1. Fluorescently-conjugated secondary antibodies were used and signals quanitified with a Li-Cor Odyssey scanner. Precleared cell lysates in RIPA buffer (1 mg total protein) were immunoprecipitated with 1 ∝g of antibodies against TGFBR1 with 30 ∝L of protein G Sepharose beads. Immunoprecipitated proteins were incubated with 100 ng of Smad3 substrate followed by immunoblotting with pSmad3 antibody. For immunofluorescence, 5 x 104 PHM1 cells expressing FNDC3B or control vector were plated on 0.2% gelatin coated coverslips (Corning No. 1½) in 6-well plates and grown overnight in reduced serum conditions (1% serum), washed with PBS, fixed using 4% paraformaldehyde (Sigma) for 15 min at room temperature, permeabilized with 0.1% Triton-X, blocked in 1% BSA (Aurion)/10% serum for 6 h, incubated in the primary antibody overnight at 4°C and processed for standard indirect immunofluorescence using fluorescent secondary antibodies (Invitrogen). The slides were examined using a Zeiss Confocal Microscope 510. For FACS analysis, PHM1 cells expressing FNDC3B or control vector were washed, trypsinized and stained with TGFBR1 antibody (Abcam; 1:200) followed by staining with secondary antibody, Alexa Fluor‑633 (Invitrogen). The cells were analyzed on BD-LSRII FACS analyzer and data was processed using FlowJo FACS analysis software.
Additional material
Download Zip (3 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank S. Hearn for assistance with microscopy; L. Bianco for help with animals; J. Duffy for his help preparing figures; C. Johns for microarray hybridizations; K.L. Scott, L. Chin and members of the Powers laboratory for stimulating discussions. This work was supported by NIH grant CA124648 (S.P.).
References
- Stratton MR. Exploring the genomes of cancer cells: progress and promise. Science 2011; 331:1553 - 8; http://dx.doi.org/10.1126/science.1204040; PMID: 21436442
- Lander ES. Initial impact of the sequencing of the human genome. Nature 2011; 470:187 - 97; http://dx.doi.org/10.1038/nature09792; PMID: 21307931
- Sawey ET, Chanrion M, Cai C, Wu G, Zhang J, Zender L, et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by Oncogenomic screening. Cancer Cell 2011; 19:347 - 58; http://dx.doi.org/10.1016/j.ccr.2011.01.040; PMID: 21397858
- Zender L, Xue W, Zuber J, Semighini CP, Krasnitz A, Ma B, et al. An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell 2008; 135:852 - 64; http://dx.doi.org/10.1016/j.cell.2008.09.061; PMID: 19012953
- Scott KL, Nogueira C, Heffernan TP, van Doorn R, Dhakal S, Hanna JA, et al. Proinvasion metastasis drivers in early-stage melanoma are oncogenes. Cancer Cell 2011; 20:92 - 103; http://dx.doi.org/10.1016/j.ccr.2011.05.025; PMID: 21741599
- Tominaga K, Kondo C, Johmura Y, Nishizuka M, Imagawa M. The novel gene fad104, containing a fibronectin type III domain, has a significant role in adipogenesis. FEBS Lett 2004; 577:49 - 54; http://dx.doi.org/10.1016/j.febslet.2004.09.062; PMID: 15527760
- Nishizuka M, Kishimoto K, Kato A, Ikawa M, Okabe M, Sato R, et al. Disruption of the novel gene fad104 causes rapid postnatal death and attenuation of cell proliferation, adhesion, spreading and migration. Exp Cell Res 2009; 315:809 - 19; http://dx.doi.org/10.1016/j.yexcr.2008.12.013; PMID: 19138685
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646 - 74; http://dx.doi.org/10.1016/j.cell.2011.02.013; PMID: 21376230
- Kendall J, Liu Q, Bakleh A, Krasnitz A, Nguyen KC, Lakshmi B, et al. Oncogenic cooperation and coamplification of developmental transcription factor genes in lung cancer. Proc Natl Acad Sci U S A 2007; 104:16663 - 8; http://dx.doi.org/10.1073/pnas.0708286104; PMID: 17925434
- Shayesteh L, Lu Y, Kuo WL, Baldocchi R, Godfrey T, Collins C, et al. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet 1999; 21:99 - 102; http://dx.doi.org/10.1038/5042; PMID: 9916799
- Nanjundan M, Cheng KW, Zhang F, Lahad J, Kuo WL, Schmandt R, et al. Overexpression of SnoN/SkiL, amplified at the 3q26.2 locus, in ovarian cancers: a role in ovarian pathogenesis. Mol Oncol 2008; 2:164 - 81; http://dx.doi.org/10.1016/j.molonc.2008.05.001; PMID: 19383336
- Linxweiler M, Linxweiler J, Barth M, Benedix J, Jung V, Kim YJ, et al. Sec62 bridges the gap from 3q amplification to molecular cell biology in non-small cell lung cancer. Am J Pathol 2012; 180:473 - 83; http://dx.doi.org/10.1016/j.ajpath.2011.10.039; PMID: 22197383
- Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet 2009; 41:1238 - 42; http://dx.doi.org/10.1038/ng.465; PMID: 19801978
- Pollack JR, Sørlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, et al. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc Natl Acad Sci U S A 2002; 99:12963 - 8; http://dx.doi.org/10.1073/pnas.162471999; PMID: 12297621
- Kim RH, Wang D, Tsang M, Martin J, Huff C, de Caestecker MP, et al. A novel smad nuclear interacting protein, SNIP1, suppresses p300-dependent TGF-beta signal transduction. Genes Dev 2000; 14:1605 - 16; PMID: 10887155
- Kolligs FT, Kolligs B, Hajra KM, Hu G, Tani M, Cho KR, et al. gamma-catenin is regulated by the APC tumor suppressor and its oncogenic activity is distinct from that of beta-catenin. Genes Dev 2000; 14:1319 - 31; PMID: 10837025
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119:1420 - 8; http://dx.doi.org/10.1172/JCI39104; PMID: 19487818
- Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J, et al. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol 2002; 156:299 - 313; http://dx.doi.org/10.1083/jcb.200109037; PMID: 11790801
- Tobin NP, Sims AH, Lundgren KL, Lehn S, Landberg G. Cyclin D1, Id1 and EMT in breast cancer. BMC Cancer 2011; 11:417; http://dx.doi.org/10.1186/1471-2407-11-417; PMID: 21955753
- Wattenberg B, Lithgow T. Targeting of C-terminal (tail)-anchored proteins: understanding how cytoplasmic activities are anchored to intracellular membranes. Traffic 2001; 2:66 - 71; http://dx.doi.org/10.1034/j.1600-0854.2001.20108.x; PMID: 11208169
- Gao YS, Alvarez C, Nelson DS, Sztul E. Molecular cloning, characterization, and dynamics of rat formiminotransferase cyclodeaminase, a Golgi-associated 58-kDa protein. J Biol Chem 1998; 273:33825 - 34; http://dx.doi.org/10.1074/jbc.273.50.33825; PMID: 9837973
- Chen YG. Endocytic regulation of TGF-beta signaling. Cell Res 2009; 19:58 - 70; http://dx.doi.org/10.1038/cr.2008.315; PMID: 19050695
- Mendelsohn R, Cheung P, Berger L, Partridge E, Lau K, Datti A, et al. Complex N-glycan and metabolic control in tumor cells. Cancer Res 2007; 67:9771 - 80; http://dx.doi.org/10.1158/0008-5472.CAN-06-4580; PMID: 17942907
- Sotillo R, Hernando E, Díaz-Rodríguez E, Teruya-Feldstein J, Cordón-Cardo C, Lowe SW, et al. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 2007; 11:9 - 23; http://dx.doi.org/10.1016/j.ccr.2006.10.019; PMID: 17189715
- Huesken D, Lange J, Mickanin C, Weiler J, Asselbergs F, Warner J, et al. Design of a genome-wide siRNA library using an artificial neural network. Nat Biotechnol 2005; 23:995 - 1001; http://dx.doi.org/10.1038/nbt1118; PMID: 16025102
- Barrett JC, Crawford BD, Mixter LO, Schechtman LM, Ts’o PO, Pollack R. Correlation of in vitro growth properties and tumorigenicity of Syrian hamster cell lines. Cancer Res 1979; 39:1504 - 10; PMID: 427793
- Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, et al. A public genome-scale lentiviral expression library of human ORFs. Nat Methods 2011; 8:659 - 61; http://dx.doi.org/10.1038/nmeth.1638; PMID: 21706014
- Kumar A, Agarwal S, Heyman JA, Matson S, Heidtman M, Piccirillo S, et al. Subcellular localization of the yeast proteome. Genes Dev 2002; 16:707 - 19; http://dx.doi.org/10.1101/gad.970902; PMID: 11914276
- Pelech S. Tracking cell signaling protein expression and phosphorylation by innovative proteomic solutions. Curr Pharm Biotechnol 2004; 5:69 - 77; http://dx.doi.org/10.2174/1389201043489666; PMID: 14965210
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004; 10:789 - 99; http://dx.doi.org/10.1038/nm1087; PMID: 15286780
- Manning AL, Dyson NJ. pRB, a tumor suppressor with a stabilizing presence. Trends Cell Biol 2011; 21:433 - 41; http://dx.doi.org/10.1016/j.tcb.2011.05.003; PMID: 21664133
- Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003; 113:685 - 700; http://dx.doi.org/10.1016/S0092-8674(03)00432-X; PMID: 12809600
- O’Connor PM, Jackman J, Bae I, Myers TG, Fan S, Mutoh M, et al. Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res 1997; 57:4285 - 300; PMID: 9331090
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102:15545 - 50; http://dx.doi.org/10.1073/pnas.0506580102; PMID: 16199517
- Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 2006; 439:353 - 7; http://dx.doi.org/10.1038/nature04296; PMID: 16273092
- Györffy B, Schäfer R. Biomarkers downstream of RAS: a search for robust transcriptional targets. Curr Cancer Drug Targets 2010; 10:858 - 68; http://dx.doi.org/10.2174/156800910793357916; PMID: 20718707
- Turner N, Lambros MB, Horlings HM, Pearson A, Sharpe R, Natrajan R, et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene 2010; 29:2013 - 23; http://dx.doi.org/10.1038/onc.2009.489; PMID: 20101236
- Kao J, Pollack JR. RNA interference-based functional dissection of the 17q12 amplicon in breast cancer reveals contribution of coamplified genes. Genes Chromosomes Cancer 2006; 45:761 - 9; http://dx.doi.org/10.1002/gcc.20339; PMID: 16708353
- Lutterbach B, Zeng Q, Davis LJ, Hatch H, Hang G, Kohl NE, et al. Lung cancer cell lines harboring MET gene amplification are dependent on Met for growth and survival. Cancer Res 2007; 67:2081 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-06-3495; PMID: 17332337
- Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res 2010; 70:2085 - 94; http://dx.doi.org/10.1158/0008-5472.CAN-09-3746; PMID: 20179196
- Guan Y, Kuo WL, Stilwell JL, Takano H, Lapuk AV, Fridlyand J, et al. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin Cancer Res 2007; 13:5745 - 55; http://dx.doi.org/10.1158/1078-0432.CCR-06-2882; PMID: 17908964