Abstract
The biological aging process is commonly associated with increased risk of cardiovascular diseases. Several theories have been put forward for aging-associated deterioration in ventricular function, including attenuation of growth hormone (insulin-like growth factors and insulin) signaling, loss of DNA replication and repair, histone acetylation and accumulation of reactive oxygen species. Recent evidence has depicted a rather unique role of autophagy as another important pathway in the regulation of longevity and senescence. Autophagy is a predominant cytoprotective (rather than self-destructive) process. It carries a prominent role in determination of lifespan. Reduced autophagy has been associated with aging, leading to accumulation of dysfunctional or damaged proteins and organelles. To the contrary, measures such as caloric restriction and exercise may promote autophagy to delay aging and associated comorbidities. Stimulation of autophagy using rapamycin may represent a novel strategy to prolong lifespan and combat aging-associated diseases. Rapamycin regulates autophagy through inhibition of the nutrient-sensing molecule mammalian target of rapamycin (mTOR). Inhibition of mTOR through rapamycin and caloric restriction promotes longevity. The purpose of this review is to recapitulate some of the recent advances in an effort to better understand the interplay between rapamycin-induced autophagy and decelerating cardiovascular aging.
Keywords: :
Background
Human life expectancy has risen significantly over the last century, and consequently, the elderly represent the fastest-growing segment of the population. It is projected that the elderly population (> 65 y of age) will increase from 10% of the total population in 2000 to ~22% by 2050 and 32% in 2100.Citation1 Not surprisingly, the prevalence of aging-related chronic diseases is on the horizon. Aging is a major independent risk factor for cardiovascular disease, the leading cause of morbidity and mortality in the United States and rest of the world.Citation2-Citation4 The prevalence of cardiovascular disease increases rapidly with age and ranks the highest in the elderly population.Citation5 Together with the adverse health implications, cardiovascular disease also represents a huge economic burden on the society.Citation6,Citation7 It is therefore pertinent to understand the molecular mechanisms behind declined cardiac function with biological aging, with an ultimate goal to develop effective strategies to manage or halt the progression of cardiac aging.
Aging is a rather complicated pathophysiological process accompanied by a wide array of biological adaptations, including progressive myocardial remodeling and deteriorated cardiac reserve.Citation8-Citation11 With aging, myocardial contractile capacity progressively declines as manifested by increased left ventricular wall thickness and chamber size as well as change in diastolic filling pattern, such as prolonged diastole.Citation11,Citation12 In addition, aging leads to stiffening and loss of elasticity of the ventricular wall and coronary vasculature, possibly due to increase in after-load in the elderly.Citation10,Citation13,Citation14 This increased workload for the heart often triggers unfavorable remodeling, leading to a much greater heart mass (or cardiac hypertrophy).Citation15 Although an initial adaptive process develops, the aging heart will eventually transit from compensatory to decompensatory stage, resulting in further impairment of contractile function. In addition, aging is also associated with increased cardiomyocyte cell death via apoptosis. Furthermore, with aging there is a decline in the response of myocardium to certain endogenous autonomic (e.g., adrenergic) stimulation. Advanced age is accompanied with overt calcification and fibrosis of ventricular structure (such as free walls and valves), further deteriorating cardiac geometry and contractile function. In addition to the aforementioned physiological changes, many factors have been identified to play a role in the increased incidence of cardiovascular diseases with aging. For example, sympathetic over-activation, hypertension, dyslipidemia, oxidative stress, chronic low-grade inflammation and sustained obesity may all play a role toward compromised cardiac structure and function in the elderly.Citation11,Citation16,Citation17 However, none of these theories has been fully validated by clinical findings to explain the complex cardiac aging process.
Several theories of aging have been articulated at length elsewhereCitation18-Citation20 and, thus, will not be emphasized here. Many of these classical theories of aging have also been implicated in cardiac aging.Citation21 The “free radical theory of aging” attributes cumulative exposure of biomolecules to oxygen-derived free radical species as the cause of aging.Citation12 Indeed, the accumulation of lipofuscin pigments in the heart has been considered as the “tell-tale sign” of free radical damage to the heart.Citation15 This theory was consolidated by the evidence of extended lifespan following overexpression of endogenous antioxidants such as superoxide dismutase, catalase and metallothionein.Citation5,Citation22-Citation25 Build-up of advanced glycation end products (AGEs) associated with aging forms the basis for the “glycation theory of aging.”Citation26 AGEs may trigger oxidative stress, thus abridging the free radical theory with the glycation theory of aging.Citation27,Citation28 To maintain physiological contractile function of the heart, cardiomyocytes have to rely on a constant supply of high-energy phosphates from mitochondria.Citation29 The “mitochondrial decline theory” postulates that the aging is associated with decline in mitochondrial function (hence ATP supply), reduction in mitochondrial numbers and the presence of functionally compromised enlarged mitochondria.Citation30 Mitochondria is also the primary source of reactive oxygen species (ROS) produced by the electron transport chain during mitochondrial respiration, which triggers mitochondrial-derived (endogenous) apoptosis, recognized as a leading cause of cell death in cardiomyocytes.Citation30 Recent evidence suggested that mitochondrial expression of Nox4 a member of the NADPH oxidase family, is upregulated in aging hearts, which serves as a major source of oxidative stress to promote cardiac aging.Citation31 More evidence has implicated that compromised mitochondrial function in aging hearts may likely result from mitochondrial permeability transition pore (MPTP) opening through a NAD+-dependent deacetylase SIRT3-cyclophilin D-dependent manner.Citation32 These observations have caught much attention, as MPTP is associated with release of Ca2+ and ROS in cardiomyocytes.Citation33 Telomere shortening also contributes to cellular senescence in replicating cells, while defective telomere in cardiac stem cells has been recently identified as a useful marker of cardiac aging.Citation34,Citation35 Genome-wide transcriptome analysis suggests that age-associated alterations in transcripts govern the cardiac aging process.Citation8,Citation36 The role of epigenetic modifications (such as differential levels of DNA methylation, microRNA) or activation of certain protein kinase cascade has been implicated in cardiac aging.Citation37-Citation39 Despite this enriched knowledge of the molecular basis of cardiac aging, the precise aging process remains rather complex and cannot be explained in a unifying manner. As a result, novel molecular mechanisms are being explored, and the role of the autophagic process is gaining prominence as a converging point in the biological aging process. This mini-review will summarize some of the recent advances in the understanding of the interplay between autophagy and cardiac aging.
Autophagy is a highly conserved cytoprotective, rather than self-destructive, process involving degradation and recycling of intracellular organelles and proteins.Citation40,Citation41 A variety of cellular stress conditions such as nutrient or growth factor deprivation, ROS accumulation, aggregation of the long-lived or damaged proteins and organelles and hypoxia usually turn on autophagy. Three distinct forms of autophagy have been identified (1) microautophagy, wherein the lysosomal membrane invaginates to engulf cytosolic components, (2) chaperone-mediated autophagy, wherein chaperones, such as heat-shock proteins, help deliver macromolecules to the lysosomes via direct binding to the lysosomes through specific receptors on the lysosomal membrane and (3) macroautophagy (commonly referred to as autophagy), wherein double membrane vesicles, referred to a autophagosomes, are first formed, which eventually fuse with the lysosomes. The autophagic process is initiated with the formation of a phagophore (from the endoplasmic reticulum, mitochondria or other cellular components), which engulfs cytosolic components and organelles to from the autophagosome (). The autophagy initiation process is mediated by activation of the class III phosphatidylinositol-3-kinase (PI-3K) and beclin-1, which help recruit autophagic proteins. These autophagosomes then undergo an elongation process mediated by several autophagy-related genes (ATG) that are evolutionarily conserved.Citation42 The Atg genes are also responsible for the recruitment of microtubule light chain-3 (LC3) to the autophagosome. Completion of autophagosome formation is characterized by the proteolytic cleavage of LC3, which undergoes lipidation to LC3-II, the form that associates with autophagic membrane. At this stage, the outer membrane of the autophagosome fuses with a lysosomes to form autophagolysosome. Lysosomal contents subsequently degrade the cytosolic components inside autophagosomes. In addition to the ATG-genes, the class 3 PI-3K, Beclin-1, Unc-51-like kinase (Ulk), lysosome membrane protein (LAMP-2) and a small GTPase, Rab7, are all involved in various process of autophagy.Citation17
Figure 1. Stages involved in the formation of autolysosome from phagophore for various cellular components.
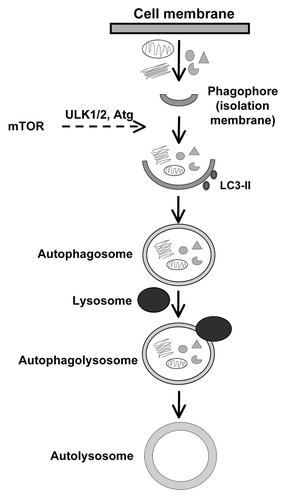
Autophagy plays a pivotal role in survival and longevity of cells and organisms. This is particularly true for the permanently differentiated cells, such as cardiomyocytes, which rely on autophagic process to remove and recycle unwanted, damaged or long-lived cellular components or proteins in order to maintain homeostasis. Although autophagy is a mechanism by which the cell destroys itself, paradoxically, it also serves as a protective function and a means of meeting metabolic requirements during conditions of energy/nutrient deficit, such as starvation, ischemia reperfusion and heart failure. Autophagy is therefore considered a “double-edged sword,” in that low baseline levels of regulated autophagy is beneficial to help maintain cardiac structure and function, although uncontrolled or excessive autophagy can cause extensive cell death and pathological state.Citation43 It has been shown that autophagy declines with aging, whereas pharmacological intervention and genetic manipulations to induce longevity are associated with autophagy induction.Citation44,Citation45 Conversely, suppression of autophagy by knockdown of autophagy related-genes induces cell death and displays the opposite consequence for lifespan. In addition, loss of autophagy was demonstrated to accelerate aging.Citation46-Citation50 In particular, lessened autophagy with aging may be responsible for aging-induced accumulation of damaged intracellular components, resulting in altered cellular homeostasis and loss of function associated with aging.Citation44,Citation49,Citation51 Autophagy is upregulated by AMP-dependent protein kinase (AMPK) while being negatively regulated by Akt and mammalian target of rapamycin (mTOR) through phosphorylation of its downstream targets, such as p70s6k.Citation52,Citation53 mTOR signaling has been considered the main driving force for aging, interruption of which triggers reduced lifespan.Citation54,Citation55 Nonetheless, the precise mechanism(s) through which autophagy contributes to longevity and improved cardiac function with aging still remains elusive. Here, we are attempting to explore some of the emerging theories and controversies involving the role of the autophagic pathway in cellular aging in general and aging-associated heart disease with a special emphasis on the mTOR signaling pathway, as it is gaining prominence as a key regulator of autophagy.
Autophagy plays a critical role in cardiac homeostasis and pathologies, including myocardial hypertrophy, diabetic and alcoholic cardiomyopathy as well as ischemic heart disease.Citation56-Citation61 Fatal hypertrophic cardiomyopathy is well recognized as a consequence of lysosomal storage defect, such as the Danon disease, a defect of LAMP-2. Tanaka and coworkers demonstrated that LAMP-2 deficiency increases mortality in mice.Citation62 Interestingly, surviving mice with LAMP-2 deficiency exhibit abnormal cardiomyocyte morphology, cardiac hypertrophy, impaired contractility and accumulation of autophagic vacuoles. These findings favored a critical role of autophagy in the maintenance of heart morphology and function, the reduction of which predisposes the heart to pathological changes. The presence of autophagic vacuoles in cardiomyocytes was also demonstrated by Kostin and coworkers in hearts from patients with idiopathic dilated cardiomyopathy.Citation63 In these patients, autophagic vacuoles were present in cardiomyocytes in association with depleted contractile elements and disintegrated nuclei distinct from apoptosis. Cells with autophagic vacuoles exhibited polyubiquitinated protein accumulation, suggesting a role of disrupted ubiquitin/proteasomal pathway with aging. Recent findings from Zheng and coworkers demonstrate that inhibition of the proteasome is sufficient to trigger autophagy in cardiomyocytes.Citation64 Vigliano and colleagues examined endomyocardial biopsy specimens from 100 patients with idiopathic dilated cardiomyopathy and advanced heart failure and found a positive correlation between autophagic vacuoles and idiopathic dilated cardiomyopathy.Citation65 It is likely that the autophagic process in the heart is upregulated under conditions of pathological stress. To better distinguish the role of autophagy in normal vs. stressed heart, Nakai and coworkers established a cardiac-specific knockout murine model of Atg5 gene essential for autophagosome formation.Citation66 In adult mice, conditional knockdown of Atg5 resulted in reduced autophagy in cardiomyocytes followed by cardiac hypertrophy, left ventricular dilation, contractile dysfunction and heart failure. Interestingly, embryonic deletion of Atg5 failed to exhibit any cardiac phenotype and the mice displayed normal survival. However, Atg5-knockout mice were much more susceptible to pressure overload-induced heart failure. It is noteworthy that both the embryonic Atg5-knockout mice and the control mice had similar extent of hypertrophy, indicating that autophagy is not critical for the development of hypertrophy.
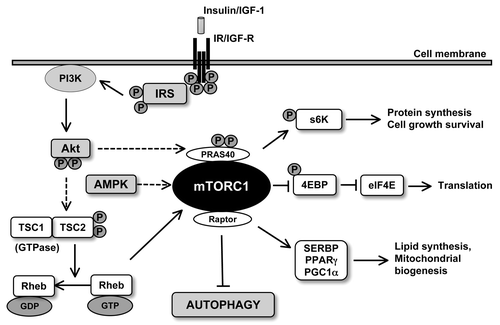
The serine threonine kinase mTOR is an evolutionarily conserved protein kinase with a central role in cell growth (hypertrophy), cell survival and autophagy in response to nutrients such as amino acids, fatty acids and growth factors, such as insulin and IGF‑1, and energy.Citation67-Citation69 mTOR exists as two complexes, as mTORC1 and mTORC2, each with distinct structures and activities. mTORC1 is a complex of mTOR with Raptor, mLST8, PRAS40 and is rapamycin-sensitive, whereas mTORC2 consists of Rictor, mSin1, mLST8 and is rapamycin-insensitive (). Of the two complexes, the rapamycin-sensitive mTORC1 has been shown to be essential for the regulation of translation and autophagy. Recent evidence has depicted that inhibition of the TOR pathway extends lifespan in yeast,Citation70 C. elegansCitation71 and Drosophila.Citation72 Harrison and coworkers found that inhibition of mTOR with rapamycin dramatically extends maximal lifespan in mice.Citation73 This study was performed in three different sites with genetically heterogeneous mice, and rapamycin was administered late in life (20 mo of age). In a follow-up study, the authors demonstrated that extension of median and maximal lifespan in mice can be achieved by providing rapamycin from 9 mo of age.Citation74 Along the same line, Chen and coworkers found elevated mTOR activity in hematopoietic stem cells from aged mice compared with young mice. Interestingly, activation of mTOR using conditional deletion of TSC1 in young mice mimicked the aging phenotypes (decrease in lymphopoiesis, impaired capacity to reconstitute hematopoietic cells) in young hematopoietic stem cells.Citation75 A series of elegant studies from Blagosklonny and coworkers has enriched our understanding of the role of mTOR in cellular senescence. In immortalized, cultured cells (e.g., human retinal pigment cells, human fibrosarcoma cells and rodent fibroblasts), inhibition of mTOR by rapamycin partly reconciled the loss of proliferative potential elicited by ectopic p21 or p16 expression or sodium butyrate-induced p21 expression.Citation76 Rapamycin successfully re-induced proliferation, only after removal of cell cycle inhibition, thus transforming “irreversible arrest into a reversible condition” without forcing cells to proliferate if the cell cycle was arrested.Citation76 These in vitro studies were extended to in vivo setting, wherein the effect of rapamycin on longevity in cancer-prone mice was tested. Interestingly, rapamycin prolonged lifespan and suppressed carcinogenesis in transgenic HER-2/neu cancer-prone mice, further substantiating the in vitro finding.Citation77 Furthermore, these authors showed that intermittent (2 weeks per month), life-long administration of rapamycin extends lifespan and inhibits age-related weight gain.Citation78 In a recent review, Menendez and coworkersCitation79 elegantly discussed the published evidence to support the hypothesis that mTOR inhibition and subsequently autophagy induction can decelerate cellular senescence by upregulating the reprogramming of somatic cells to induced pluripotent stem cells. These studies have convincingly favored a role of TORC pathway in aging and provided the rationale for mTOR inhibition in lifespan extension. However, as calorie restriction is “inconvenient” and may lead to malnutrition in the elderly population, Blagosklonny proposed the usefulness of mTOR inhibitors as “gerosuppressants” to curb senescence and senescence-related comorbidities.Citation80
Figure 2. Signaling pathways regulating the activity of mTORC1. Growth factor (such as insulin and IGF1) stimulation activates Akt which inactivates TSC2 via phosphorylation leading to the conversion of Rheb-GTP to Reb-GD preventing inhibition of mTORC1. On the other hand, low levels of ATP can result in the activation of AMPK which can inhibit mTORC1. Activated mTORC1 can induce translation of mRNA via the phosphorylation of 4E-BP1 and p70S6 kinase. mTORC1 also results in suppression of autophagy.
More recently, data from our group revealed that chronic cardiac-specific Akt activation accentuates aging-induced cardiac geometric and functional changes, possibly through deteriorated autophagy. Akt stimulates mTOR to inhibit autophagic activity. Results from our lab revealed cardiac hypertrophy, fibrosis, decreased myocardial contractility, prolonged diastole along with compromised intracellular Ca2+ release and clearance in aged mice compared with young mice, the effects of which were accentuated by chronic Akt activation. Aging enhanced Akt and mTOR phosphorylation while reducing that of PTEN, AMPK and ACC with a more pronounced response in Akt transgenic mice. Levels of beclin-1, Atg5 and LC3-II-to-LC3-I ratio were decreased in aged hearts, the effect of which, with the exception of Atg 5, was exacerbated by Akt over-activation. Levels of the autophagosome adaptor protein p62 were enhanced in aged mice, with a more pronounced rise in Akt mice. In addition, rapamycin reduced aging-induced cardiomyocyte contractile and intracellular Ca2+ dysfunction, while Akt activation suppressed autophagy in young but not aged cardiomyocytes. These data favored that notion that Akt may accentuate aging-induced cardiac geometric and contractile defects through a loss of autophagic regulation.Citation81
What makes mTOR an attractive target is the fact that many essential genes involved in lifespan control, including components of insulin/IGF signaling, FOXO transcription factors, AMPK and sitruins all converge at mTOR.Citation50 Additionally, extension of lifespan associated with caloric restriction has been shown to be mediated through downregulation of mTOR, supporting the notion of mTORC as a bona fide pharmacological target for lifespan extension. Given the essential role of mTOR in the regulation of lifespan and aging, how does rapamycin or inhibition of mTORC1 extend lifespan? It appears that a number of mediators of TORC1 may affect lifespan. Activated TORC1 mediates protein synthesis by phosphorylating a variety of downstream substrates, including S6Kinase (S6K) and the eukaryotic initiation factor 4E binding protein 1 (4EBP1) eIF4E involved in transcription and translation processes (). It may therefore be argued that mTORC1 inhibition extends lifespan through inhibition of protein synthesis. Interestingly, deletion of S6K1, the downstream effector of mTOR, extends lifespan and resists aging-related pathologies.Citation82 Conversely, rapamycin fails to extend lifespan in flies overexpressing S6 kinase.Citation83 Furthermore, inhibition of the translation initiation factor eIF4E may extend lifespan in C. elegans.Citation84 These pieces of evidence suggest that protein synthesis regulated by TORC1 signaling seems to play an important role in the aging process, while its inhibition mediates lifespan extension. An alternative explanation would be the autophagic pathway that is inhibited TORC1.Citation85,Citation86 Recent studies have shown that mTORC1 phosphorylates the serine thyronine kinase unc-51-like kinase 1 (ULK1 or Atg1), an initiator of autophagy ().Citation87,Citation88 Treatment with rapamycin upregulates Atg1 and promotes autophagy, even in the presence of a surplus of nutrients, suggesting a direct link between mTORC1 and autophagy induction. In addition to early stages of autophagy, mTORC also contributes to late stages of recycling lysosomes.Citation86
Caloric restriction without malnutrition has been shown to slow aging, extend lifespan and counter age-related diseases in a variety of species.Citation89-Citation91 Autophagy helps in recycling intracellular constituents to provide amino acids during nutrient deprivation. Interestingly, recent studies by Wohlgemuth and coworkers failed to observe any alteration of autophagy in aging hearts, whereas age combined with calorie restriction resulted in elevated autophagy, indicating that caloric restriction may be responsible for its beneficial effects through autophagy induction in the heart.Citation92 Shinmura and coworkers recently demonstrated that the beneficial effects of caloric restriction on cardiac dysfunction associated with aging may be a consequence of mTOR suppression and autophagy induction.Citation93 Not only can autophagy be an essential mediator for caloric restriction-elicited beneficial cardiac response, it also participates in the neuroendocrine regulation of the satiety hormone, leptin.Citation94 Pharmacological autophagy induction by rapamycin, spermidine and resveratrol may reduce serum leptin levels, whereas genetic inactivation of leptin promotes autophagy in peripheral tissues, including skeletal muscle, heart and liver. Paradoxically, administration of recombinant leptin triggered autophagy, the effect of which was mediated through AMPK activation and mTOR inhibition. These findings depict an important role of leptin in the neuroendocrine control of autophagy, possibly en route to leptin-induced regulation of food intake.Citation94
Autophagy is also critical for energy regulation and has been demonstrated to play an important role in lipolysis, a pathway termed as macrolipophagy.Citation95 Decreased autophagy during aging has been attributed to lipid accumulation in metabolic syndrome and aging.Citation44 Similar to the hepatic tissues, autophagic pathway also plays a critical role in the formation of lipid droplets in the heart.Citation96 Inhibition of autophagy dramatically increases the build-up of triglycerides and lipid droplets, indicating a potential role of autophagy in pathologies associated with lipid accumulation.Citation95 Therefore it is tempting to hypothesize that increased autophagy-mediated lipolysis may change bioenergetics and substrate availability in the heart, which may restore the use of fatty acids as the primary source of energy in the heart under stress conditions.
How do we fit the mTORC-senescence paradigm in the aging heart? Unfortunately, not many reports are currently available to address the role of mTORC in cardiac aging. Shinmura and coworkers examined the effect of long-term caloric restriction on cardiac senescence and found that caloric restriction inhibits age-associated decline in diastolic function in the heart.Citation93 Interestingly, caloric restriction was associated with the suppression of mTOR and elevation of autophagic response the heart, suggesting a pivotal role of mTOR suppression-mediated autophagy induction in caloric restriction-elicited retardation of cardiac senescence. Inuzuka and coworkers found that inhibition of PI-3K preserved cardiac function, enhanced autophagy and inhibited senescence markers in murine hearts.Citation97 Furthermore, inhibition of mTOR with rapamycin prevented lipofuscin accumulation in aged murine hearts. Shinde and coworkers recently demonstrated that cardiac-specific ablation of raptor resulted in overt dilated cardiomyopathy and accelerated heart failure in response to increased load following dynamic exercise or aortic constriction without exhibiting adaptive hypertrophy.Citation98 Similar to knockdown of raptor, ablation of mTORC1 in the heart resulted in cardiomyopathy and accelerated heart failure along with impaired hypertrophic response.Citation60 Adaptive hypertrophy of cardiomyocytes is commonly present with pressure overload due to hypertension or hemodynamic stress associated with aging. In a mouse model of pressure overload-induced cardiac hypertrophy, rapamycin either prevented or reversed cardiac hypertrophy.Citation99,Citation100 These findings confirmed a role of mTORC in the regulation of cardiac geometry and function under pathological conditions, such as pressure overload and aging ().
Table 1. Upregulation or downregulation of autophagy in the heart under various pathophysiological conditions
Conclusion
In summary, autophagy is a cellular protective process through recycling macromolecules in times of stress or through removal of damaged proteins and organelles, although excessive autophagy causes cell death. The mTOR pathway appears to represent a common pathway at which several well-known cell survival pathways converge, leading to autophagy and cell survival. Clinical trials have depicted that structural analogs of rapamycin, such as everolimus, prevent endomyocardial remodeling following heart transplantation.Citation101 Future strategies may be engaged on mTOR-related autophagy regulation to retard or halt aging-associated cardiac anomalies.
Acknowledgment
This work was supported in part by a grant from NIH P20RR016474.
Related Research Data
References
- Lutz W, Sanderson W, Scherbov S. The coming acceleration of global population ageing. Nature 2008; 451:716 - 9; http://dx.doi.org/10.1038/nature06516; PMID: 18204438
- Lakatta EG. Cardiovascular regulatory mechanisms in advanced age. Physiol Rev 1993; 73:413 - 67; PMID: 8475195
- Boengler K, Schulz R, Heusch G. Loss of cardioprotection with ageing. Cardiovasc Res 2009; 83:247 - 61; http://dx.doi.org/10.1093/cvr/cvp033; PMID: 19176601
- Rich MW, Quality of Care Committee, Heart Failure Society of America. The year in quality of care in heart failure. J Card Fail 2011; 17:443 - 50; http://dx.doi.org/10.1016/j.cardfail.2011.04.003; PMID: 21624731
- Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, et al, American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics--2011 update: a report from the American Heart Association. Circulation 2011; 123:e18 - 209; http://dx.doi.org/10.1161/CIR.0b013e3182009701; PMID: 21160056
- Gajarsa JJ, Kloner RA. Left ventricular remodeling in the post-infarction heart: a review of cellular, molecular mechanisms, and therapeutic modalities. Heart Fail Rev 2011; 16:13 - 21; http://dx.doi.org/10.1007/s10741-010-9181-7; PMID: 20623185
- Jugdutt BI. Heart failure in the elderly: advances and challenges. Expert Rev Cardiovasc Ther 2010; 8:695 - 715; http://dx.doi.org/10.1586/erc.10.36; PMID: 20450303
- Dai Q, Escobar GP, Hakala KW, Lambert JM, Weintraub ST, Lindsey ML. The left ventricle proteome differentiates middle-aged and old left ventricles in mice. J Proteome Res 2008; 7:756 - 65; http://dx.doi.org/10.1021/pr700685e; PMID: 18166010
- Strait JB, Lakatta EG. Aging-associated cardiovascular changes and their relationship to heart failure. Heart Fail Clin 2012; 8:143 - 64; http://dx.doi.org/10.1016/j.hfc.2011.08.011; PMID: 22108734
- Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a “set up” for vascular disease. Circulation 2003; 107:139 - 46; http://dx.doi.org/10.1161/01.CIR.0000048892.83521.58; PMID: 12515756
- Yang X, Sreejayan N, Ren J. Views from within and beyond: narratives of cardiac contractile dysfunction under senescence. Endocrine 2005; 26:127 - 37; http://dx.doi.org/10.1385/ENDO:26:2:127; PMID: 15888924
- Ferrari AU, Radaelli A, Centola M. Invited review: aging and the cardiovascular system. J Appl Physiol 2003; 95:2591 - 7; PMID: 14600164
- Fleg JL, Shapiro EP, O’Connor F, Taube J, Goldberg AP, Lakatta EG. Left ventricular diastolic filling performance in older male athletes. JAMA 1995; 273:1371 - 5; http://dx.doi.org/10.1001/jama.1995.03520410065028; PMID: 7715063
- Sussman MA, Anversa P. Myocardial aging and senescence: where have the stem cells gone?. Annu Rev Physiol 2004; 66:29 - 48; http://dx.doi.org/10.1146/annurev.physiol.66.032102.140723; PMID: 14977395
- Dhalla NS, Rangi S, Babick AP, Zieroth S, Elimban V. Cardiac remodeling and subcellular defects in heart failure due to myocardial infarction and aging. Heart Fail Rev 2011; In press http://dx.doi.org/10.1007/s10741-011-9278-7; PMID: 21850540
- Lakatta EG. Cardiovascular aging research: the next horizons. J Am Geriatr Soc 1999; 47:613 - 25; PMID: 10323658
- Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol 2009; 335:1 - 32; http://dx.doi.org/10.1007/978-3-642-00302-8_1; PMID: 19802558
- Cefalu CA. Theories and mechanisms of aging. Clin Geriatr Med 2011; 27:491 - 506; http://dx.doi.org/10.1016/j.cger.2011.07.001; PMID: 22062437
- Jin K. Modern Biological Theories of Aging. Aging Dis 2010; 1:72 - 4; PMID: 21132086
- Martin GM. The biology of aging: 1985-2010 and beyond. FASEB J 2011; 25:3756 - 62; http://dx.doi.org/10.1096/fj.11-1102.ufm; PMID: 22046003
- Dai DF, Chen T, Johnson SC, Szeto HH, Rabinovitch PS. Cardiac Aging: From Molecular Mechanisms to Significance in Human Health and Disease. Antioxid Redox Signal 2012; In press http://dx.doi.org/10.1089/ars.2011.4179; PMID: 22229339
- Chen Z, Siu B, Ho YS, Vincent R, Chua CC, Hamdy RC, et al. Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. J Mol Cell Cardiol 1998; 30:2281 - 9; http://dx.doi.org/10.1006/jmcc.1998.0789; PMID: 9925365
- Fang CX, Doser TA, Yang X, Sreejayan N, Ren J. Metallothionein antagonizes aging-induced cardiac contractile dysfunction: role of PTP1B, insulin receptor tyrosine phosphorylation and Akt. Aging Cell 2006; 5:177 - 85; http://dx.doi.org/10.1111/j.1474-9726.2006.00201.x; PMID: 16626396
- Ge W, Zhang Y, Han X, Ren J. Cardiac-specific overexpression of catalase attenuates paraquat-induced myocardial geometric and contractile alteration: role of ER stress. Free Radic Biol Med 2010; 49:2068 - 77; http://dx.doi.org/10.1016/j.freeradbiomed.2010.10.686; PMID: 20937379
- Ren J, Li Q, Wu S, Li SY, Babcock SA. Cardiac overexpression of antioxidant catalase attenuates aging-induced cardiomyocyte relaxation dysfunction. Mech Ageing Dev 2007; 128:276 - 85; http://dx.doi.org/10.1016/j.mad.2006.12.007; PMID: 17250874
- Smit AJ, Hartog JW, Voors AA, van Veldhuisen DJ. Advanced glycation endproducts in chronic heart failure. Ann N Y Acad Sci 2008; 1126:225 - 30; http://dx.doi.org/10.1196/annals.1433.038; PMID: 18448821
- Li SY, Du M, Dolence EK, Fang CX, Mayer GE, Ceylan-Isik AF, et al. Aging induces cardiac diastolic dysfunction, oxidative stress, accumulation of advanced glycation endproducts and protein modification. Aging Cell 2005; 4:57 - 64; http://dx.doi.org/10.1111/j.1474-9728.2005.00146.x; PMID: 15771609
- Shih H, Lee B, Lee RJ, Boyle AJ. The aging heart and post-infarction left ventricular remodeling. J Am Coll Cardiol 2011; 57:9 - 17; http://dx.doi.org/10.1016/j.jacc.2010.08.623; PMID: 21185495
- Judge S, Leeuwenburgh C. Cardiac mitochondrial bioenergetics, oxidative stress, and aging. Am J Physiol Cell Physiol 2007; 292:C1983 - 92; http://dx.doi.org/10.1152/ajpcell.00285.2006; PMID: 17344313
- Wojtovich AP, Nadtochiy SM, Brookes PS, Nehrke K. Ischemic preconditioning: the role of mitochondria and aging. Exp Gerontol 2012; 47:1 - 7; http://dx.doi.org/10.1016/j.exger.2011.11.001; PMID: 22100642
- Ago T, Matsushima S, Kuroda J, Zablocki D, Kitazono T, Sadoshima J. The NADPH oxidase Nox4 and aging in the heart. Aging (Albany NY) 2010; 2:1012 - 6; PMID: 21212466
- Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, et al. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010; 2:914 - 23; PMID: 21212461
- Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 2000; 192:1001 - 14; http://dx.doi.org/10.1084/jem.192.7.1001; PMID: 11015441
- Cesselli D, Beltrami AP, D’Aurizio F, Marcon P, Bergamin N, Toffoletto B, et al. Effects of age and heart failure on human cardiac stem cell function. Am J Pathol 2011; 179:349 - 66; http://dx.doi.org/10.1016/j.ajpath.2011.03.036; PMID: 21703415
- Wong LS, de Boer RA, Samani NJ, van Veldhuisen DJ, van der Harst P. Telomere biology in heart failure. Eur J Heart Fail 2008; 10:1049 - 56; http://dx.doi.org/10.1016/j.ejheart.2008.08.007; PMID: 18815070
- Bodyak N, Kang PM, Hiromura M, Sulijoadikusumo I, Horikoshi N, Khrapko K, et al. Gene expression profiling of the aging mouse cardiac myocytes. Nucleic Acids Res 2002; 30:3788 - 94; http://dx.doi.org/10.1093/nar/gkf497; PMID: 12202764
- Chu MW, Siegmund KD, Eckstam CL, Kim JY, Yang AS, Kanel GC, et al. Lack of increases in methylation at three CpG-rich genomic loci in non-mitotic adult tissues during aging. BMC Med Genet 2007; 8:50; http://dx.doi.org/10.1186/1471-2350-8-50; PMID: 17672908
- Liu N, Olson EN. MicroRNA regulatory networks in cardiovascular development. Dev Cell 2010; 18:510 - 25; http://dx.doi.org/10.1016/j.devcel.2010.03.010; PMID: 20412767
- Enns LC, Pettan-Brewer C, Ladiges W. Protein kinase A is a target for aging and the aging heart. Aging (Albany NY) 2010; 2:238 - 43; PMID: 20448293
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717 - 21; http://dx.doi.org/10.1126/science.290.5497.1717; PMID: 11099404
- Mizushima N. Autophagy: process and function. Genes Dev 2007; 21:2861 - 73; http://dx.doi.org/10.1101/gad.1599207; PMID: 18006683
- Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 2009; 10:458 - 67; http://dx.doi.org/10.1038/nrm2708; PMID: 19491929
- Takemura G, Miyata S, Kawase Y, Okada H, Maruyama R, Fujiwara H. Autophagic degeneration and death of cardiomyocytes in heart failure. Autophagy 2006; 2:212 - 4; PMID: 16874110
- Cuervo AM, Bergamini E, Brunk UT, Dröge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy 2005; 1:131 - 40; http://dx.doi.org/10.4161/auto.1.3.2017; PMID: 16874025
- Vellai T. Autophagy genes and ageing. Cell Death Differ 2009; 16:94 - 102; http://dx.doi.org/10.1038/cdd.2008.126; PMID: 19079287
- Alvers AL, Fishwick LK, Wood MS, Hu D, Chung HS, Dunn WA Jr., et al. Autophagy and amino acid homeostasis are required for chronological longevity in Saccharomyces cerevisiae. Aging Cell 2009; 8:353 - 69; http://dx.doi.org/10.1111/j.1474-9726.2009.00469.x; PMID: 19302372
- Hars ES, Qi H, Ryazanov AG, Jin S, Cai L, Hu C, et al. Autophagy regulates ageing in C. elegans. Autophagy 2007; 3:93 - 5; PMID: 17204841
- Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008; 4:176 - 84; PMID: 18059160
- Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 2010; 6:600 - 6; http://dx.doi.org/10.4161/auto.6.5.11947; PMID: 20431347
- Tóth ML, Sigmond T, Borsos E, Barna J, Erdélyi P, Takács-Vellai K, et al. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 2008; 4:330 - 8; PMID: 18219227
- Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med 2008; 14:959 - 65; http://dx.doi.org/10.1038/nm.1851; PMID: 18690243
- Goswami SK, Das DK. Autophagy in the myocardium: Dying for survival?. Exp Clin Cardiol 2006; 11:183 - 8; PMID: 18651029
- Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ 2009; 16:46 - 56; http://dx.doi.org/10.1038/cdd.2008.110; PMID: 18636076
- Blagosklonny MV. Aging: ROS or TOR. Cell Cycle 2008; 7:3344 - 54; http://dx.doi.org/10.4161/cc.7.21.6965; PMID: 18971624
- Kaeberlein M, Powers RW 3rd, Steffen KK, Westman EA, Hu D, Dang N, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 2005; 310:1193 - 6; http://dx.doi.org/10.1126/science.1115535; PMID: 16293764
- Gottlieb RA, Finley KD, Mentzer RM Jr.. Cardioprotection requires taking out the trash. Basic Res Cardiol 2009; 104:169 - 80; http://dx.doi.org/10.1007/s00395-009-0011-9; PMID: 19242643
- McCormick J, Knight RA, Barry SP, Scarabelli TM, Abounit K, Latchman DS, et al. Autophagy in the stress-induced myocardium. Front Biosci (Elite Ed) 2012; 4:2131 - 41; http://dx.doi.org/10.2741/530; PMID: 22202025
- Gustafsson AB, Gottlieb RA. Autophagy in ischemic heart disease. Circ Res 2009; 104:150 - 8; http://dx.doi.org/10.1161/CIRCRESAHA.108.187427; PMID: 19179668
- Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH, et al. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U S A 2005; 102:13807 - 12; http://dx.doi.org/10.1073/pnas.0506843102; PMID: 16174725
- Zhang D, Contu R, Latronico MV, Zhang J, Rizzi R, Catalucci D, et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest 2010; 120:2805 - 16; http://dx.doi.org/10.1172/JCI43008; PMID: 20644257
- Rothermel BA, Hill JA. Autophagy in load-induced heart disease. Circ Res 2008; 103:1363 - 9; http://dx.doi.org/10.1161/CIRCRESAHA.108.186551; PMID: 19059838
- Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lüllmann-Rauch R, et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 2000; 406:902 - 6; http://dx.doi.org/10.1038/35022595; PMID: 10972293
- Kostin S, Pool L, Elsässer A, Hein S, Drexler HC, Arnon E, et al. Myocytes die by multiple mechanisms in failing human hearts. Circ Res 2003; 92:715 - 24; http://dx.doi.org/10.1161/01.RES.0000067471.95890.5C; PMID: 12649263
- Zheng Q, Su H, Tian Z, Wang X. Proteasome malfunction activates macroautophagy in the heart. Am J Cardiovasc Dis 2011; 1:214 - 26; PMID: 22081794
- Vigliano CA, Cabeza Meckert PM, Diez M, Favaloro LE, Cortés C, Fazzi L, et al. Cardiomyocyte hypertrophy, oncosis, and autophagic vacuolization predict mortality in idiopathic dilated cardiomyopathy with advanced heart failure. J Am Coll Cardiol 2011; 57:1523 - 31; http://dx.doi.org/10.1016/j.jacc.2010.09.080; PMID: 21453830
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 2007; 13:619 - 24; http://dx.doi.org/10.1038/nm1574; PMID: 17450150
- Balasubramanian S, Johnston RK, Moschella PC, Mani SK, Tuxworth WJ Jr., Kuppuswamy D. mTOR in growth and protection of hypertrophying myocardium. Cardiovasc Hematol Agents Med Chem 2009; 7:52 - 63; http://dx.doi.org/10.2174/187152509787047603; PMID: 19149544
- Weichhart T. Mammalian target of rapamycin: a signaling kinase for every aspect of cellular life. Methods Mol Biol 2012; 821:1 - 14; http://dx.doi.org/10.1007/978-1-61779-430-8_1; PMID: 22125056
- Glazer HP, Osipov RM, Clements RT, Sellke FW, Bianchi C. Hypercholesterolemia is associated with hyperactive cardiac mTORC1 and mTORC2 signaling. Cell Cycle 2009; 8:1738 - 46; http://dx.doi.org/10.4161/cc.8.11.8619; PMID: 19395857
- Powers RW 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev 2006; 20:174 - 84; http://dx.doi.org/10.1101/gad.1381406; PMID: 16418483
- Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Müller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature 2003; 426:620; http://dx.doi.org/10.1038/426620a; PMID: 14668850
- Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol 2004; 14:885 - 90; http://dx.doi.org/10.1016/j.cub.2004.03.059; PMID: 15186745
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009; 460:392 - 5; PMID: 19587680
- Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci 2011; 66:191 - 201; http://dx.doi.org/10.1093/gerona/glq178; PMID: 20974732
- Chen C, Liu Y, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal 2009; 2:ra75; http://dx.doi.org/10.1126/scisignal.2000559; PMID: 19934433
- Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle 2009; 8:1888 - 95; http://dx.doi.org/10.4161/cc.8.12.8606; PMID: 19471117
- Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, et al. Rapamycin extends maximal lifespan in cancer-prone mice. Am J Pathol 2010; 176:2092 - 7; http://dx.doi.org/10.2353/ajpath.2010.091050; PMID: 20363920
- Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, et al. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle 2011; 10:4230 - 6; http://dx.doi.org/10.4161/cc.10.24.18486; PMID: 22107964
- Menendez JA, Vellon L, Oliveras-Ferraros C, Cufí S, Vazquez-Martin A. mTOR-regulated senescence and autophagy during reprogramming of somatic cells to pluripotency: a roadmap from energy metabolism to stem cell renewal and aging. Cell Cycle 2011; 10:3658 - 77; http://dx.doi.org/10.4161/cc.10.21.18128; PMID: 22052357
- Blagosklonny MV. Calorie restriction: decelerating mTOR-driven aging from cells to organisms (including humans). Cell Cycle 2010; 9:683 - 8; http://dx.doi.org/10.4161/cc.9.4.10766; PMID: 20139716
- Hua Y, Zhang Y, Ceylan-Isik AF, Wold LE, Nunn JM, Ren J. Chronic Akt activation accentuates aging-induced cardiac hypertrophy and myocardial contractile dysfunction: role of autophagy. Basic Res Cardiol 2011; 106:1173 - 91; http://dx.doi.org/10.1007/s00395-011-0222-8; PMID: 21901288
- Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009; 326:140 - 4; http://dx.doi.org/10.1126/science.1177221; PMID: 19797661
- Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, et al. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab 2010; 11:35 - 46; http://dx.doi.org/10.1016/j.cmet.2009.11.010; PMID: 20074526
- Pan KZ, Palter JE, Rogers AN, Olsen A, Chen D, Lithgow GJ, et al. Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging Cell 2007; 6:111 - 9; http://dx.doi.org/10.1111/j.1474-9726.2006.00266.x; PMID: 17266680
- Beugnet A, Tee AR, Taylor PM, Proud CG. Regulation of targets of mTOR (mammalian target of rapamycin) signalling by intracellular amino acid availability. Biochem J 2003; 372:555 - 66; http://dx.doi.org/10.1042/BJ20021266; PMID: 12611592
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010; 465:942 - 6; http://dx.doi.org/10.1038/nature09076; PMID: 20526321
- Chan EY, Tooze SA. Evolution of Atg1 function and regulation. Autophagy 2009; 5:758 - 65; PMID: 19411825
- Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 2010; 22:132 - 9; http://dx.doi.org/10.1016/j.ceb.2009.12.004; PMID: 20056399
- Anderson RM, Weindruch R. Metabolic reprogramming, caloric restriction and aging. Trends Endocrinol Metab 2010; 21:134 - 41; http://dx.doi.org/10.1016/j.tem.2009.11.005; PMID: 20004110
- Han X, Ren J. Caloric restriction and heart function: is there a sensible link?. Acta Pharmacol Sin 2010; 31:1111 - 7; http://dx.doi.org/10.1038/aps.2010.146; PMID: 20729873
- Madeo F, Tavernarakis N, Kroemer G. Can autophagy promote longevity?. Nat Cell Biol 2010; 12:842 - 6; http://dx.doi.org/10.1038/ncb0910-842; PMID: 20811357
- Wohlgemuth SE, Julian D, Akin DE, Fried J, Toscano K, Leeuwenburgh C, et al. Autophagy in the heart and liver during normal aging and calorie restriction. Rejuvenation Res 2007; 10:281 - 92; http://dx.doi.org/10.1089/rej.2006.0535; PMID: 17665967
- Shinmura K, Tamaki K, Sano M, Murata M, Yamakawa H, Ishida H, et al. Impact of long-term caloric restriction on cardiac senescence: caloric restriction ameliorates cardiac diastolic dysfunction associated with aging. J Mol Cell Cardiol 2011; 50:117 - 27; http://dx.doi.org/10.1016/j.yjmcc.2010.10.018; PMID: 20977912
- Malik SA, Mariño G, BenYounès A, Shen S, Harper F, Maiuri MC, et al. Neuroendocrine regulation of autophagy by leptin. Cell Cycle 2011; 10:2917 - 23; http://dx.doi.org/10.4161/cc.10.17.17067; PMID: 21857156
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature 2009; 458:1131 - 5; http://dx.doi.org/10.1038/nature07976; PMID: 19339967
- Shibata M, Yoshimura K, Furuya N, Koike M, Ueno T, Komatsu M, et al. The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem Biophys Res Commun 2009; 382:419 - 23; http://dx.doi.org/10.1016/j.bbrc.2009.03.039; PMID: 19285958
- Inuzuka Y, Okuda J, Kawashima T, Kato T, Niizuma S, Tamaki Y, et al. Suppression of phosphoinositide 3-kinase prevents cardiac aging in mice. Circulation 2009; 120:1695 - 703; http://dx.doi.org/10.1161/CIRCULATIONAHA.109.871137; PMID: 19822807
- Shende P, Plaisance I, Morandi C, Pellieux C, Berthonneche C, Zorzato F, et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation 2011; 123:1073 - 82; http://dx.doi.org/10.1161/CIRCULATIONAHA.110.977066; PMID: 21357822
- McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, et al. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation 2004; 109:3050 - 5; http://dx.doi.org/10.1161/01.CIR.0000130641.08705.45; PMID: 15184287
- Shioi T, McMullen JR, Tarnavski O, Converso K, Sherwood MC, Manning WJ, et al. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation 2003; 107:1664 - 70; http://dx.doi.org/10.1161/01.CIR.0000057979.36322.88; PMID: 12668503
- Hiemann NE, Wellnhofer E, Lehmkuhl HB, Knosalla C, Hetzer R, Meyer R. Everolimus prevents endomyocardial remodeling after heart transplantation. Transplantation 2011; 92:1165 - 72; http://dx.doi.org/10.1097/TP.0b013e3182332886; PMID: 21956201