Abstract
Blocking tumor angiogenesis is an important goal of cancer therapy, but clinically approved anti-angiogenic agents suffer from limited efficacy and adverse side effects, fueling the need to identify alternative angiogenesis regulators. Tumor endothelial marker 8 (TEM8) is a highly conserved cell surface receptor overexpressed on human tumor vasculature. Genetic disruption of Tem8 in mice revealed that TEM8 is important for promoting tumor angiogenesis and tumor growth but dispensable for normal development and wound healing. The induction of TEM8 in cultured endothelial cells by nutrient or growth factor deprivation suggests that TEM8 may be part of a survival response pathway that is activated by tumor microenvironmental stress. In preclinical studies, antibodies targeted against the extracellular domain of TEM8 inhibited tumor angiogenesis and blocked the growth of multiple human tumor xenografts. Anti-TEM8 antibodies augmented the activity of other anti-angiogenic agents, vascular targeting agents and conventional chemotherapeutic agents and displayed no detectable toxicity. Thus, anti-TEM8 antibodies provide a promising new tool for selective blockade of neovascularization associated with cancer and possibly other angiogenesis-dependent diseases.
Introduction
Angiogenesis plays an important role in normal tissue expansion, wound healing and remodeling and also facilitates tumor growth and spread. Many endogenous anti-angiogenic factors are known to inhibit tumor angiogenesis, such as thrombospondin-1 and endostatin,Citation1-Citation3 and proangiogenic factors, including fibroblast growth factor-2 and vascular endothelial growth factor (VEGF), promote tumor angiogenesis.Citation4,Citation5 Among the known angiogenic regulators, VEGF stands out due to its proven ability to facilitate tumorigenesis in both preclinical models and patients. Indeed, bevacizumab, an anti-VEGF blocking antibody, is clinically approved for the treatment of patients with colorectal, brain, lung and kidney cancer, and receptor tyrosine kinase (RTK) inhibitors that block VEGFR2 activity, including sorafenib, sunitinib, pazopanib, vandetanib and axitinib have also proven efficacious, particularly in treating kidney cancer.Citation5,Citation6 Despite these encouraging inroads, two major problems remain with current anti-angiogenic agents used for cancer therapy. The first is limited efficacy. Clinical trials have revealed that anti-VEGF antibodies typically work best in combination with other anticancer agents, displaying minimal to no efficacy on their own. Furthermore, angiogenesis usually persists, albeit at a reduced rate, in the face of continued VEGF/VEGFR blockade.Citation7 This has led to an intense search for alternative angiogenesis pathways responsible for persistent tumor angiogenesis in the face of VEGF/VEGFR pathway inhibition. The second problem with current anti-angiogenic therapy is toxicity. An increasing number of adverse side effects are associated with VEGF pathway blockade. Although the more common side effects, such as hypertension and proteinuria are usually manageable, there are also some rare serious adverse side effects, such as thromboembolic events and severe bleeding complications that are potentially life-threatening.Citation8,Citation9 The toxicities of VEGF-inhibitor therapy may be due to off-target or on-target activity of the anti-angiogenic agent employed.Citation9 With respect to the latter, it is now clear that VEGF plays critical roles in normal adult physiology that extend far beyond its angiogenesis-promoting activity.Citation10-Citation13
One approach to overcoming these obstacles is the development of new agents that target alternative pathways required for tumor angiogenesis. To minimize toxicity, an ideal anti-angiogenic agent should be able to discriminate tumor angiogenesis from normal physiological angiogenesis. In other words, an ideal target is one that is required for tumor angiogenesis but is dispensable for all normal tissue functions, including, for example, neovascularization associated with ovulation, endometrial regeneration and wound healing. The central importance of VEGF for developmental, physiological and pathological angiogenesis raises the question as to whether or not such a discriminating target exists. However, tumor vasculature is morphologically distinct from normal proliferating vasculature, being disorganized, tortuous and leaky,Citation13 and molecular profiling has revealed some promising tumor-induced vascular markers that may have the desired specificity.
The first systematic profiling of gene expression in human tumor vs. normal endothelium revealed a series of tumor endothelial markers (TEMs) upregulated in tumor vasculaure.Citation14 Further validation revealed that the majority of genes overexpressed in tumor endothelium were also highly expressed in the angiogenic vessels of the ovaries, an indicator of physiological angiogenesis. One notable exception was tumor endothelial marker (TEM8), a cell surface receptor undetectable in the angiogenic vessels of the ovaries at both the mRNA and protein level.Citation14,Citation15 The identification of TEM8 stimulated further studies aimed at specifically identifying genes that can separate physiological and pathological angiogenesis and resulted in a small list of additional TEMs with a similar tumor-restricted vascular expression pattern, including the cell surface receptor CD276 (B7-H3).Citation16 Although several other potential targets of tumor vasculature have been identified through gene expression profiling, peptide phage display or proteomic approaches,Citation17-Citation24 most cannot separate physiological from pathological angiogenesis.
Elucidating the Function of TEM8
Soon after TEM8 was identified, it was found to be a receptor for anthrax toxin and is also called ANTXR1.Citation25 Subsequently, the closest homolog of TEM8, capillary morphogenesis gene 2 (CMG2/ANTXR2), was identified as a second receptor for anthrax toxinCitation26 and was found to have ~11-fold higher affinity for anthrax toxin than TEM8.Citation27 Human CMG2 and TEM8 share 57% amino acid identify overall. CMG2 appears to be the primary receptor mediating anthrax pathogenicity in vivo,Citation27,Citation28 which could be related to its higher affinity for PA and/ or its more ubiquitous expression. Three proteins, protective antigen (PA), lethal factor and edema factor assemble to form anthrax toxin. The protective antigen moiety is responsible for binding TEM8 and CMG2 and mediating toxin transfer into the cell. The higher affinity of PA for CMG2 has made it difficult to identify mutant PA molecules that preferentially bind TEM8 and not CMG2 Citation29 (Steve Leppla, personal communication).
TEM8 and CMG2 are both integrin-like receptors that contain a vonWillebrand factor type A domain in their extracellular region (). Although very little is known about their physiological function, TEM8 been found to bind to collagen type VICitation15 and type I,Citation30 whereas CMG2 has been shown to bind collagen type IV and laminin.Citation31,Citation32 In 6-month-old Tem8-knockout (KO) mice an accumulation of collagen was detected in certain organs, such as the ovary, uterus and skin, implicating a role for TEM8 in collagen homeostasis. Mutations in CMG2 cause hyaline fibromatosis syndrome (HFS),Citation32-Citation34 an autosomal recessive disorder characterized by the presence of non-cancerous tissue proliferations and nodules that contain an excess of amorphous ECM, including glycosaminoglycans and collagens type I and VI.Citation35-Citation40 Although the stimulus for lesion formation is unclear, their prevalence over points of mechanical stress or pressure suggests that their pathogenesis may involve disregulation of repair mechanisms after microtrauma.Citation41 The common buildup of ECM both in tissues of Tem8-KO mice and lesions from HFS patients suggests that TEM8 and CMG2 may share similar roles in regulation of ECM turnover.
Figure 1. TEM8 structure and antibody binding. (A) TEM8 is a type I membrane protein containing a vWF type A (VWA) domain in its extracellular domain. (B) The image depicts the selective binding of anti-TEM8 blocking antibodies (blue) to TEM8 receptors (orange) that are overexpressed on the surface of tumor endothelium. Tumor cells are depicted in yellow.
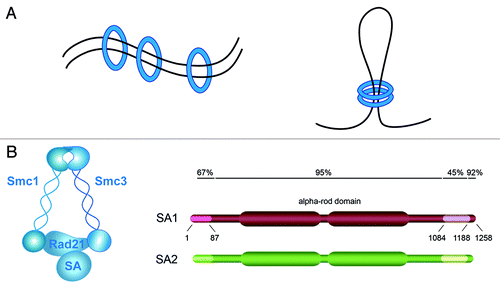
A rigorous test of the importance of TEM8 in pathological vs. physiological angiogenesis comes from studies of genetically engineered Tem8-KO mice.Citation42 Tem8-KO mice reach adulthood without any gross phenotypic abnormalities, with the exception of misaligned incisors. The tooth defect appears to originate from excessive extracellular matrix in the gums, and, as previously mentioned, a mild buildup of extracellular matrix (fibrosis) was detected in various organs of adult mice by histological examination. Evaluation of developmental angiogenesis failed to reveal any abnormalities in the Tem8 KO. Similarly, wound healing and angiogenesis associated with tissue repair were unaltered.Citation42,Citation43 However, the growth of multiple cancer types was significantly impaired in both immunocompetent and immunodeficient Tem8-KO mice, and the reduced tumor growth was associated with a reduction in tumor vessel density. These preclinical studies reveal that host-derived TEM8 is important for promoting tumor angiogenesis and tumor growth but dispensable for physiological angiogenesis.
Development of Anti-TEM8 Blocking Antibodies
Based on the functional importance of TEM8 in tumorigenesis, our group set out to develop neutralizing monoclonal anti-TEM8 antibodiesCitation43 (). Two independent anti-TEM8 full IgG antibodies, called L2 and L5, were identified and tested in preclinical transplanted tumor models. Anti-TEM8 antibodies displayed potent antitumor activity against various mouse syngeneic or human xenogenic tumor types, including melanoma, colon and lung cancers. Antitumor activity was only observed in Tem8 WT but not KO mice, demonstrating the specificity of the antibodies for host TEM8 in vivo. Importantly, TEM8 antibodies did not prevent wound healing, consistent with a primary role for this cell surface receptor in regulating pathological (tumor) angiogenesis without affecting normal tissue repair.
Naked antibodies can elicit antitumor activity in vivo through multiple mechanisms. Many clinically approved antibodies that bind cell surface receptors block target antigen function and/or promote antibody-dependent cellular cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC). A complete understanding of the mechanisms responsible for the antitumor activity of L2 and L5 in vivo awaits further studies, but so far, the evidence suggests that a function blocking activity may be of primary importance.Citation43 First, the extent of antitumor activity of the antibodies varied between the different tumors tested, but strikingly similar tumor-dependent growth patterns were also observed in comparisons of Tem8 KO and WT mice. For example, a comparison of several human tumor xenografts revealed that UACC melanoma was the most responsive to both anti-TEM8 antibody treatment and genetic disruption of host Tem8. Similarly, in immunocompetent C57BL6 mice, murine B16 melanoma tumors responded strongly to both pharmacologic and genetic ablation of TEM8, whereas Lewis lung carcinoma (LLC) tumors responded poorly to both.Citation42-Citation44 A careful evaluation of TEM8 expression in endothelial cells purified from these tumors revealed a 4-fold higher level of TEM8 expression in the LLC vs. B16 tumor endothelium.Citation42 The reason for TEM8’s decreased functional importance in LLC tumor vasculature despite its higher expression is currently unclear but could be a consequence of a lower ligand level in LLC tumors. Nevertheless, these data argue that the antibodies may work primarily through their function blocking activity rather than other mechanisms, such as ADCC and CDC, which depend primarily on the levels of target antigen expression. These results also highlight the intricate crosstalk between the tumor cells and tumor-associated stromal cells and suggest that each individual tumor may have its own intrinsic level of dependence on TEM8 expression in the host stroma. This degree of dependence may be related to the level of tumor cell expression of TEM8 ligands, such as collagen I or collagen VI, the aggressiveness of the tumor or tumor-specific influences on the biophysical properties of the tumor microenvironment. Nevertheless, these studies highlight the fact that a high level of TEM8 expression in tumor vasculature may not necessarily predict a high level of responsiveness to anti-TEM8 antibody therapy.
Importantly, when used as a monotherapy, anti-TEM8 antibodies were unable to regress pre-established tumors or halt tumor growth completely, similar to other anti-angiogenic agents. However, in combination trials, the antibodies significantly augmented the activity of other anticancer agents, including VEGF pathway blockers, vascular disrupting agents and chemotherapeutic agents.Citation43 The combination of anti-TEM8 antibodies with irinotecan proved particularly efficacious, resulting in complete tumor regressions and cures in 41% of the mice. No cures were observed with either agent alone. Importantly, toxicology studies failed to reveal any detectable toxicity associated with anti-TEM8 antibody treatment, even following 6 weeks of antibody treatment. This is not too surprising given the mild phenotype of the Tem8-KO mice. Based on these studies, we would predict that anti-TEM8 antibodies may prove most useful in combination with other anticancer agents. However, because anti-angiogenic agents may need to be administered for long periods of time, there is still potential concern for toxicity, especially for patients prone to fibrosis.
Key Unanswered Questions
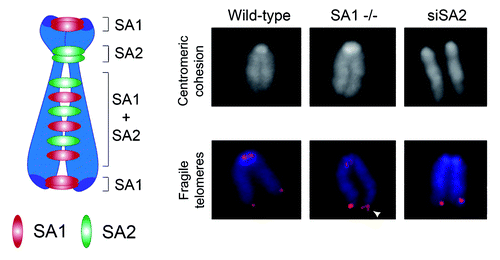
For the remainder of this article, we focus on key unanswered questions regarding TEM8 and tumor angiogenesis. One important question is, what are the TEM8 host cells that are most important for promoting tumor growth? Are tumor endothelial cells the only important cell type, or do other tumor-associated vascular cells, such as pericytes or non-vascular cells, also play a role? Some studies indicate that TEM8 may also be expressed by tumor-associated pericytes and possibly other unidentified perivascular stromal cells.Citation43,Citation45 Future studies using conditional Tem8-KO mice should help to address these issues. One thing that is urgently needed is better antibodies for histological detection of TEM8 protein. Early anti-TEM8 antibodies used for histology were extremely sensitive to initial sample preparation conditions and required tedious antigen retrieval strategies, limiting the number of samples that could be analyzed.Citation15 The recently described L2 anti-TEM8 antibodies recognize the native TEM8 protein with high affinity but are also suboptimal for histology due to an extreme sensitivity to sample preparation and fixation methods.Citation44 Although there are now several commercially available anti-TEM8 antibodies, none of the ones tested in our laboratory have proven suitable for IHC. Bonuccelli and coworkers reported that TEM8 is widely expressed in normal human and mouse tissues, particularly in the epithelial cells lining the lung, skin and intestine.Citation46 Their conclusions were based on immunohistochemistry (IHC) and western blotting results obtained using a commercially available rabbit polyclonal antibody raised against a 16 aa peptide sequence that is identical between mouse and human and resides in the N-terminal extracellular domain of TEM8. When we tested the same rabbit polyclonal antibody by immunofluorescence staining of normal tissues from Tem8 WT and KO mice, we were unable to detect any specific staining.Citation44 Although we could confirm reactivity of the antibody against the ~85 kDa form of TEM8 in TEM8-transfected 293 cells by western blotting, when tested against various normal tissue lysates from Tem8 WT and KO mice, the antibody appeared to react with proteins of unexpected sizes, and no differences were observed between WT and KO mice (). The unusual size products were not detected by other anti-TEM8 monoclonal antibodies, such as clone SB5, suggesting that they likely represent non-specific proteins rather than alternative splice variants of TEM8, as originally proposed by Bonuccelli et al.Citation46 Although the identities of the putative cross-reactive antigens are currently unclear, the antibody is unlikely to recognize CMG2, because the particular peptide selected for immunization does not show significant homology to CMG2. Furthermore, western blotting with the polyclonal antibody failed to detect CMG2 in comparisons of CHO/PR230 cells and CHO/PR230 cells transfected with either mouse or human CMG2.Citation44 TEM8s high level of conservation among species makes it difficult to break immunological tolerance using conventional hybridoma technologies, which has likely contributed to the challenge in identifying robust anti-TEM8 antibodies for histology.
Figure 2. TEM8 antibodies recognize proteins in both Tem8 WT and KO mice. (A) The rabbit anti-TEM8 polyclonal antibody from AbCAM (cat # ab19387) generated against the TEM8 extracellular domain peptide LMKLTEDREQIRQGLE was used to detect TEM8 in various normal tissues from Tem8 WT and KO mice. Although the polyclonal antibody recognized both mouse TEM8 (mTEM8) and human TEM8 (hTEM8) in 293 cells transfected with the corresponding Tem8 genes (asterisk), when tested against lysates from various normal mouse tissues it reacted predominantly with proteins that were present in both the Tem8 WT and KO mice. (B) PCR genotyping assay used to confirm the Tem8 WT and KO status of the samples used in A.
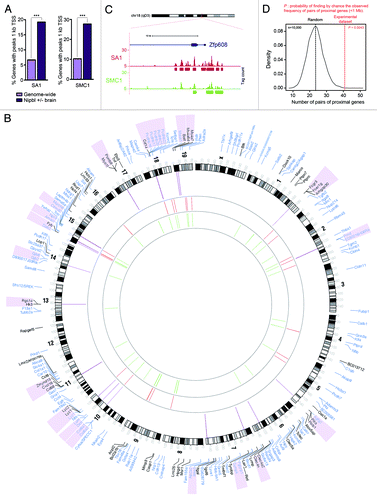
A more basic question is: what is the normal physiological function of TEM8? TEM8 is highly conserved among species, with mice and men sharing 96% amino acid identity overall. This implies that TEM8 plays some fundamental role in normal physiology, yet mice with a disrupted TEM8 gene appear remarkably normal. Furthermore, functional redundancy seems unlikely, given that TEM8 has only one obvious paralog, CMG2, and TEM8/CMG2 double mutant mice also reach adulthood and are remarkably normal.Citation27 A possible explanation for this apparent paradox is that TEM8 function in normal tissues requires a particular stress or stimulus for full manifestation, analogous to p53 and other stress-induced genes. Many stressors, such as hypoxia, exist in the tumor microenvironment and can also occur in non-tumor settings, for example, during tissue ischemia. To determine if TEM8 may be involved in a stress-mediated response, we evaluated TEM8 expression in cultured endothelial cells exposed to a variety of stressors found in the tumor microenvironment. Although TEM8 expression was not affected by various factors, including low pH and hypoxia, TEM8 expression was found to depend strongly on growth factor availability.Citation43 Thus, endothelial cells deprived of growth factors gradually increased both TEM8 mRNA and protein expression by 5-fold over 10 d. More recently, we have found that amino acid deprivation also leads to TEM8 overexpression (). An important question is whether or not tumor endothelial cells in vivo experience significant variations in growth factor or nutrient availability. Local variations in endothelial proliferation in different regions of the tumorCitation47 suggest this may be the case. Furthermore, blood flow through tumor vessels is frequently sluggish, erratic, sometimes static and not always unidirectional, which may lead to transient fluctuations in nutrient availability. Furthermore, tumor cells frequently overexpress receptors for endothelial growth factors, such as FGFRs and VEGFRsCitation48-Citation51 as well as amino acid transporters,Citation52 and may compete with tumor blood vessels for local growth factors or nutrients. Finally, soluble VEGFR1, a well-known VEGFR decoy, is frequently elevated in tumors,Citation53,Citation54 which may be due to its upregulation by hypoxia.Citation55
Figure 3. Amino acid starvation induces TEM8 expression. (A) Real-time quantitative PCR revealed a gradual increase in TEM8 mRNA expression following amino acid (AA) depletion from the growth medium. (B) Immunoblotting (IB) with anti-TEM8 antibodies (clone SB5) revealed an increase in TEM8 protein 24 h following removal of amino acids from the growth medium. β actin was used as a loading control.
Interestingly, an increased level of apoptosis was found in tumor endothelial cells following L2 treatment.Citation43 This suggests that TEM8 may normally function to block apoptosis in tumor endothelial cells that are growth factor- or nutrient-deprived. Because growth factor and amino acid starvation are also potent inducers of autophagy,Citation56,Citation57 one possibility is that TEM8 functions to promote autophagy, allowing the tumor vasculature to survive until growth factors are replenished. This, in turn, could help explain why TEM8 blocking antibodies complement the activity of other anticancer agents, including other anti-angiogenic agents, in preclinical drug combination trials. The primary cells targeted by TEM8 antibodies may be tumor endothelial cells that are transiently deprived of angiogenic growth factors and utilizing alternative pathways for survival. Such cells may be intrinsically resistant to VEGF pathway inhibitors and other anti-angiogenic therapies that depend on growth factor stimulation for activity. According to this model, any anticancer agent that targets the actively dividing tumor endothelial cells or the tumor cells themselves may work better in combination with anti-TEM8 antibodies.
In summary, TEM8 is a cell surface receptor that plays an important role promoting tumor angiogenesis, making it a potential target of anti-angiogenic therapy. Although much has been learnt about TEM8 over the past decade, still very little is known about its normal physiological function. It will be important to determine if TEM8 is involved in angiogenesis associated with other pathologies such as ischemia and tissue reperfusion, diabetic retinopathy or age-related macular degeneration. It will also be important to decipher the role of TEM8 in stress-induced response pathway and to determine if collagen binding is important for such responses. A better understanding of the biological pathways regulated by TEM8 should aid in development of more effective approaches for controlling cancer and possibly other diseases that depend on pathological angiogenesis.
Acknowledgments
We thank our collaborators especially Drs. Xiaoyan M. Zhang and Saurabh Saha from BioMed Valley Discoveries, and Drs. Susan Stevenson and Tony Fleming from Novartis. The work discussed was supported in part by a Cooperative Research and Development Agreement (CRADA) between the Novartis Institutes for BioMedical Research and the intramural research program of the National Cancer Institute (NCI), National Institutes of Health, Department of Health and Social Services (DHSS), and with federal funds from the NCI under Contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the DHSS nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Related Research Data
References
- Taraboletti G, Rusnati M, Ragona L, Colombo G. Targeting tumor angiogenesis with TSP-1-based compounds: rational design of antiangiogenic mimetics of endogenous inhibitors. Oncotarget 2010; 1:662 - 73; PMID: 21317461
- Kaur S, Roberts DD. CD47 applies the brakes to angiogenesis via vascular endothelial growth factor receptor-2. Cell Cycle 2011; 10:10 - 2; http://dx.doi.org/10.4161/cc.10.1.14324; PMID: 21191182
- Shichiri M, Tanaka Y. Inhibition of cancer progression by rifampicin: involvement of antiangiogenic and anti-tumor effects. Cell Cycle 2010; 9:64 - 8; http://dx.doi.org/10.4161/cc.9.1.10354; PMID: 20016271
- Acevedo VD, Ittmann M, Spencer DM. Paths of FGFR-driven tumorigenesis. Cell Cycle 2009; 8:580 - 8; http://dx.doi.org/10.4161/cc.8.4.7657; PMID: 19182515
- Kerbel RS. Tumor angiogenesis. N Engl J Med 2008; 358:2039 - 49; http://dx.doi.org/10.1056/NEJMra0706596; PMID: 18463380
- Samant RS, Shevde LA. Recent advances in anti-angiogenic therapy of cancer. Oncotarget 2011; 2:122 - 34; PMID: 21399234
- Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol 2011; 8:210 - 21; http://dx.doi.org/10.1038/nrclinonc.2011.21; PMID: 21364524
- Chen HX, Cleck JN. Adverse effects of anticancer agents that target the VEGF pathway. Nat Rev Clin Oncol 2009; 6:465 - 77; http://dx.doi.org/10.1038/nrclinonc.2009.94; PMID: 19581909
- Verheul HM, Pinedo HM. Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nat Rev Cancer 2007; 7:475 - 85; http://dx.doi.org/10.1038/nrc2152; PMID: 17522716
- Eremina V, Cui S, Gerber H, Ferrara N, Haigh J, Nagy A, et al. Vascular endothelial growth factor a signaling in the podocyte-endothelial compartment is required for mesangial cell migration and survival. J Am Soc Nephrol 2006; 17:724 - 35; http://dx.doi.org/10.1681/ASN.2005080810; PMID: 16436493
- Sung HK, Michael IP, Nagy A. Multifaceted role of vascular endothelial growth factor signaling in adult tissue physiology: an emerging concept with clinical implications. Curr Opin Hematol 2010; 17:206 - 12; PMID: 20216210
- Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell 2007; 130:691 - 703; http://dx.doi.org/10.1016/j.cell.2007.06.054; PMID: 17719546
- Rouhi P, Lee SL, Cao Z, Hedlund EM, Jensen LD, Cao Y. Pathological angiogenesis facilitates tumor cell dissemination and metastasis. Cell Cycle 2010; 9:913 - 7; http://dx.doi.org/10.4161/cc.9.5.10853; PMID: 20160500
- St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, et al. Genes expressed in human tumor endothelium. Science 2000; 289:1197 - 202; http://dx.doi.org/10.1126/science.289.5482.1197; PMID: 10947988
- Nanda A, Carson-Walter EB, Seaman S, Barber TD, Stampfl J, Singh S, et al. TEM8 interacts with the cleaved C5 domain of collagen alpha 3(VI). Cancer Res 2004; 64:817 - 20; http://dx.doi.org/10.1158/0008-5472.CAN-03-2408; PMID: 14871805
- Seaman S, Stevens J, Yang MY, Logsdon D, Graff-Cherry C, St Croix B. Genes that distinguish physiological and pathological angiogenesis. Cancer Cell 2007; 11:539 - 54; http://dx.doi.org/10.1016/j.ccr.2007.04.017; PMID: 17560335
- Oh P, Li Y, Yu J, Durr E, Krasinska KM, Carver LA, et al. Subtractive proteomic mapping of the endothelial surface in lung and solid tumours for tissue-specific therapy. Nature 2004; 429:629 - 35; http://dx.doi.org/10.1038/nature02580; PMID: 15190345
- Beaty RM, Edwards JB, Boon K, Siu IM, Conway JE, Riggins GJ. PLXDC1 (TEM7) is identified in a genome-wide expression screen of glioblastoma endothelium. J Neurooncol 2007; 81:241 - 8; http://dx.doi.org/10.1007/s11060-006-9227-9; PMID: 17031559
- Parker BS, Argani P, Cook BP, Liangfeng H, Chartrand SD, Zhang M, et al. Alterations in vascular gene expression in invasive breast carcinoma. Cancer Res 2004; 64:7857 - 66; http://dx.doi.org/10.1158/0008-5472.CAN-04-1976; PMID: 15520192
- Carson-Walter E, St. Croix B. Molecular Targets of Tumor Vasculature. In: Aird WC, ed. Endothelial Cell Phenotypes in Health and Disease: Marcel Dekker, Inc., 2005.
- Madden SL, Cook BP, Nacht M, Weber WD, Callahan MR, Jiang Y, et al. Vascular gene expression in nonneoplastic and malignant brain. Am J Pathol 2004; 165:601 - 8; http://dx.doi.org/10.1016/S0002-9440(10)63324-X; PMID: 15277233
- Roesli C, Neri D. Methods for the identification of vascular markers in health and disease: from the bench to the clinic. J Proteomics 2010; 73:2219 - 29; http://dx.doi.org/10.1016/j.jprot.2010.05.017; PMID: 20541635
- Neri D, Bicknell R. Tumour vascular targeting. Nat Rev Cancer 2005; 5:436 - 46; http://dx.doi.org/10.1038/nrc1627; PMID: 15928674
- Hajitou A, Pasqualini R, Arap W. Vascular targeting: recent advances and therapeutic perspectives. Trends Cardiovasc Med 2006; 16:80 - 8; http://dx.doi.org/10.1016/j.tcm.2006.01.003; PMID: 16546688
- Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature 2001; 414:225 - 9; http://dx.doi.org/10.1038/n35101999; PMID: 11700562
- Scobie HM, Rainey GJ, Bradley KA, Young JA. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci U S A 2003; 100:5170 - 4; http://dx.doi.org/10.1073/pnas.0431098100; PMID: 12700348
- Liu S, Crown D, Miller-Randolph S, Moayeri M, Wang H, Hu H, et al. Capillary morphogenesis protein-2 is the major receptor mediating lethality of anthrax toxin in vivo. Proc Natl Acad Sci U S A 2009; 106:12424 - 9; http://dx.doi.org/10.1073/pnas.0905409106; PMID: 19617532
- Liu S, Miller-Randolph S, Crown D, Moayeri M, Sastalla I, Okugawa S, et al. Anthrax toxin targeting of myeloid cells through the CMG2 receptor is essential for establishment of Bacillus anthracis infections in mice. Cell Host Microbe 2010; 8:455 - 62; http://dx.doi.org/10.1016/j.chom.2010.10.004; PMID: 21075356
- Chen KH, Liu S, Bankston LA, Liddington RC, Leppla SH. Selection of anthrax toxin protective antigen variants that discriminate between the cellular receptors TEM8 and CMG2 and achieve targeting of tumor cells. J Biol Chem 2007; 282:9834 - 45; http://dx.doi.org/10.1074/jbc.M611142200; PMID: 17251181
- Hotchkiss KA, Basile CM, Spring SC, Bonuccelli G, Lisanti MP, Terman BI. TEM8 expression stimulates endothelial cell adhesion and migration by regulating cell-matrix interactions on collagen. Exp Cell Res 2005; 305:133 - 44; http://dx.doi.org/10.1016/j.yexcr.2004.12.025; PMID: 15777794
- Bell SE, Mavila A, Salazar R, Bayless KJ, Kanagala S, Maxwell SA, et al. Differential gene expression during capillary morphogenesis in 3D collagen matrices: regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J Cell Sci 2001; 114:2755 - 73; PMID: 11683410
- Dowling O, Difeo A, Ramirez MC, Tukel T, Narla G, Bonafe L, et al. Mutations in capillary morphogenesis gene-2 result in the allelic disorders juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am J Hum Genet 2003; 73:957 - 66; http://dx.doi.org/10.1086/378781; PMID: 12973667
- Hanks S, Adams S, Douglas J, Arbour L, Atherton DJ, Balci S, et al. Mutations in the gene encoding capillary morphogenesis protein 2 cause juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am J Hum Genet 2003; 73:791 - 800; http://dx.doi.org/10.1086/378418; PMID: 14508707
- Deuquet J, Lausch E, Superti-Furga A, van der Goot FG. The dark sides of capillary morphogenesis gene 2. EMBO J 2012; 31:3 - 13; http://dx.doi.org/10.1038/emboj.2011.442; PMID: 22215446
- Tanaka K, Ebihara T, Kusubata M, Adachi E, Arai M, Kawaguchi N, et al. Abnormal collagen deposition in fibromas from patient with juvenile hyaline fibromatosis. J Dermatol Sci 2009; 55:197 - 200; http://dx.doi.org/10.1016/j.jdermsci.2009.06.005; PMID: 19592224
- Murata K, Motoyama T, Suka M, Ohno M, Kuboki Y. High production of type VI collagen in multiple fibromatosis with multiple articular dysplasia. Biochem Biophys Res Commun 1987; 147:275 - 81; http://dx.doi.org/10.1016/S0006-291X(87)80117-1; PMID: 3632670
- Glover MT, Lake BD, Atherton DJ. Infantile systemic hyalinosis: newly recognized disorder of collagen?. Pediatrics 1991; 87:228 - 34; PMID: 1702887
- Katagiri K, Takasaki S, Fujiwara S, Kayashima K, Ono T, Shinkai H. Purification and structural analysis of extracellular matrix of a skin tumor from a patient with juvenile hyaline fibromatosis. J Dermatol Sci 1996; 13:37 - 48; http://dx.doi.org/10.1016/0923-1811(95)00492-0; PMID: 8902652
- Iwata S, Horiuchi R, Maeda H, Ishikawa H. Systemic hyalinosis or juvenile hyaline fibromatosis. Ultrastructural and biochemical study of cultured skin fibroblasts. Arch Dermatol Res 1980; 267:115 - 21; http://dx.doi.org/10.1007/BF00569097; PMID: 6157366
- Tzellos TG, Dionyssopoulos A, Klagas I, Karakiulakis G, Lazaridis L, Papakonstantinou E. Differential glycosaminoglycan expression and hyaluronan homeostasis in juvenile hyaline fibromatosis. J Am Acad Dermatol 2009; 61:629 - 38; http://dx.doi.org/10.1016/j.jaad.2009.03.042; PMID: 19559501
- Stucki U, Spycher MA, Eich G, Rossi A, Sacher P, Steinmann B, et al. Infantile systemic hyalinosis in siblings: clinical report, biochemical and ultrastructural findings, and review of the literature. Am J Med Genet 2001; 100:122 - 9; http://dx.doi.org/10.1002/1096-8628(20010422)100:2<122::AID-AJMG1236>3.0.CO;2-0; PMID: 11298373
- Cullen M, Seaman S, Chaudhary A, Yang MY, Hilton MB, Logsdon D, et al. Host-derived tumor endothelial marker 8 promotes the growth of melanoma. Cancer Res 2009; 69:6021 - 6; http://dx.doi.org/10.1158/0008-5472.CAN-09-1086; PMID: 19622764
- Chaudhary A, Hilton MB, Seaman S, Haines DC, Stevenson S, Lemotte PK, et al. TEM8/ANTXR1 blockade inhibits pathological angiogenesis and potentiates tumoricidal responses against multiple cancer types. Cancer Cell 2012; 21:212 - 26; http://dx.doi.org/10.1016/j.ccr.2012.01.004; PMID: 22340594
- Chaudhary A, St. Croix B. Unpublished Observations.
- Yang MY, Chaudhary A, Seaman S, Dunty J, Stevens J, Elzarrad MK, et al. The cell surface structure of tumor endothelial marker 8 (TEM8) is regulated by the actin cytoskeleton. Biochim Biophys Acta 2011; 1813:39 - 49; http://dx.doi.org/10.1016/j.bbamcr.2010.11.013; PMID: 21129411
- Bonuccelli G, Sotgia F, Frank PG, Williams TM, de Almeida CJ, Tanowitz HB, et al. ATR/TEM8 is highly expressed in epithelial cells lining Bacillus anthracis’ three sites of entry: implications for the pathogenesis of anthrax infection. Am J Physiol Cell Physiol 2005; 288:C1402 - 10; http://dx.doi.org/10.1152/ajpcell.00582.2004; PMID: 15689409
- Eberhard A, Kahlert S, Goede V, Hemmerlein B, Plate KH, Augustin HG. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies. Cancer Res 2000; 60:1388 - 93; PMID: 10728704
- Dallas NA, Fan F, Gray MJ, Van Buren G 2nd, Lim SJ, Xia L, et al. Functional significance of vascular endothelial growth factor receptors on gastrointestinal cancer cells. Cancer Metastasis Rev 2007; 26:433 - 41; http://dx.doi.org/10.1007/s10555-007-9070-2; PMID: 17786539
- Masood R, Cai J, Zheng T, Smith DL, Hinton DR, Gill PS. Vascular endothelial growth factor (VEGF) is an autocrine growth factor for VEGF receptor-positive human tumors. Blood 2001; 98:1904 - 13; http://dx.doi.org/10.1182/blood.V98.6.1904; PMID: 11535528
- Liu B, Earl HM, Baban D, Shoaibi M, Fabra A, Kerr DJ, et al. Melanoma cell lines express VEGF receptor KDR and respond to exogenously added VEGF. Biochem Biophys Res Commun 1995; 217:721 - 7; http://dx.doi.org/10.1006/bbrc.1995.2832; PMID: 8554590
- Slongo ML, Molena B, Brunati AM, Frasson M, Gardiman M, Carli M, et al. Functional VEGF and VEGF receptors are expressed in human medulloblastomas. Neuro Oncol 2007; 9:384 - 92; http://dx.doi.org/10.1215/15228517-2007-032; PMID: 17704359
- Fuchs BC, Bode BP. Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime?. Semin Cancer Biol 2005; 15:254 - 66; http://dx.doi.org/10.1016/j.semcancer.2005.04.005; PMID: 15916903
- Yamaguchi T, Bando H, Mori T, Takahashi K, Matsumoto H, Yasutome M, et al. Overexpression of soluble vascular endothelial growth factor receptor 1 in colorectal cancer: Association with progression and prognosis. Cancer Sci 2007; 98:405 - 10; http://dx.doi.org/10.1111/j.1349-7006.2007.00402.x; PMID: 17214745
- Lamszus K, Ulbricht U, Matschke J, Brockmann MA, Fillbrandt R, Westphal M. Levels of soluble vascular endothelial growth factor (VEGF) receptor 1 in astrocytic tumors and its relation to malignancy, vascularity, and VEGF-A. Clin Cancer Res 2003; 9:1399 - 405; PMID: 12684411
- Gerber HP, Condorelli F, Park J, Ferrara N. Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes. Flt-1, but not Flk-1/KDR, is up-regulated by hypoxia. J Biol Chem 1997; 272:23659 - 67; http://dx.doi.org/10.1074/jbc.272.38.23659; PMID: 9295307
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280 - 93; http://dx.doi.org/10.1016/j.molcel.2010.09.023; PMID: 20965422
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005; 120:237 - 48; http://dx.doi.org/10.1016/j.cell.2004.11.046; PMID: 15680329