Abstract
Huntington disease (HD) is a neurodegenerative disorder caused by an expansion of a polyglutamine repeat within the HD gene product, huntingtin. Huntingtin, a large (347 kDa) protein containing multiple HEAT repeats, acts as a scaffold for protein-protein interactions. Huntingtin-induced toxicity is believed to be mediated by a conformational change in expanded huntingtin, leading to protein misfolding and aggregation, aberrant protein interactions and neuronal cell death. While many non-systematic studies of huntingtin interactions have been reported, they were not designed to identify and quantify the changes in the huntingtin interactome induced by polyglutamine expansion. We used tandem affinity purification and quantitative proteomics to compare and quantify interactions of normal or expanded huntingtin isolated from a striatal cell line. We found that proteins preferentially interacting with expanded huntingtin are enriched for intrinsically disordered proteins, consistent with previously suggested roles of such proteins in neurodegenerative disorders. Our functional analysis indicates that proteins related to energy production, protein trafficking, RNA post-transcriptional modifications and cell death were significantly enriched among preferential interactors of expanded huntingtin. Expanded huntingtin interacted with many mitochondrial proteins, including AIFM1, consistent with a role for mitochondrial dysfunction in HD. Furthermore, expanded huntingtin interacted with the stress granule-associated proteins Caprin-1 and G3BP and redistributed to RNA stress granules under ER-stress conditions. These data demonstrate that a number of key cellular functions and networks may be disrupted by abnormal interactions of expanded huntingtin and highlight proteins and pathways that may be involved in HD cellular pathogenesis and that may serve as therapeutic targets.
Introduction
Huntington disease (HD) is a neurodegenerative disorder caused by an expansion of a CAG repeat coding for polyglutamine (polyQ) within the HD gene product huntingtin (Htt).Citation1 Expansion of the polyQ in Htt beyond a threshold of 36 repeats causes HD, and there is an inverse correlation between the length of the polyQ and the age of onset.Citation2-Citation4 Htt is a large (347 kDa) protein containing multiple HEAT repeats.Citation5 HEAT repeat proteins can act as scaffolds for protein-protein interactions, and Htt has emerged as a major protein interaction hub.Citation6
The mechanisms of Htt-induced toxicity are still uncertain, but a critical initial event is believed to be a conformational change in mutant Htt protein, or its N-terminal proteolytic fragments, leading to protein misfolding and aggregation and aberrant protein interactions.Citation7-Citation9 In addition, interactions important for normal Htt function may be lost as a result of the polyQ expansion. Comparable to HD, in the case of ataxin 1 (ATXN1, another CAG expanded protein, associated with spinocerebellar ataxia 1, SCA1), polyQ expansion favors the particular interactions, including the formation of a complex containing RBM17 (RNA-binding motif protein 17), contributing to the gain-of-function mechanism of SCA1 pathology. By contrast, glutamine expansion in ATXN1 attenuates its interaction with other complexes, resulting in a partial loss of function.Citation10,Citation11 Thus changes in protein interactions are likely to be critical for polyQ pathogenesis in several situations.
Many non-systematic studies of Htt interactors have been reported, beginning with the first report of a protein interacting with Htt, termed Htt-associated protein-1 (HAP-1), identified by our group using the yeast two-hybrid system.Citation12-Citation16 In several cases, the strength of Htt protein interactions has been shown to be affected by the length of polyQ tract, including the interaction with CREB binding protein,Citation7,Citation9 HAP-1,Citation17 nuclear receptor co-repressor N-CoR,Citation18 Sp1Citation19 and others.Citation20,Citation21 Rhes, a striatal-specific guanine nucleotide-binding protein, interacts preferentially with polyQ-expanded Htt and induces its sumoylation, leading to cytotoxicity.Citation22 Interactors of Htt include several proteins related to RNA processing;Citation23,Citation24 however, their relation to the polyQ expansion has not been elucidated. Two systematic studies of the Htt interactome have been reported to date: Wanker and coworkers used yeast two-hybrid screens to describe a protein interactions network for HD.Citation6 This network includes 165 potential interactions, 32 of which were confirmed by independent binding experiments. Hughes et al.Citation25 used high-throughput yeast two-hybrid screen and affinity pull-downs followed by mass spectrometry (MS). They identified 234 Htt-associated protein candidates, 104 of which were found by two-hybrid screen, and 130 by pull-downs. Although presenting compelling evidence that both normal and expanded Htt are involved in multiple protein interactions, the above studies did not use current methods of quantitative proteomics to identify and quantify the changes in Htt interactome induced by polyQ expansion.
To establish whether amyloid-like aggregates sequester proteins with specific sequence features, Olzscha and coworkers designed artificial β-sheet proteins forming amyloid-like fibrils. In this study using quantitative proteomics, it was found that such amyloid-like aggregates sequester proteins enriched in intrinsically disordered (unstructured) regions, a feature linked to multifunctionality.Citation26
We have now used quantitative proteomics to assess the changes in Htt interactions caused by polyQ expansion that occur in a striatal neuronal precursor cell line. We used tandem affinity purification (TAP) and isobaric tags for relative and absolute quantification (iTRAQ) approaches to compare and quantify the composition of normal or expanded Htt-associated proteins isolated from a striatal precursor cell line. We found that proteins preferentially interacting with expanded Htt were enriched for intrinsically disordered proteins, consistent with previously suggested roles of such proteins in neurodegenerative disorders.Citation26,Citation27 Our functional analysis indicated that a number of key cellular functions and networks may be altered by abnormal interactions of polyQ-expanded Htt. In particular, proteins related to energy production, protein trafficking, RNA post-transcriptional modifications and cell death were significantly enriched among preferential interactors of expanded Htt. Expanded Htt interacted with many mitochondrial proteins, including AIFM1, consistent with a role for mitochondrial dysfunction in HD pathogenesis. In addition, expanded Htt interacted with the stress granule (SG)-associated proteins Caprin-1 and G3BP and redistributed to SGs under ER-stress conditions. These data highlight proteins and pathways that may be involved in HD cellular pathogenesis and that may serve as therapeutic targets.
Results
Purification of Htt protein complexes and iTRAQ procedure.
For isolation of Htt complexes, we have implemented a TAP procedure based on the InterPlay mammalian TAP system (Stratagene). This method allows purification of protein complexes under mild native conditions to preserve protein interactions. The TAP procedure includes two consecutive steps of affinity purification using streptavidin (SBP) and calmodulin (CBP) tags (). As bait we chose the Htt fragment of 586 amino acids, corresponding to the putative caspase 6 generated fragment, described by Hayden’s group, and potentially a pathologic fragment in vivo.Citation28 Previous studies have suggested that most of the relevant protein interaction domains of Htt lie within the N-terminal region, where the polyQ repeat is located. For the expanded polyQ construct, we chose a repeat length of 50 glutamines (50Q), which is well within the expanded range, and usually yields juvenile or young adult onset of HD and does not cause aggregation as rapidly in cell transfection experiments as longer polyQ expansions. The N-terminal tag was chosen, because the N586 fragment of Htt is likely to undergo further N-terminal proteolysis.Citation29,Citation30 Short N-terminal fragments of mutant Htt are believed to mediate its toxicity, possibly through aberrant protein interactions. By using a C-terminal tag, such interactions may be overlooked. The resulting TAP-N586-20Q and TAP-N586-50Q expression constructs were generated as described in the Materials and Methods.
Figure 1. Htt expression and purification of Htt complexes and iTRAQ procedure. (A) Htt constructs, used in the study, comprising the first 586 amino acids of Htt with either 20 or 50 polyQ and two N-terminal tags (streptavidin, SBP and calmodulin, CBP) for affinity purification. (B) HEK293 cells were transfected with the TAP-Htt constructs, and Htt complexes were purified using the InterPlay mammalian TAP system (as described in the Materials and Methods); Protein complexes were eluted from the calmodulin agarose in two steps: first- with 2M NaCl, and then with 2% SDS and aliquots of the samples (~10%) were separated on SDS-PAGE and stained with Silver Stain; C-non-transfected cells. (C and D) STHdh Q7/Q7 cells were transiently transfected with normal (TAP-N586–20Q) or expanded (TAP-N586–50Q) Htt fragments: Highly expressed tagged normal or mutant Htt N586 fragments (migrating at 80 or 100 kDa respectively) were detected with both MAB2166 antibody to Htt (left panel) and a specific N586 neo-epitope antibody (right panel), the low levels of endogenous full-length Htt (migrating at 300 kDa) were also detected with MAB2166 (C). Immunostaining with Htt specific MAB2166 (green) and DAPI (blue) (D). (E) Purification of Htt protein complexes from STHdh cells. SDS-PAGE analysis of the aliquots of the samples prepared for MS. Normal (20Q) and expanded (50Q) Htt complexes from STHdh cells were expressed, purified and analyzed (as described in B). The inputs (before purification) are shown. (F) The iTRAQ workflow. Each sample was labeled with a unique tag, consisting of a reporter and a balance region, and then all samples were combined, fractionated and analyzed by LC-MS/MS.
To test the new purification protocol, we conducted a pilot experiment in HEK293 cells, transfected with normal (TAP-N586-20Q) and expanded (TAP-N586-50Q) Htt fragments, followed by isolation of Htt complexes and associated proteins under native conditions by TAP. As seen from , the control lanes (C) contained virtually no proteins that non-specifically bound to the affinity matrix. The elution step presented a technical challenge: the standard elution method with calmodulin peptide is not compatible with the downstream MS analysis. Elution with 2% SDS has released more protein complexes from the beads than elution with NaCl (), so this method was used, followed by precipitation of protein complexes with trichloro-acetic acid (TCA, ).
We sought to assess changes in expanded Htt interactions in a striatal cell line, since the striatum is most relevant for HD. We employed the iTRAQ method, which allows a direct comparison of the relative amount of individual proteins in multiple complex protein mixtures.Citation31 Neuronal progenitor cells STHdh Q7/Q7 generated in the MacDonald lab from E14 striatal primordia wild-type mouse embryos,Citation32 were transiently transfected with normal (TAP-N586-20Q) or polyQ expanded (TAP-N586-50Q) Htt fragments, which produced an adequately high yield of Htt complexes required for downstream purification and iTRAQ analysis. The endogenous expression of Htt in these cells was much lower than of transfected Htt as demonstrated by western blotting (): highly expressed tagged normal or mutant Htt N586 fragments (migrating at 80 or 100 kDa, respectively) were detected with both MAB2166 antibody to Htt (left) and a specific N586 neo-epitope antibody, which reacts only with N586 fragment (right); the low levels of endogenous full-length Htt (migrating at 300 kDa) were also detected with MAB2166. Based on the immunofluorescent staining (with Htt specific antibody) of striatal cells transfected with tagged Htt constructs, we estimated the transfection efficiency to be around 40–50% (). Transfection efficiency and levels of expression were equivalent for normal and mutant Htt.
Htt complexes isolated from striatal cells using TAP procedure were eluted with 2% SDS, and small aliquots of IP material were resolved by SDS-PAGE and silver-stained to monitor the quality of the samples (). The main portion of the eluted Htt complexes after TCA-precipitation was subjected to the iTRAQ analysis, as described in the Materials and Methods: after trypsin digestion, the N termini of peptides from each sample were labeled with a unique isobaric tag with the following reporters: 114 and 115 for Htt-20Q, 116 and 117 for Htt-50Q. All samples were combined, fractionated and analyzed by liquid chromatography coupled to tandem mass spectrometry (LC-MS/ MS) (). Two independent transfections and TAPs (biological replicates) were prepared for each normal and expanded Htt sample.
Quantitative analysis of normal and expanded Htt interactome.
The MS/MS spectra were extracted and searched against the RefSeq 40 database using “Mascot” software. Peptide identifications from “Mascot” searches were processed within the “Proteome Discoverer” to identify peptides with a confidence threshold 5% false discovery rate (FDR), based on a concatenated decoy database search. A protein’s ratio is the median ratio of all unique peptides identifying the protein at a 5% FDR. The iTRAQ ratios were normalized by total protein (i.e., the average of all the ratios). Only proteins identified with median ratios > 1.2 or < 0.8 between 50Q and 20Q were considered as potential differential interactors, based on technical variation of less than 20%, as empirically determined from an eight-sample technical replicate 8-plex iTRAQ experiment. To identify proteins that were reproducibly more abundant either within normal (20Q) or expanded (50Q) Htt complexes, we quantified coefficient of variation (CV) calculated from iTRAQ ratios between two parallel biological replicates. 349 proteins, identified as differential (preferential) interactors (200 proteins—more abundant within Htt-50Q complexes, and 149-within Htt-20Q complexes), based on the above criteria and with CV values below 0.35 were included in further functional analysis (Table S1).
The effectiveness of the quantitative proteomics approach used in this study is validated by identification of 22 previously reported Htt interacting proteins, such as such as GAPDH, p53, RAC1,TCP1, caspase 2, caveolin 1, ATP synthase, cytochrome C, MAP/ERK kinase kinase 1, β catenin, CBP, BASP1, HSP70, elongation factor 1α, clathrin, dynein, actin and tubulin.
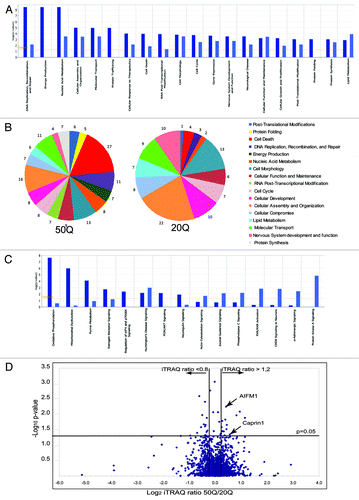
To identify main cellular functions, pathways and protein networks that may be disrupted by abnormal interactions of expanded Htt, we used Ingenuity Pathway Analysis (IPA) tools. First, we sought to determine whether Htt-20Q and Htt-50Q complexes were significantly enriched for proteins related to specific cellular functions and pathways (). We found that differential interactors are enriched in proteins involved in a variety of essential cellular functions, designated by IPA as gene expression, nucleic acid metabolism, protein synthesis, folding and posttranslational modifications, molecular transport, cell cycle and cell death. Interestingly, proteins that were more abundant within expanded Htt complexes are more enriched in molecules related to DNA replication, recombination and repair, energy production, protein trafficking, RNA post-transcriptional modifications and cell death (). A more stringent analysis (using only proteins with CV < 0.25) revealed even more striking differences between functions enriched within normal (20Q) or expanded (50Q) Htt complexes (). Thus, proteins related to cell death were overrepresented within 50Q complexes and comprised 27% of all preferential Htt-50Q interactors (vs. only 4% of preferential Htt-20Q interactors). On the contrary, proteins related to normal cellular functioning—cellular function and maintenance, cellular assembly and organization and nervous system development and function—were overrepresented within 20Q complexes, compared with 50Q complexes. Thus, IPA-based analysis of functions and pathways suggests that aberrant protein interactions of expanded Htt may interfere with established normal functions of Htt, such as protein translation, and cellular transport, while its toxic gain of function may be also induced by abnormal interactions within energy production and mitochondria-mediated intrinsic apoptotic pathways.
Figure 2. Quantitative analysis of normal and expanded Htt interactome. (A) Top cellular functions (as defined by IPA) most significantly enriched within Htt-50Q (dark blue) and Htt-20Q (light blue) preferential interactors, based on the protein data set shown in Table S1 (CV < 0.35 between duplicate samples). The graph shows the enrichment of particular functional categories based on the observed number of proteins for each group, relative to the number expected by chance. Log10 p-values are calculated by IPA. (B) The pie diagram, showing selected functional categories most significantly enriched within Htt-20Q and Htt-50Q preferential interactors, based on the IPA analysis of differential interactors with CV < 0.25 between duplicate samples. The numbers represent the percentage of differential interactors involved in a specific function, relative to the number of all differential interactors combined identified for all functional categories shown. (C) Top Canonical pathways (as defined by IPA) most significantly enriched within Htt-50Q (dark blue) and Htt-20Q (light blue) preferential interactors using the protein data set shown in Table S1. The graph shows the enrichment of particular canonical pathways based on the observed number of proteins for each pathway, relative to the number expected by chance. Log10 p-values are calculated by IPA. (D) Volcano plot showing the distribution of proteins differentially interacting with normal (20Q) and expanded (50Q) Htt based on iTRAQ ratios and p-values. The x-axis shows the Log2 of the median normalized iTRAQ ratios between 50Q and 20Q. The vertical lines indicate the threshold iTRAQ ratios required for a protein to be considered a differential interactor (> 1.2 or < 0.8).The x-axis shows the –Log10 of p-values, obtained using ANOVA approach, described in the Results. The horizontal line represents p-value of 0.05.
We also investigated whether any potential specific structural features exist that distinguish proteins preferentially interacting with expanded Htt. One hypothesis is that these might be enriched for intrinsically disordered (naturally unfolded) proteins, since intrinsically disordered proteins are involved in several kinds of diseases, especially neurodegenerative diseases, including HD.Citation26,Citation27,Citation33 We found that 76% of 50 randomly selected preferential interactors of Htt-50Q contain regions of disorder of ≥ 30 residues, as predicted by at least three out of four PONDR predictors used, compared with 47% in the total pool of eukaryotic proteins in SwissProt database.Citation27
To identify proteins most significantly changed in abundance between normal and expanded Htt complexes, we used statistical analysis of variance (ANOVA) approach, which considers both biologic and experimental sources of variability. Relative peptide and protein abundances were estimated based on collected reporter ion peak areas from all observed tandem mass spectra using linear mixed effects modelsCitation34-Citation36 (and Herbrich et al. in preparation). This allows to correct for different amount of material loaded in the channels (as a random effect) and enables a comparison between the relative protein abundances (as random effects), comparing proteins present within expanded and normal Htt complexes. Multiple comparisons were addressed by controlling the family-wise error rate via Bonferroni correction. shows a Volcano plot demonstrating the distribution of proteins differentially interacting with normal (20Q) and expanded (50Q) Htt based on iTRAQ ratios and p-values. The vertical lines indicate the threshold iTRAQ ratios required for a protein to be considered a differential interactor (> 1.2 or < 0.8). Among 33 most significant differential interactors (with ANOVA p-value less than 0.05, Table S2) we found seven proteins with mitochondrial localization or function (ACACA, MCCC2, ATPC1, IRGM, Bit-1/PTRH2, TIMM50 and AIFM1) and five RNA-binding regulators of RNA processing and transport (MCM3AP/GANP, RBM18, RPS7, SART1 and Caprin-1). These findings further emphasize a potential involvement of mitochondrial dysfunction and RNA processing pathways in expanded Htt cell toxicity.
Abnormal interactions of expanded Htt may cause mitochondrial dysfunction.
Pathways implicated in intrinsic cell death are often associated with mitochondrial processes (e.g., signaling, energy production, mitochondrial movement). Among cell death-related proteins more abundant within Htt-50Q complexes, several proteins were assigned by IPA mitochondrial location and function (e.g., IMMT, HSPE1, HSPD1, ATP5A1, VDAC1, NDUFS2, RHOT1); furthermore, several proteins related to mitochondria-mediated cell death—IRGM, Bit-1/PTRH2, TIMM50 and AIFM1—were among the most significant differential interactors (Table S2).
Analysis of abnormal Htt-50Q interactions within protein networks (as defined by IPA) also revealed potential alteration of processes related to energy production and oxidative phosphorylation, including electron transport chain and ATP synthesis (Fig. S1). In fact, oxidative phosphorylation and mitochondrial dysfunction pathways were the top two canonical pathways (as defined by IPA) most significantly enriched within Htt-50Q complexes but not enriched within Htt-20Q complexes (). Five different subunits of ATP synthases (ATP5A1, ATP5B, ATP5C1, ATP5F1 and ATP5L), components of mitochondrial complex I (four NADH dehydrogenase subunits-NDUFA8, NDUFAF4, NDUFS2, NDUFS7), complex III (two ubiquinone-cytochrome C reductase-related proteins-UQCR10, UQCRB) and complex IV (Cytochrome C-1-CYC1, cytochrome C oxidase subunit-COX4I1) were all more abundant within Htt-50Q purifications, compared with Htt-20Q purifications.
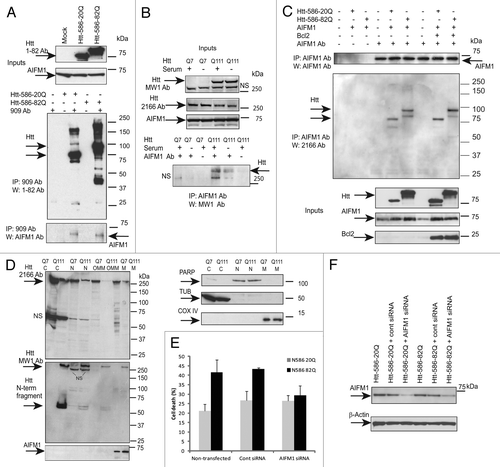
Apoptosis-inducing factor 1 (AIFM1, AIF1) was among the proteins most significantly enriched within Htt-50Q complexes (). AIFM1 has a dual function in cell: it is involved in both energy production and apoptosis,Citation37 both processes enriched within preferential expanded Htt interactors. Hence, we further conducted functional studies to elucidate potential involvement of AIFM1 in HD pathology. First, we confirmed AIFM1/Htt interactions identified by MS. demonstrates that transfected Htt-586-20Q and Htt-586-82Q co-immunoprecipitated endogenous AIFM1 in striatal STHdh Q7/Q7 cells. (The reverse experiment-IP with AIFM1 antibody and blotting with 2166 antibody to Htt is shown on ). In addition, using AIFM1 antibody for immunoprecipitations, we found that endogenous expanded Htt (detected with polyQ-specific MW1 antibody) co-precipitated with endogenous AIFM1 in STHdh Q111/Q111 knock-in cells (). Notably, the Htt/AIFM1 interaction was preserved when mitochondrial cell death was suppressed by Bcl2 overexpression ().
Figure 3. AIFM1 interacts with Htt and mediates Htt toxicity. (A) STHdh Q7/Q7 cells were transiently transfected with normal (Htt-N586–20Q) or expanded (Htt-N586–82Q) Htt fragments, lysed 48 h after transfection, and Htt complexes were immunoprecipitated using a specific antibody to Htt (909). AIFM1 was detected in the IPs from transfected cells, but not in non-transfected cells or in control samples without the primary antibody (bottom panel). IPs were also analyzed for the presence of Htt using 1–82 antibody (middle panel). The inputs are shown on the top panels. (B) STHdh Q7/Q7 and Q111/Q111 cells were grown with and without FBS for 48 h, and AIFM1 complexes were immunoprecipitated using a specific antibody to AIFM1. Expanded Htt proteins were detected in the IPs from STHdh Q111/Q111 cells using MW1 antibody recognizing expanded polyQ, but not in control samples without the primary antibody (bottom panel). The inputs are shown on the top panels: expanded Htt is detected using MW1 antibody, both normal and expanded Htt are detected with 2166 Ab; NS - non-specific bands. (C) STHdh Q7/Q7 cells were transiently co-transfected with normal (Htt-N586–20Q) or expanded (Htt-N586–82Q) Htt fragments, AIFM1 and Bcl-2, as indicated. Cells were lysed 48 h after transfection, and AIFM1 complexes were immunoprecipitated using a specific antibody to AIFM1. Htt was detected in the IPs from transfected cells, but not in mock transfections or in control samples without the primary antibody (middle panel). IPs were also analyzed for the presence of AIFM1 (top panel). The inputs are shown on the bottom panels. (D) Sub-cellular fractionation of STHdh Q7/Q7 and Q111/Q111 cells was performed as described in the Materials and Methods to obtain cytoplasmic (C), nuclear (N), outer mitochondrial membrane (OMM) and mitochondrial (M) fractions. Htt proteins were detected with 2166 MAB to Htt (top panel), or with MW1 antibody (middle panel); AIFM1 protein was detected exclusively in the mitochondrial fractions. Cytoplasmic (β tubulin, TUB), nuclear (PARP) and mitochondrial (COXIV) markers are shown. (E) AIFM1 knock-down in striatal cells attenuates toxicity of expanded Htt-N585–82Q fragment. STHdh Q7/Q7 cells were co-transfected with indicated plasmids and siRNAs, fixed 48 h later, co-stained with an antibody to Htt (1–82) and with Hoechst 33258, and the nuclei staining intensity was analyzed as described in Materials and Methods. The data are presented as percentage of surviving cells among transfected cells. About 250–600 transfected cells were counted for each condition, and the experiment was repeated 3 times (*n = 3, p = 0.01; **n = 3, p = 0.03). (F) blots demonstrating AIFM1 knock-down upon transfection with AIFM1 siRNA, but not with control siRNA
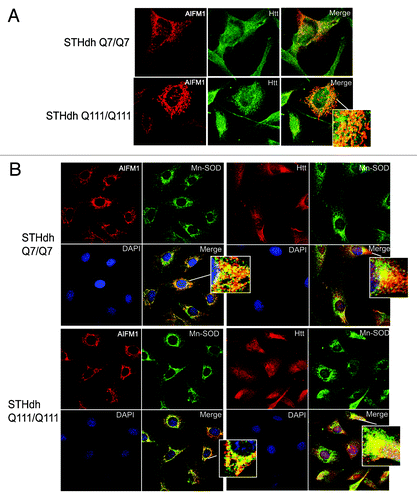
Our cell fractionation experiments indicate that in healthy cells, both normal (Q7) and polyQ-expanded (Q111) Htt were present mostly in cytoplasm, with considerable nuclear and mitochondrial localization as well (, top part). Additional fractionation of mitochondrial fraction using digitonin (see Materials and Methods) demonstrated that normal (Q7) and polyQ-expanded (Q111) Htt were present in both outer mitochondrial membrane fraction (OMM) and within remaining mitochondrial pellet (M), which included intermembrane space (IMS), inner membrane (IMM) and matrix. Notably, using MW1 antibody specific for polyQ, we were able to detect the N-terminal fragments of expanded Htt present in cytoplasm, nucleus and OMM but not within the mitochondria, where only full-length Htt was detected (, middle part). AIFM1 was almost exclusively found in mitochondrial fraction M, but not in OMM (, bottom part). We further examined the subcellular co-localization of AIFM1 and Htt using immunofluorescent confocal microscopy (). Co-staining of AIFM1 and endogenous Htt in STHdh cells demonstrated their partial co-localization (). Both proteins also partially co-localized with mitochondrial protein Mn-SOD in both STHdh Q7/Q7 and Q111/Q111 cells (). These two sets of experiments suggest that AIFM1 and Htt may interact within mitochondria, and that full-lenth Htt but not Htt fragments may be the primary species involved.
Figure 4. Htt and AIFM1 partially co-localize in association with mitochondria. (A) STHdh Q7/Q7 (top) and Q111/Q111 (bottom) cells were fixed 48 h after plating. Confocal immunofluorescent detection of Htt with 2166 monoclonal antibody is shown in green (Alexa Fluor 488); detection of AIFM1 with polyclonal specific antibody is shown in red (Alexa Fluor 555); Yellow staining in merged images demonstrates partial co-localization. (B) Confocal immunofluorescent images of STHdh Q7/Q7 (top) and Q111/Q111 (bottom) cells. Detection of AIFM1 with polyclonal specific antibody and of Htt with rabbit polyclonal antibody to Htt epitope 1–17 are shown in red (Alexa Fluor 555); detection of Mn-SOD with mouse monoclonal antibody is shown in green (Alexa Fluor 488); The nuclear staining (DAPI) is shown in blue; Yellow staining in merged images demonstrates partial co-localization of Htt and AIFM1 with mitochondrial marker Mn-SOD.
The AIFM1-mediated pathway is not critical for all cell death mechanisms.Citation37 AIFM1 plays an important role in cell death only in certain cell types, such as neurons and some tumor cells. We used RNAi silencing to determine whether AIFM1-mediated cell death pathway is activated in HD models and can mediate mutant Htt toxicity. demonstrates that AIFM1 knockdown in striatal cells attenuates toxicity of expanded Htt-N585-82Q fragment in striatal cells.
Expanded Htt complexes are enriched in proteins related to RNA processing and regulation of translation.
Figure S2 shows a more extended network (merged two top networks defined by IPA) that includes differential interactors of Htt. This network combines proteins related to gene expression, RNA post-transcriptional modifications and protein synthesis. Among proteins that were more abundant within Htt-50Q complexes (shown in red), we found a number of chromatin-associated proteins/transcriptional regulators—SMARCC1, SMARCA4, SIN3A and SUZ12. Interestingly, mSin3A immunoreactivity was previously observed in Htt-positive intranuclear inclusions.Citation18
As shown in , Htt-50Q preferential interactors were more enriched in proteins related to RNA post-transcriptional modifications. Twenty-eight proteins related to RNA processing and regulation of translation were identified as preferential interactors of Htt-50Q (Table S1). The network analysis (Fig. S2) also showed that several regulators of RNA transcription and splicing regulatory proteins (CTNNB1, SF3B4, SF3B14, SRPK2, SMNDC1, PRPF4) bound to Htt-50Q at the greater degree as well as a number of RNA-binding translational regulators—EIF4G1, NOL6, Caprin-1 and G3BP.
Caprin-1 (RNG 105, RNA granule protein 105) is expressed in brain in postsynaptic stress granules (SG) in dendrites in the hippocampus and neocortex, where it acts as a translational repressor by directly binding mRNAs and via induction of phosphorylation of eIF-2.Citation38,Citation39 Caprin-1 was shown to heteromerize and co-localize with G3BP1, a SG marker and an effector of SG assembly, in cytoplasmic RNA granules associated with microtubules. Furthermore, Caprin-1/G3BP1 complex is implicated in the regulation of the transport and translation of mRNAs of proteins involved in the regulation of synaptic plasticity in neurons.Citation39 Since both Caprin-1 and G3BP1 came up in our screen as preferential interactors of Htt-50Q, we further investigated whether expanded Htt may associate with and affect the assembly of SGs in striatal cells.
Htt redistributes to stress granules under the ER-stress conditions, where it co-localizes with Caprin-1 and a SG marker G3BP1.
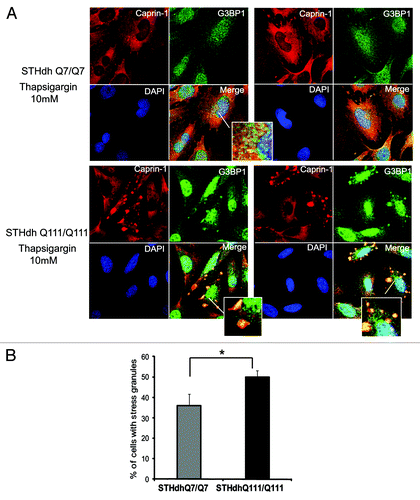
First, we confirmed Caprin-1/Htt interactions, identified by MS (). We demonstrated that transfected Htt‑586‑20Q and Htt-586-82Q co-immunoprecipitated endogenous Caprin-1 in striatal STHdh Q7/Q7 cells (). In addition, using Caprin-1 antibody, we found that endogenous expanded Htt (detected with polyQ-specific MW1 antibody) co-precipitated with endogenous Caprin-1 in STHdh Q111/Q111 knock-in cells (). Next we examined the subcellular co-localization of Caprin-1, G3BP1 and Htt using immunofluorescent confocal microscopy (). Co-staining of Caprin-1 and G3BP1 with endogenous Htt in STHdh cells under normal conditions showed very little or no co-localization ( and data not shown). The formation of SGs in cells is induced by stresses such as heat, arsenite and the ER stress response. We used thapsigargin (10 μM) to induce ER stress and the formation of SGs in STHdh Q7/Q7 and Q111/Q111 cells. As expected, under the ER stress conditions, both Caprin-1 and G3BP1 redistributed to the SGs in both STHdh Q7/Q7 and Q111/Q111 cells (). We further found that in cells treated with thapsigargin, both normal and expanded Htt also redistriduted to the SGs, where it co-localized with both Caprin-1 and G3BP1 (). Interestingly, cells expressing expanded Htt formed SGs more robustly than normal cells: we observed more cells containing Caprin-1- and G3BP1-positive SGs (), and SGs in these cells were generally larger (). Thus, our findings suggest that Htt is involved in the regulation of RNA processing and translation under the stress conditions, and that expanded Htt may alter this process via its abnormal interaction with RNA-binding SG-associated proteins Caprin-1 and G3BP.
Figure 5. Confirmation of Caprin- 1/Htt interaction by IP-Western blotting. (A) STHdh Q7/Q7 cells were transiently transfected with normal (Htt-N586–20Q) or expanded (Htt-N586–82Q) Htt fragments, lysed 48 h after transfection, and Htt complexes were immunoprecipitated using a specific antibody to Htt (909). Endogenous Caprin -1 was detected in the IPs from transfected cells, but not in non-transfected cells or in control samples without the primary antibody (bottom panel). IPs were also analyzed for the presence of Htt using 1–82 antibody (middle panel). The inputs are shown on the top panels. (B) STHdh Q7/Q7 and Q111/Q111 cells were grown for 48 h with or without incubation with 10 μM thapsigargin for 50 min, and Caprin-1 complexes were immunoprecipitated using a specific antibody to Caprin -1. Expanded Htt proteins were detected in the IPs from STHdh Q111/Q111 cells using MW1 antibody recognizing expanded polyQ, but not in control samples without the primary antibody (bottom panel). NS- non-specific bands . The inputs are shown on the top panels: Normal and expanded Htt proteins were detected using 2166 antibody.
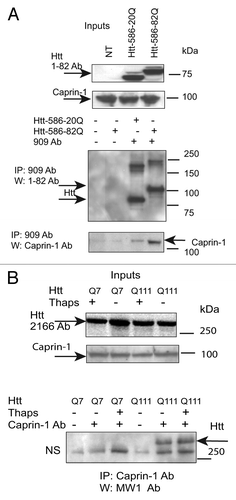
Figure 6. Htt redistributes to cytoplasmic stress granules (SG) under the ER-stress conditions, where it co-localizes with Caprin-1 and a SG marker G3BP1. (A) STHdh Q7/Q7 (left) and Q111/Q111 (right) cells were fixed 48 h after plating. Confocal immunofluorescent detection of Htt with 2166 monoclonal antibody is shown in green (Alexa Fluor 488); detection of Caprin-1 with polyclonal specific antibody is shown in red (Alexa Fluor 555); The nuclear staining (DAPI) is shown in blue; Merged images demonstrates little or no co-localization. (B) STHdh Q7/Q7 and Q111/Q111 cells were treated with 10 μM thapsigargin for 50 min before fixing, to induce ER stress. Htt and Caprin-1 were detected as described above. Yellow dots in merged images demonstrate Htt and Caprin-1 co-localization in stress granules. (C) STHdh Q7/Q7 and Q111/Q111 cells were treated with 10 μM thapsigargin for 50 min before fixing, to induce ER stress. Confocal immunofluorescent detection of Htt with rabbit polyclonal antibody to epitope 1–17 is shown in red (Alexa Fluor 555); detection of G3BP1 with mouse monoclonal antibody is shown in green (Alexa Fluor 488); The nuclear staining (DAPI) is shown in blue; Yellow dots in merged images demonstrates Htt and G3BP1 co-localization in stress granules.
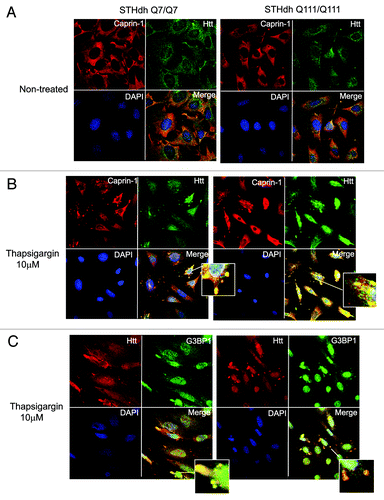
Figure 7. STHdh Q111/Q111 cells expressing expanded Htt form stress granules more robustly than normal Q7/Q7 cells. (A) STHdh Q7/Q7 (top) and Q111/Q111 (bottom) cells were treated with 10 μM thapsigargin for 50 min before fixing, to induce ER stress. Caprin-1 and G3BP1 were detected as described in the legend to . (B) Graph shows percentage of Q7/Q7 and Q111/Q111 cells containing stress granules (Caprin-1 and G3BP1- positive) upon treatment with 10 μM thapsigargin for 50 min before fixing, as determined by the presence of yellow dots in the merged images as shown in A. Total 150 cells were counted for each cell line (*n = 3, p = 0.02).
Discussion
The mechanism of polyQ-expanded Htt induced toxicity is believed to involve a conformational change in mutant Htt protein or its N-terminal proteolytic fragments, which leads to abnormal protein interactions, resulting in cellular dysfunction and death. Previous studies of the Htt interactome, based on yeast two-hybrid screens and affinity pull-downs followed by MS, were not designed to compare normal and expanded Htt protein interactions. Here, we used, for the first time, quantitative proteomics to assess and quantify the changes in Htt interactome induced by polyQ expansion in striatal neuronal precursor cell line, the most relevant for HD cell type. Our findings support the hypothesis that polyQ expansion induces abnormal interactions of Htt, which may disrupt key cellular functions and networks. We showed that among the most altered are Htt interactions with several mitochondrial proteins, including AIFM1, consistent with a role for mitochondrial dysfunction in HD pathogenesis. Furthermore, our data suggest a novel role of mutant Htt in RNA processing and regulation of translation via its interaction and co-localization with SG-associated RNA-binding proteins Caprin-1 and G3BP1 in striatal cells under the ER-stress conditions.
The N-terminal segment of Htt possesses a mix of hydrophobic and hydrophilic amino acids and is predicted to have both significant α helix potential and a tendency toward a compact structure in the presence of a binding partner.Citation40,Citation41 CD spectra and molecular dynamics simulationsCitation42 suggest that the N-terminal sequence by itself has a tendency to take on some α-helical secondary structure, while expanded polyQ was shown to aquire a β-sheet conformation.Citation43 Formation of amyloid-like fibrils with β-sheet structure is observed with many protein sequences and is linked to many neurodegenerative conditions, including Parkinson, Huntington and Alzheimer diseases. If the underlying mechanism is mediated by aberrant interactions of aggregates with other cellular proteins, resulting in their sequestration and functional impairment, are there specific sequence structural features which make cellular proteins especially prone to such sequestration? In a recent report, the authors used quantitative proteomics to study interactions of artificial β-sheet proteins forming amyloid-like fibrils.Citation26 They found that these interactors are particularly enriched in intrinsically disordered (unstructured) regions, a feature linked to multifunctionality. Consistent with these data, we found that preferential interactors of expanded Htt-N586 were also enriched in such metastable disordered regions, which further supports previously suggested role of intrinsically disordered proteins in neurodegenerative disorders.Citation26,Citation27
The hypothesis that a structural conformational change in expanded Htt leads to abnormal protein interactions was recently challenged in a study designed to assess the influence of Htt aggregation on its interactions.Citation44 The authors found that in the absence of aggregation, normal and expanded Htt interacts with endophillin-3 with similar affinity, as measured by surface plasmon resonance, suggesting that expanded polyQ per se does not alter interactions. The authors demonstrate that Htt aggregates strongly affect the outcome of the pull-down experiments and propose that aggregates are forming molecular platforms that influence the Htt-interacting network. Although we did not specifically control for the absence of aggregates in our experimental system, we observed neither visible aggregate formation in striatal cells transfected with expanded Htt, nor accumulation of SDS-insoluble material on the top of the gel. However, we have observed high molecular weight Htt species formed in cells transfected with Htt constructs ( and , middle parts). Although these species were more evident after immunoprecipitation, we often observed trace amonts of similar material also in the analysis of inputs. We think that these may represent soluble oligomeric species of expanded Htt formed in cells, thus presenting efficient molecular platforms that perturb Htt interactions,Citation44 and, consequently, our results may partially reflect binding to these oligomers. Whether abnormal interactions of expanded Htt are mediated by expanded polyQ, per se, or by certain aggregated or soluble oligomeric forms of expanded Htt, the understanding of such changes in Htt interactome helps to illuminate the pathways and molecular targets affected, with a potential to develop new HD therapies.
Mitochondrial dysfunction has been previously implicated in HD pathogenesis (reviewed in ref. Citation45), although the mechanism of Htt involvement is still elusive. We found that expanded Htt complexes were particularly enriched in proteins assigned (by IPA) mitochondrial function and localization, while protein network analysis revealed potential disruption of processes related to energy production and oxidative phosphorylation. The biochemical interaction of N-terminal Htt fragments with mitochondria was previously demonstrated in HD knock-in mouse brainCitation46 and mutant Htt oligomers co-localize with mitochondrial proteins COX1 and cytochrome C in brain sections of HD patients.Citation47 However, it is not clear whether Htt or its fragments can enter mitochondria. Our cell fractionation experiments demonstrated that that normal (Q7) and polyQ-expanded (Q111) Htt were present in OMM and within mitochondrial fraction, which included intermembrane space, inner membrane and matrix; however, the N-terminal fragments of expanded Htt were only detected in OMM (). We further demonstrated that expanded Htt interacted and co-localized with mitochondrial protein AIFM1. AIFM1 is attached to the inner mitochondrial membrane, where it exerts NADH oxidase activity,Citation48 while mutations in this gene cause oxidative phosphorylation deficiency.Citation49-Citation51 Our biochemical fractionation confirmed the exclusive mitochondrial localization of AIFM1 in our experimental conditions. Subcellular co-localization of Htt and AIFM1 was also associated with mitochondria, with both proteins co-localizing with mitochondrial marker MnSOD (), further supporting the notion that mitochondria may be the predominant site of Htt/AIFM1 interaction. We found only the full-length Htt, but not its cleavage fragments, in the mitochondria, indicating specificity and suggesting that full-length Htt may be selectively transported into mitochondria, which may warrant further study.
AIFM1 plays a critical role in mitochondria-mediated cell death in certain cell types, including neurons.Citation37 Induction of apoptosis results in the translocation of AIFM1 to the nucleus, where it binds DNA and affects chromosome condensation and fragmentation, although the precise mechanism of AIFM1 nuclear function is unknown.Citation48,Citation52-Citation55 We showed that AIFM1 knockdown in striatal cells attenuated toxicity of expanded Htt-N585-82Q fragment (). However, we found no evidence of substantial nuclear co-localization of normal or expanded Htt with AIFM1 in our experimental conditions. To further clarify the possible role of Htt/AIFM1 interaction in mitochondria-mediated cell death, we examined whether this interaction is altered when mitochondrial death is suppressed. Bcl-2 is a multifunctional inhibitor of apoptosis that has been suggested to guard mitochondrial integrity and to control the release of mitochondrial proteins into the cytoplasm.Citation56 Mitochondrial release of AIFM1 was also shown to be blocked by Bcl-2 at the cleavage step in the IMS.Citation49 Our results indicate that Htt/AIFM1 interaction is preserved when mitochondrial death (and possibly the release of AIFM1 to the cytoplasm) is suppressed by Bcl2 overexpression. This is consistent with the predominant Htt/AIFM1 interaction within mitochondria. Taken together, these results suggest that AIFM1-mediated cell death pathway is activated in HD models, and that Htt and AIFM1 interaction within or in association with mitochondria, in part mediates mutant Htt toxicity.
Multiple transcript variants of AIFM1 arise from alternative splicing. Existence of a brain-specific isoform, AIF2, utilizing an alternative exon 2b, was demonstrated recently in reference Citation55. AIF2 has a different IMM sorting signal that results in a stronger membrane attachment, thus AIF2 is less likely (than AIFM1) to translocate to the nucleus and mediate cell death. Since AIF2 dimerizes with AIFM1, potentially also preventing a release of AIFM1 from mitochondria, AIF2 may have a neuroprotective function. Thus it is possible that the loss of the brain-specific isoform, AIF2 (but not AIFM1), mediates CNS defects in AIF-difficient models. This hypothesis may also explain why only neurons are affected in Harlequin (Hq) mice,Citation49 expressing less than 20% of normal levels of both AIFM1 and AIF2.Citation55 These mice develop blindness, ataxia and neurodegeneration.Citation49 Expression of AIF2 increases dramatically as neuronal precursor cells differentiate. Furthermore, AIF2 expression is restricted to developing and adult brain, with AIF2/AIFM1 ratio particularly increased in caudate nucleus and nucleus accumbens.Citation55 These findings are of particular interest, since striatum is the most affected brain region in HD, and this may signify a possible interplay between brain-specific isoforms of AIF and mutant Htt.
In our iTRAQ experiments we identified two unique peptides (corresponding to AIFM), both downstream of exon 2, which is alternatively utilized in AIFM1/AIF2 isoforms. AIFM1 siRNA used in this study did not specifically target either splice variant. Thus both interaction/co-localization with Htt and attenuation of mutant Htt toxicity may be attributed to either or both AIFM1 and AIF2 isoforms. Future studies utilizing AIF2-specific reagents (antibodies and exon 2b-specific siRNAs) will elucidate possible roles of AIF isoforms in mutant Htt pathogenesis.
We found that Htt interacted and co-localized with SG-associated RNA-binding proteins Caprin-1 and G3BP1 in striatal cells under the ER-stress conditions. Both proteins were enriched within expanded Htt complexes, as shown by quantitative proteomics analysis. Caprin-1/G3BP1 physical and functional interaction has been shown to be involved in synaptic plasticity and neuronal network formation.Citation57
In the brain, Caprin-1 (RNG 105) is expressed in postsynaptic granules (stress granules, SGs) in dendrites in the hippocampus and neocortex.Citation38 It is found in messenger ribonucleoprotein particles (mRNPs) that also contain RNA-binding proteins like hnRNPK, PABP-1 Staufen, β tubulin and the motor protein dynein.Citation58 SG formation occurs in cells exposed to environmental stress and is usually induced by stalled preinitiation complexes accumulating due to phosphorylation of eukatyotic translation initiation factor 2α (eIF2α), which blocks translation initiation. SGs contain mRNAs associated with small ribosomal subunit and certain translation factors, and SG assembly is promoted by any one of the RNA-binding proteins, many of which are able to oligomerize. Such proteins include TIA-1, FXR1, G3BP1 and others.Citation59 Caprin-1 was shown to bind directly to mRNAs and repress translation via induction of phosphorylation of eIF2α;Citation38,Citation39 Caprin-1 associates and colocalizes with G3BP-1 in cytoplasmic RNA granules associated with microtubules.Citation39 RasGAP-associated phosphorylation-dependent endoribonuclease G3BP (RasGAP SH3 domain binding protein-1) is a marker for SG and an effector of SG assembly.Citation60,Citation61 Caprin-1-localizing granules are specifiically associated with CaMKII, CREB, MAP2, BDNF and TrkB mRNAs.Citation38 Thus Caprin-1/G3BP-1 complex is likely to regulate the transport and translation of mRNAs of proteins involved with synaptic plasticity in neurons, including BDNF.
BDNF has been linked to HD pathogenesis, since its levels are reduced in HD patients (reviewed in ref. Citation62). The deregulation of BDNF gene transcription and defects in the microtubule-dependent transport of vesicles containing BDNF were also reported in HD neurons.Citation63,Citation64 A recent study from Tanese’s group demonstrates co-localization of BDNF mRNA with Htt, Ago2, CPEB and dynein in neuronal granules of cultured cortical neurons and the rat cortex.Citation65 The authors propose that Htt may play a role in post-transcriptional transport/targeting of mRNA for BDNF, thus contributing to neuronal survival. Our new discovery that Htt associates with Caprin-1/G3BP1 complex in the SGs of striatal cells supports these ideas and suggests a new mechanism of regulation of BDNF mRNA processing and translation.
Neurons contain several types of RNA granules, which may contain specific and shared components. Mutant Htt associates with Ago2 in P bodies and contributes to RNA-mediated gene silencing.Citation23,Citation24 P bodies, unlike SG, are not associated with ribosome and mostly serve for storage and degradation of repressed mRNAs. P bodies contain decapping enzymes and endonucleases and may associate with components of RISC complex (argonaute, DICER). In our experiments, Htt associated with neuronal granules mostly resembling SG, since their formation was induced by ER stress, and they were positive for Caprin-1 and G3BP1, markers for SG. We also found other RNA-binding proteins associated with SG-PABP1, FXR1, Eif4G, dynein, FUS/TLS and TDP-43 among Htt-associated proteins (data not shown). In neurons, transport of translationally silenced mRNAs to dendritic synapses for translation occurs in the neuronal post-synaptic RNA granules, containing both small and large ribosomal subunits and translation initiation factors. Future studies will determine if normal or mutant Htt is also involved in these processes.
RNA metabolism has been increasingly implicated in a variety of motor neuron and neurodegenerative disorders. The mechanisms include all steps of RNA processing, such as RNA synthesis, splicing, localization, transport and translation.Citation66-Citation70 A major contribution to the field was a discovered link between two RNA-binding proteins, TDP-43 and FUS/TLS, and the pathogenesis of amyotrophic lateral sclerosis (ALS) and other neurodegenerative disorders. Postmortem analysis of patients with different polyQ diseases, including HD, showed the association of TDP-43 and FUS/TLS with intranuclear and cytoplasmic inclusions and co-localization with Htt (reviewed in ref. Citation71). RNA processing has been previously implicated in HD based on several expression profiling studies in cells, mouse, yeast and fly models of HD.Citation72-Citation76 These studies demonstrate enrichment in ribosomal and RNA-processing proteins. In line with this, we showed that expanded Htt complexes were enriched in proteins related to RNA processing and regulation of translation and identified several novel Htt interactors within these pathways. It should be noted that many interactions of Htt identified in our studies may be indirect and are perhaps mediated by other proteins or even RNAs. The exact composition of Htt protein and RNA complexes will be determined by further studies.
In summary, our work presents the first systematic assessment of cellular processes that may be disrupted by abnormal interactions of expanded Htt. This information will serve as a platform for emerging studies that may pinpoint the key pathways and molecules involved, with a potential to identify new therapeutic targets for HD.
Materials and Methods
Plasmids and mutagenesis.
To generate the TAP-N586-20Q and the TAP-N586-50Q expression plasmids, we amplified the N586-20Q and the N586-50Q fragments of Htt using the Htt N586-20Q and the Htt-N586-50Q constructs as templates and the primers (forward-GTA AAG ATC CAT GGC GAC CCT GGA AAA G; reverse-CGC GGT CGA CTT AGT CTA ACA CAA TTT C) incorporating BamHI and SalI restriction sites and Start and Stop codons for sub-cloning in the pNTAP-B vector (Stratagene). The Htt N586-20Q plasmid was a kind gift from Michael Hayden and was described previously in reference Citation28. The Htt-N586-82Q plasmid was a gift from David Borchelt (University of Florida). The Htt-N586-50Q construct was produced from N511-82Q by random contractions of polyQ repeat in bacterial cells.
Cell culture and transfection.
Human embryonic kidney (HEK) 293FT cells are from Invitrogen. Striatal STHdh neuronal progenitor lines were generated in the MacDonald lab from E14 striatal primordia of wild-type (Q7) or HdhQ111 knock-in mouse embryos.Citation32 Cells were grown in DMEM (with 4.5 g/L D-Glucose, Invitrogen) supplemented with 10% FBS, 100 μg/ ml Geneticin, 100 units/ml penicillin and 100 units/ml streptomycin. All cells were transfected with constructs encoding Htt using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s protocol. To induce ER stress, cells were treated with 10 μM thapsigargin for 50 min and harvested or fixed immediately after that.
Purification of Htt complexes for mass spectrometry.
Htt complexes expressed in HEK293 and STHdhQ7/Q7 cells were purified using the InterPlay mammalian TAP system kit (Stratagene) according to the manufacturer’s protocol. Total cell extracts were prepared 48 h after transfection using a buffer provided in the kit (Stratagene) and manufacturer’s recommended protocol (three freeze/thaw cycles). The last elution step after purification was performed with 2% SDS (10 min, 80°C). Eluted proteins were TCA-precipitated prior to iTRAQ analysis using 2-D Clean-Up kit (GE Healthcare). Sample preparation was monitored by SDS-PAGE, followed by silver staining using SilverQuest Silver Staining Kit (Invitrogen) according to manufacturer’s protocol.
iTRAQ procedure and LC-MS analysis.
The iTRAQ proteomic technology is based on amine-reactive isobaric tagging reagents that label peptides in a mixture of proteolysed proteins. After proteolysis using trypsin, samples were dried to 40 μl, and peptides were labeled with an isobaric tag by adding 100 ul of an iTRAQ reagent (dissolved in isopropanol) at room temperature for 2 h. After labeling, all samples were mixed and dried to a volume of 200 μL and fractionated by strong cation exchange (SCX) chromatography on an Agilent 1200 Capillary HPLC system using a PolySulfethyl A column. Each SCX fraction was redissolved in 0.2% TFA and separated on a C18 column with an 8 μm emitter tip using 5–40% B (90% acetonitrile in 0.1% formic acid) gradient over 60 min at 300 nl/min. Eluting peptides were sprayed directly into an LTQ Orbitrap Velos mass spectrometer (ThermoScientific, www.thermo.com/orbitrap) through an 1 μm emitter tip at 1.6 kV. Survey scans (full ms) were acquired from 350–1800 m/z with up to 10 peptide masses (precursor ions) individually isolated with a 1.2 Da window and fragmented (MS/MS) using a collision energy of 45 and 30 sec dynamic exclusion. Precursor and the fragment ions were analyzed at 30,000 and 7,500, respectively.
Data analysis.
The MS/MS spectra were extracted and searched against the RefSeq 40 database using Mascot (Matrix Science) through Proteome Discoverer software (v1.3, Thermo Scientific) specifying sample’s species, trypsin as the enzyme allowing one missed cleavage to identify peptides with a confidence threshold 5% false discovery rate, based on concatenated decoy database search. A protein’s ratio is the median ratio of all unique peptides identifying the protein at a 5% FDR. The iTRAQ ratios were normalized by total protein, i.e., the average of all the ratios. Only proteins identified with ratios > 1.2 or < 0.8 were considered potential differential interactors (technical variation is less than 20%, as empirically determined from an eight-sample technical replicate 8-plex iTRAQ experiment). Functional analysis was performed using Ingenuity Pathways Analysis (IPA) software with Ingenuity Knowledge Base reference set (genes only). Network analysis included direct and indirect relationships with set 35 molecules per network and 25 networks per analysis. Statistical analysis was performed using ANOVA approach. Relative peptide and protein abundances were estimated based on collected reporter ion peak areas from all observed tandem mass spectra using linear mixed effects modelsCitation34,Citation35 (and Herbrich et al. manuscript in preparation). Multiple comparisons were addressed by controlling the family-wise error rate via Bonferroni correction. Analysis for intrinsically disordered regions in proteins was performed using Predictor of Naturally Disordered Regions (PONDR) using VLXT, XL1_XT, CAN_XT and VL3-BA predictors.
Western blotting and immunoprecipitation.
For western blotting analysis, cells were lysed 48 h after transfection in M-PER buffer (Pierce) with protease inhibitors (PIC, Protease Inhibitor Cocktail III, Calbiochem), and protein concentrations were estimated using BCA method (Biorad). Lysates were fractionated on NuPAGE 4–12% Bis-Tris polyacrylamide gels (Invitrogen), transferred to nitrocellulose membranes and probed with antibodies to Htt, AIFM1, Caprin-1 and cytoplasmic, nuclear and mitochondrial markers. Immunoblots were developed with peroxidase-conjugated secondary antibodies (Amersham) and enhanced chemi-luminescence (ECL-Plus detection reagent, Amersham). Protein bands were quantitated using Molecular Imager Gel Doc XR System and Quantity One software (Biorad). For immunoprecipitation, cells were lysed 48 h after plating or after transfection in a lysis buffer (50 mM Tris, pH 7.0, 150 mM NaCl, 5 mM EDTA, 50 mM MgCl2, 0.5% Triton X100 and PIC), followed by centrifugation at 13,000 g. Lysates were pre-cleared by incubating with Protein G-Sepharose beads (GE Healthcare) for 1 h at 4°C, followed by the incubation (ON at 4°C) with primary antibodies to Htt (909 antibody), to AIFM1 and to Caprin-1 and then were incubated with Protein G-Sepharose for 1 h at 4°C. The immunoprecipitates were washed three times with the lysis buffer, and protein complexes were eluted from the beads with 2xSDS Laemmli sample buffer (Biorad) and fractionated on SDS-PAGE as described above.
Antibodies.
Polyclonal antibody to Htt-Htt 1–17 (against residues 1–17) was described previously in reference Citation77; goat polyclonal 909 antibody, prepared against the N-terminal Htt exon-1 fragment was described previously in reference Citation78; Htt monoclonal 2166 antibody (against residues 181–810 of Htt) and 1–82 antibody (against residues 1–82 of Htt) were from Millipore; a specific N586 neo-epitope antibody was a gift from Michael Hayden; MW1 monoclonal antibody to expanded polyQ was a gift from Paul Patterson;Citation79 AIFM1 polyclonal antibody was from Cell Signaling Technologies; Caprin-1 polyclonal antibody was from Sigma; G3BP1 monoclonal antibody was from Santa-Cruz Biotechnology; monoclonal antibody to Mn-SOD was from Santa-Cruz Biotechnology; PARP antibody was from Cell Signaling Technologies; β-tubulin antibody was from Sant-Cruz Biotechnology; COXIV polyclonal antibody was from Cell Signaling Technologies.
Preparation of subcellular fractions.
STHdh Q7/Q7 and Q111/Q111 cells were harvested and washed twice with ice-cold PBS followed by resuspending the cell pellet in a hypotonic buffer A (300 mM sucrose, 10 mM HEPES-K+ pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 0.5 DTT) in the presence of PIC. Cells were then lysed with Dounce homogenizer and centrifuged for 10 min at 600 g. The nuclear pellets were washed with buffer A, followed by resuspending in buffer C (20 mM HEPES-K+ pH 7.9, 420 mM NaCl, 0.2 mM EDTA, 1.5 mM MgCl2, 0.5 DTT, 25% Glycerol) with PIC. Nuclei were incubated on ice for 30 min and vortexed periodically. Supernatants containing nuclear proteins (fraction N) were collected by spinning at 20,000 g for 10 min at 4°C. The supernatants from the low speed centrifugation were transferred to a fresh tube and centrifuged for an additional 15 min at 7,000 g. The subsequent supernatants were centrifuged at 12,000 g for 10 min at 4°C, and the supernatants were used as cytoplasmic fractions (C). The pellets from the 7,000 g spin, containing mitochondrial proteins, were washed with buffer A and resuspended in 0.4 mg/ml digitonin (prepared in buffer A), followed by incubation with stirring for 15 min at 4°C to solubilize the outer mitochondrial membrane.Citation80 The supernatants (outer mitochondrial membrane fraction OMM) were collected by centrifugation (12,000 g, 10 min). The pellets were washed with buffer A and lysed in Laemmli SDS buffer (BioRad) and used as mitochondrial fraction (M).
Cell toxicity assay in transfected STHdh cells.
The assay is based on nuclear condensation measured by the Hoechst staining intensity of cell nuclei. STHdh Q7/Q7 cells were grown in 24-well plates, transfected with either normal (N586-20Q) and expanded (N586-82Q) Htt plasmids and co-stained with 1–82 antibody to Htt and with Hoechst 33,258 (Invitrogen) nuclear stain. Images were acquired using the Axiovision imaging software on an Axiovert 100 microscope (Carl Zeiss) with the automated Mozaix function Analysis was performed using the Velocity software (Perkin-Elmer). Cells were considered as not viable when their DAPI intensity was higher than 200% of the control intensity.
Immunofluorescence.
STHdh Q7/Q7 and Q111/Q111 cells were fixed (48 h after plating) with 4% paraformaldehyde for 15 min, permealized with 0.5% Triton X-100 (Sigma) for 10 min, blocked in 10% normal donkey serum (Sigma) for 30 min, and incubated with the following primary antibodies (ON at 4°C): for Htt and AIFM1 co-localization studies, with monoclonal antibody 2166 to Htt and rabbit polyclonal antibody to AIFM1; for Htt and Mn-SOD co-localization, with rabbit polyclonal antibody to Htt epitope 1–17 and mouse monoclonal antibody to Mn-SOD; for Htt and Caprin-1 co-localization, with monoclonal antibody 2166 to Htt and rabbit polyclonal antibody to Caprin-1; for Htt and G3BP1 co-localization, with rabbit polyclonal antibody to Htt epitope 1–17 and mouse monoclonal antibody to G3BP1. Secondary antibodies used: donkey anti-rabbit Alexa Flour 555 and donkey anti-mouse Alexa Flour 488. Nuclei were stained with Hoechst 33,258 (Invitrogen). Confocal microscopy was performed using a Zeiss Axiovert 200 inverted microscope with 510-Meta confocal module and 63x objective.
| Abbreviations: | ||
| HD | = | Huntington disease |
| Htt | = | huntingtin |
| polyQ | = | polyglutamine |
| aa | = | amino acid |
| DMEM | = | Dulbecco’s modified Eagle’s medium |
| FBS | = | fetal bovine serum |
| HEK | = | human embryonic kidney |
| AD | = | Alzheimer disease |
| iTRAQ | = | isobaric tags for relative and absolute quantification |
| LC-MS | = | liquid chromatography-mass spectrometry |
| SG | = | stress granules |
| ER | = | endoplasmic reticulum |
| IMM | = | inner mitochondrial membrane |
| OMM | = | outer mitochondrial membrane |
| IMS | = | intermembrane space |
| PIC | = | protease inhibitors cocktail |
| IP | = | immunoprecipitate |
| TCA | = | trichloro-acetic acid |
Additional material
Download Zip (444.8 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This research was supported by NINDS grant NS16375.
Acknowledgments
We thank David Borchelt (University of Florida) for providing us with the Htt N586–82Q expression construct, Michael Hayden (University of British Columbia) for the Htt-N586 expression constructs and N586 neo-epitope antibody, Paul Patterson (California Institute of Technology) for MW1 monoclonal antibody, and Marcy MacDonald (Massachusetts General Hospital) for STHdh striatal cell lines.
References
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993; 72:971 - 83; http://dx.doi.org/10.1016/0092-8674(93)90585-E; PMID: 8458085
- Ross CA, Margolis RL, Rosenblatt A, Ranen NG, Becher MW, Aylward E. Huntington disease and the related disorder, dentatorubral-pallidoluysian atrophy (DRPLA). Medicine (Baltimore) 1997; 76:305 - 38; http://dx.doi.org/10.1097/00005792-199709000-00001; PMID: 9352736
- Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 2011; 10:83 - 98; http://dx.doi.org/10.1016/S1474-4422(10)70245-3; PMID: 21163446
- Walker FO. Huntington’s disease. Lancet 2007; 369:218 - 28; http://dx.doi.org/10.1016/S0140-6736(07)60111-1; PMID: 17240289
- Li W, Serpell LC, Carter WJ, Rubinsztein DC, Huntington JA. Expression and characterization of full-length human huntingtin, an elongated HEAT repeat protein. J Biol Chem 2006; 281:15916 - 22; http://dx.doi.org/10.1074/jbc.M511007200; PMID: 16595690
- Goehler H, Lalowski M, Stelzl U, Waelter S, Stroedicke M, Worm U, et al. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington’s disease. Mol Cell 2004; 15:853 - 65; http://dx.doi.org/10.1016/j.molcel.2004.09.016; PMID: 15383276
- Nucifora FC Jr., Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 2001; 291:2423 - 8; http://dx.doi.org/10.1126/science.1056784; PMID: 11264541
- Ross CA, Poirier MA. Opinion: What is the role of protein aggregation in neurodegeneration?. Nat Rev Mol Cell Biol 2005; 6:891 - 8; http://dx.doi.org/10.1038/nrm1742; PMID: 16167052
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, et al. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A 2000; 97:6763 - 8; http://dx.doi.org/10.1073/pnas.100110097; PMID: 10823891
- Lim J, Crespo-Barreto J, Jafar-Nejad P, Bowman AB, Richman R, Hill DE, et al. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 2008; 452:713 - 8; http://dx.doi.org/10.1038/nature06731; PMID: 18337722
- Zoghbi HY, Orr HT. Pathogenic mechanisms of a polyglutamine-mediated neurodegenerative disease, spinocerebellar ataxia type 1. J Biol Chem 2009; 284:7425 - 9; http://dx.doi.org/10.1074/jbc.R800041200; PMID: 18957430
- Li XJ, Li SH, Sharp AH, Nucifora FC Jr., Schilling G, Lanahan A, et al. A huntingtin-associated protein enriched in brain with implications for pathology. Nature 1995; 378:398 - 402; http://dx.doi.org/10.1038/378398a0; PMID: 7477378
- Li XJ, Sharp AH, Li SH, Dawson TM, Snyder SH, Ross CA. Huntingtin-associated protein (HAP1): discrete neuronal localizations in the brain resemble those of neuronal nitric oxide synthase. Proc Natl Acad Sci U S A 1996; 93:4839 - 44; http://dx.doi.org/10.1073/pnas.93.10.4839; PMID: 8643490
- Kalchman MA, Graham RK, Xia G, Koide HB, Hodgson JG, Graham KC, et al. Huntingtin is ubiquitinated and interacts with a specific ubiquitin-conjugating enzyme. J Biol Chem 1996; 271:19385 - 94; http://dx.doi.org/10.1074/jbc.271.32.19385; PMID: 8702625
- Kalchman MA, Koide HB, McCutcheon K, Graham RK, Nichol K, Nishiyama K, et al. HIP1, a human homologue of S. cerevisiae Sla2p, interacts with membrane-associated huntingtin in the brain. Nat Genet 1997; 16:44 - 53; http://dx.doi.org/10.1038/ng0597-44; PMID: 9140394
- Wanker EE, Rovira C, Scherzinger E, Hasenbank R, Wälter S, Tait D, et al. HIP-I: a huntingtin interacting protein isolated by the yeast two-hybrid system. Hum Mol Genet 1997; 6:487 - 95; http://dx.doi.org/10.1093/hmg/6.3.487; PMID: 9147654
- Li SH, Gutekunst CA, Hersch SM, Li XJ. Interaction of huntingtin-associated protein with dynactin P150Glued. J Neurosci 1998; 18:1261 - 9; PMID: 9454836
- Boutell JM, Thomas P, Neal JW, Weston VJ, Duce J, Harper PS, et al. Aberrant interactions of transcriptional repressor proteins with the Huntington’s disease gene product, huntingtin. Hum Mol Genet 1999; 8:1647 - 55; http://dx.doi.org/10.1093/hmg/8.9.1647; PMID: 10441327
- Li SH, Cheng AL, Zhou H, Lam S, Rao M, Li H, et al. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol Cell Biol 2002; 22:1277 - 87; http://dx.doi.org/10.1128/MCB.22.5.1277-1287.2002; PMID: 11839795
- Harjes P, Wanker EE. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci 2003; 28:425 - 33; http://dx.doi.org/10.1016/S0968-0004(03)00168-3; PMID: 12932731
- Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet 2004; 20:146 - 54; http://dx.doi.org/10.1016/j.tig.2004.01.008; PMID: 15036808
- Subramaniam S, Sixt KM, Barrow R, Snyder SH. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 2009; 324:1327 - 30; http://dx.doi.org/10.1126/science.1172871; PMID: 19498170
- Savas JN, Ma B, Deinhardt K, Culver BP, Restituito S, Wu L, et al. A role for huntington disease protein in dendritic RNA granules. J Biol Chem 2010; 285:13142 - 53; http://dx.doi.org/10.1074/jbc.M110.114561; PMID: 20185826
- Savas JN, Makusky A, Ottosen S, Baillat D, Then F, Krainc D, et al. Huntington’s disease protein contributes to RNA-mediated gene silencing through association with Argonaute and P bodies. Proc Natl Acad Sci U S A 2008; 105:10820 - 5; http://dx.doi.org/10.1073/pnas.0800658105; PMID: 18669659
- Kaltenbach LS, Romero E, Becklin RR, Chettier R, Bell R, Phansalkar A, et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet 2007; 3:e82; http://dx.doi.org/10.1371/journal.pgen.0030082; PMID: 17500595
- Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH, Tartaglia GG, et al. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 2011; 144:67 - 78; http://dx.doi.org/10.1016/j.cell.2010.11.050; PMID: 21215370
- Raychaudhuri S, Dey S, Bhattacharyya NP, Mukhopadhyay D. The role of intrinsically unstructured proteins in neurodegenerative diseases. PLoS One 2009; 4:e5566; http://dx.doi.org/10.1371/journal.pone.0005566; PMID: 19440375
- Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 2006; 125:1179 - 91; http://dx.doi.org/10.1016/j.cell.2006.04.026; PMID: 16777606
- Ratovitski T, Gucek M, Jiang H, Chighladze E, Waldron E, D’Ambola J, et al. Mutant huntingtin N-terminal fragments of specific size mediate aggregation and toxicity in neuronal cells. J Biol Chem 2009; 284:10855 - 67; http://dx.doi.org/10.1074/jbc.M804813200; PMID: 19204007
- Ratovitski T, Nakamura M, D’Ambola J, Chighladze E, Liang Y, Wang W, et al. N-terminal proteolysis of full-length mutant huntingtin in an inducible PC12 cell model of Huntington’s disease. Cell Cycle 2007; 6:2970 - 81; http://dx.doi.org/10.4161/cc.6.23.4992; PMID: 18156806
- Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics 2004; 3:1154 - 69; http://dx.doi.org/10.1074/mcp.M400129-MCP200; PMID: 15385600
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, et al. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet 2000; 9:2799 - 809; http://dx.doi.org/10.1093/hmg/9.19.2799; PMID: 11092756
- Raychaudhuri S, Sinha M, Mukhopadhyay D, Bhattacharyya NP. HYPK, a Huntingtin interacting protein, reduces aggregates and apoptosis induced by N-terminal Huntingtin with 40 glutamines in Neuro2a cells and exhibits chaperone-like activity. Hum Mol Genet 2008; 17:240 - 55; http://dx.doi.org/10.1093/hmg/ddm301; PMID: 17947297
- Hill EG, Schwacke JH, Comte-Walters S, Slate EH, Oberg AL, Eckel-Passow JE, et al. A statistical model for iTRAQ data analysis. J Proteome Res 2008; 7:3091 - 101; http://dx.doi.org/10.1021/pr070520u; PMID: 18578521
- Oberg AL, Mahoney DW, Eckel-Passow JE, Malone CJ, Wolfinger RD, Hill EG, et al. Statistical analysis of relative labeled mass spectrometry data from complex samples using ANOVA. J Proteome Res 2008; 7:225 - 33; http://dx.doi.org/10.1021/pr700734f; PMID: 18173221
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 2003; 100:9440 - 5; http://dx.doi.org/10.1073/pnas.1530509100; PMID: 12883005
- Norberg E, Orrenius S, Zhivotovsky B. Mitochondrial regulation of cell death: processing of apoptosis-inducing factor (AIF). Biochem Biophys Res Commun 2010; 396:95 - 100; http://dx.doi.org/10.1016/j.bbrc.2010.02.163; PMID: 20494118
- Shiina N, Shinkura K, Tokunaga M. A novel RNA-binding protein in neuronal RNA granules: regulatory machinery for local translation. J Neurosci 2005; 25:4420 - 34; http://dx.doi.org/10.1523/JNEUROSCI.0382-05.2005; PMID: 15858068
- Solomon S, Xu Y, Wang B, David MD, Schubert P, Kennedy D, et al. Distinct structural features of caprin-1 mediate its interaction with G3BP-1 and its induction of phosphorylation of eukaryotic translation initiation factor 2alpha, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs. Mol Cell Biol 2007; 27:2324 - 42; http://dx.doi.org/10.1128/MCB.02300-06; PMID: 17210633
- Jayaraman M, Kodali R, Sahoo B, Thakur AK, Mayasundari A, Mishra R, et al. Slow amyloid nucleation via α-helix-rich oligomeric intermediates in short polyglutamine-containing huntingtin fragments. J Mol Biol 2012; 415:881 - 99; http://dx.doi.org/10.1016/j.jmb.2011.12.010; PMID: 22178474
- Mishra R, Jayaraman M, Roland BP, Landrum E, Fullam T, Kodali R, et al. Inhibiting the nucleation of amyloid structure in a huntingtin fragment by targeting α-helix-rich oligomeric intermediates. J Mol Biol 2012; 415:900 - 17; http://dx.doi.org/10.1016/j.jmb.2011.12.011; PMID: 22178478
- Kelley NW, Huang X, Tam S, Spiess C, Frydman J, Pande VS. The predicted structure of the headpiece of the Huntingtin protein and its implications on Huntingtin aggregation. J Mol Biol 2009; 388:919 - 27; http://dx.doi.org/10.1016/j.jmb.2009.01.032; PMID: 19361448
- Ross CA, Poirier MA, Wanker EE, Amzel M. Polyglutamine fibrillogenesis: the pathway unfolds. Proc Natl Acad Sci U S A 2003; 100:1 - 3; http://dx.doi.org/10.1073/pnas.0237018100; PMID: 12509507
- Davranche A, Aviolat H, Zeder-Lutz G, Busso D, Altschuh D, Trottier Y, et al. Huntingtin affinity for partners is not changed by polyglutamine length: aggregation itself triggers aberrant interactions. Hum Mol Genet 2011; 20:2795 - 806; http://dx.doi.org/10.1093/hmg/ddr178; PMID: 21518730
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006; 443:787 - 95; http://dx.doi.org/10.1038/nature05292; PMID: 17051205
- Orr AL, Li S, Wang CE, Li H, Wang J, Rong J, et al. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci 2008; 28:2783 - 92; http://dx.doi.org/10.1523/JNEUROSCI.0106-08.2008; PMID: 18337408
- Shirendeb U, Reddy AP, Manczak M, Calkins MJ, Mao P, Tagle DA, et al. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum Mol Genet 2011; 20:1438 - 55; http://dx.doi.org/10.1093/hmg/ddr024; PMID: 21257639
- Miramar MD, Costantini P, Ravagnan L, Saraiva LM, Haouzi D, Brothers G, et al. NADH oxidase activity of mitochondrial apoptosis-inducing factor. J Biol Chem 2001; 276:16391 - 8; http://dx.doi.org/10.1074/jbc.M010498200; PMID: 11278689
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, et al. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature 2002; 419:367 - 74; http://dx.doi.org/10.1038/nature01034; PMID: 12353028
- Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, et al. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 2007; 131:476 - 91; http://dx.doi.org/10.1016/j.cell.2007.08.047; PMID: 17981116
- Vahsen N, Candé C, Brière JJ, Bénit P, Joza N, Larochette N, et al. AIF deficiency compromises oxidative phosphorylation. EMBO J 2004; 23:4679 - 89; http://dx.doi.org/10.1038/sj.emboj.7600461; PMID: 15526035
- Candé C, Vahsen N, Kouranti I, Schmitt E, Daugas E, Spahr C, et al. AIF and cyclophilin A cooperate in apoptosis-associated chromatinolysis. Oncogene 2004; 23:1514 - 21; http://dx.doi.org/10.1038/sj.onc.1207279; PMID: 14716299
- Otera H, Ohsakaya S, Nagaura Z, Ishihara N, Mihara K. Export of mitochondrial AIF in response to proapoptotic stimuli depends on processing at the intermembrane space. EMBO J 2005; 24:1375 - 86; http://dx.doi.org/10.1038/sj.emboj.7600614; PMID: 15775970
- Vahsen N, Candé C, Dupaigne P, Giordanetto F, Kroemer RT, Herker E, et al. Physical interaction of apoptosis-inducing factor with DNA and RNA. Oncogene 2006; 25:1763 - 74; http://dx.doi.org/10.1038/sj.onc.1209206; PMID: 16278674
- Hangen E, De Zio D, Bordi M, Zhu C, Dessen P, Caffin F, et al. A brain-specific isoform of mitochondrial apoptosis-inducing factor: AIF2. Cell Death Differ 2010; 17:1155 - 66; http://dx.doi.org/10.1038/cdd.2009.211; PMID: 20111043
- Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2002; 2:647 - 56; http://dx.doi.org/10.1038/nrc883; PMID: 12209154
- Shiina N, Yamaguchi K, Tokunaga M. RNG105 deficiency impairs the dendritic localization of mRNAs for Na+/K+ ATPase subunit isoforms and leads to the degeneration of neuronal networks. J Neurosci 2010; 30:12816 - 30; http://dx.doi.org/10.1523/JNEUROSCI.6386-09.2010; PMID: 20861386
- Angenstein F, Evans AM, Ling SC, Settlage RE, Ficarro S, Carrero-Martinez FA, et al. Proteomic characterization of messenger ribonucleoprotein complexes bound to nontranslated or translated poly(A) mRNAs in the rat cerebral cortex. J Biol Chem 2005; 280:6496 - 503; http://dx.doi.org/10.1074/jbc.M412742200; PMID: 15596439
- Kedersha N, Anderson P. Mammalian stress granules and processing bodies. Methods Enzymol 2007; 431:61 - 81; http://dx.doi.org/10.1016/S0076-6879(07)31005-7; PMID: 17923231
- Tourrière H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, et al. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J Cell Biol 2003; 160:823 - 31; http://dx.doi.org/10.1083/jcb.200212128; PMID: 12642610
- Tourrière H, Gallouzi IE, Chebli K, Capony JP, Mouaikel J, van der Geer P, et al. RasGAP-associated endoribonuclease G3Bp: selective RNA degradation and phosphorylation-dependent localization. Mol Cell Biol 2001; 21:7747 - 60; http://dx.doi.org/10.1128/MCB.21.22.7747-7760.2001; PMID: 11604510
- Zuccato C, Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol 2009; 5:311 - 22; http://dx.doi.org/10.1038/nrneurol.2009.54; PMID: 19498435
- Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004; 118:127 - 38; http://dx.doi.org/10.1016/j.cell.2004.06.018; PMID: 15242649
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001; 293:493 - 8; http://dx.doi.org/10.1126/science.1059581; PMID: 11408619
- Ma B, Culver BP, Baj G, Tongiorgi E, Chao MV, Tanese N. Localization of BDNF mRNA with the Huntington’s disease protein in rat brain. Mol Neurodegener 2010; 5:22; http://dx.doi.org/10.1186/1750-1326-5-22; PMID: 20507609
- Lemmens R, Moore MJ, Al-Chalabi A, Brown RH Jr., Robberecht W. RNA metabolism and the pathogenesis of motor neuron diseases. Trends Neurosci 2010; 33:249 - 58; http://dx.doi.org/10.1016/j.tins.2010.02.003; PMID: 20227117
- Todd PK, Paulson HL. RNA-mediated neurodegeneration in repeat expansion disorders. Ann Neurol 2010; 67:291 - 300; PMID: 20373340
- La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet 2010; 11:247 - 58; http://dx.doi.org/10.1038/nrg2748; PMID: 20177426
- Bilen J, Liu N, Bonini NM. A new role for microRNA pathways: modulation of degeneration induced by pathogenic human disease proteins. Cell Cycle 2006; 5:2835 - 8; http://dx.doi.org/10.4161/cc.5.24.3579; PMID: 17172864
- Junn E, Mouradian MM. MicroRNAs in neurodegenerative disorders. Cell Cycle 2010; 9:1717 - 21; http://dx.doi.org/10.4161/cc.9.9.11296; PMID: 20404550
- Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 2010; 19:R1 R46 - 64; http://dx.doi.org/10.1093/hmg/ddq137; PMID: 20400460
- Doumanis J, Wada K, Kino Y, Moore AW, Nukina N. RNAi screening in Drosophila cells identifies new modifiers of mutant huntingtin aggregation. PLoS One 2009; 4:e7275; http://dx.doi.org/10.1371/journal.pone.0007275; PMID: 19789644
- Runne H, Régulier E, Kuhn A, Zala D, Gokce O, Perrin V, et al. Dysregulation of gene expression in primary neuron models of Huntington’s disease shows that polyglutamine-related effects on the striatal transcriptome may not be dependent on brain circuitry. J Neurosci 2008; 28:9723 - 31; http://dx.doi.org/10.1523/JNEUROSCI.3044-08.2008; PMID: 18815258
- Sipione S, Rigamonti D, Valenza M, Zuccato C, Conti L, Pritchard J, et al. Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Hum Mol Genet 2002; 11:1953 - 65; http://dx.doi.org/10.1093/hmg/11.17.1953; PMID: 12165557
- Tauber E, Miller-Fleming L, Mason RP, Kwan W, Clapp J, Butler NJ, et al. Functional gene expression profiling in yeast implicates translational dysfunction in mutant huntingtin toxicity. J Biol Chem 2011; 286:410 - 9; http://dx.doi.org/10.1074/jbc.M110.101527; PMID: 21044956
- Wyttenbach A, Swartz J, Kita H, Thykjaer T, Carmichael J, Bradley J, et al. Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington’s disease. Hum Mol Genet 2001; 10:1829 - 45; http://dx.doi.org/10.1093/hmg/10.17.1829; PMID: 11532992
- Sharp AH, Loev SJ, Schilling G, Li SH, Li XJ, Bao J, et al. Widespread expression of Huntington’s disease gene (IT15) protein product. Neuron 1995; 14:1065 - 74; http://dx.doi.org/10.1016/0896-6273(95)90345-3; PMID: 7748554
- Peters MF, Ross CA. Isolation of a 40-kDa Huntingtin-associated protein. J Biol Chem 2001; 276:3188 - 94; http://dx.doi.org/10.1074/jbc.M008099200; PMID: 11035034
- Ko J, Ou S, Patterson PH. New anti-huntingtin monoclonal antibodies: implications for huntingtin conformation and its binding proteins. Brain Res Bull 2001; 56:319 - 29; http://dx.doi.org/10.1016/S0361-9230(01)00599-8; PMID: 11719267
- Schnaitman C, Greenawalt JW. Enzymatic properties of the inner and outer membranes of rat liver mitochondria. J Cell Biol 1968; 38:158 - 75; http://dx.doi.org/10.1083/jcb.38.1.158; PMID: 5691970