Abstract
Diabetes results from an inadequate functional β cell mass, either due to autoimmune destruction (Type 1 diabetes) or insulin resistance combined with β cell failure (Type 2 diabetes). Strategies to enhance β cell regeneration or increase cell proliferation could improve outcomes for patients with diabetes. Research conducted over the past several years has revealed that factors regulating embryonic β cell mass expansion differ from those regulating replication ofβ cells post-weaning. This article aims to compare and contrast factors known to control embryonic and postnatal β cell replication. In addition, we explore the possibility that connective tissue growth factor (CTGF) could increase adult β cell replication. We have already shown that CTGF is required for embryonicβ cell proliferation and is sufficient to induce replication of embryonic β cells. Here we examine whether adult β cell replication and expansion of β cell mass can be enhanced by increased CTGF expression in mature β cells.
Introduction
Type 2 diabetes is a condition in which there is inadequate pancreatic β cell mass and/or insufficient insulin released by β cells, leading to hyperglycemia. β cell mass is dynamic, responding to stimuli such as pregnancy and weight gain, resulting in β cell mass expansion. Autopsy studies of obese, non-diabetic individuals reveal an expansion in β cell mass when compared with lean controls. However, this compensation is lost in individuals with Type 2 diabetes.Citation1 This inadequate β cell mass expansion in individuals with diabetes has led to an increased focus of research into β cell proliferation and mechanisms of β cell mass expansion as a potential therapy for the disease. In the recent past, Type 2 diabetes was a disease that mainly affected adults as they aged due to decreasing physical activity and increased weight gain. However, in 2011, in the United States alone, 25.8 million children and adults had diabetes, representing 8% of the total population.Citation2 Strategies to increase β cell mass would thus have an impact on a significant number of people. Given the diverse nature of this population, it is unclear that strategies developed to augment β cell mass would show beneficial effects on both young patients and older adults.
Although it is becoming increasingly clear from studies in rodents that many differences exist between the mechanisms by which embryonic and adult β cell mass are increased and maintained, many of the transcription factors important in embryonic β cell development have distinct roles in maintaining adult β cell identity and functionality (). In the embryo, β cell differentiation from multipotent pancreatic progenitor cells is established by an extensive transcription factor regulatory network. This neogenesis continues through the first few weeks after birth, concomitant with proliferation, which begins at late gestation in the mouse (). In contrast, in the adult, there is a balance between β cell replication and apoptosis which dictates β cell mass; neogenesis is not thought to contribute to β cell mass expansion under normal circumstances in adult mice, although this is still unclear in humans.
Figure 1. Schematic of β cell mass expansion throughout life. The timeline shows key events and time points in mouse pancreas development and β cell mass expansion under normal circumstances. Underlying the timeline (blue) are the approximate windows of primary and secondary endocrine differentiation. Overlying the timeline (green) are major mechanisms for β cell mass increase at those times: neogenesis early and replication later, although these overlap. The replicative phase is separated into stages (orange): “embryonic” mode and “adult” mode, indicating the switch from dependency on one set of regulatory factors to another around weaning.
Here we review what is currently known about regulation of embryonic and postnatal β cell mass. For focus and clarity, we have confined our analyses to in vivo studies using gene inactivation or transgenic overexpression studies. Within this context, we specifically explore the potential of the secreted protein, connective tissue growth factor (CTGF), to induce adult β cell mass expansion. We previously showed that CTGF is required for embryonic β cell proliferationCitation3,Citation4 and that CTGF overexpression in embryonic cells increases β cell proliferation and β cell mass.Citation4 We now examine the effect of CTGF overexpression in adult cells.
Embryonic β Cell Neogenesis
The mouse pancreas develops from ventral and dorsal evaginations of the posterior foregut endoderm at embryonic day (e) 9.5, a process dependent on the transcription factors Pdx1 and Ptf1a.Citation6,Citation7 Differentiation of all pancreatic endocrine cell types (α, β, Δ and PP) is dependent on the transcription factor, neurogenin 3 (Ngn3).Citation8 Ngn3 expression is controlled by a variety of factors, including the Notch signaling pathway and the transcriptional regulators pancreatic and duodenal homeobox 1 (Pdx1), SRY-box 9 (Sox9) and hepatic nuclear factor 6 (Hnf6).Citation9-Citation11 Although β cell neogenesis begins at e10.5, these early insulin-positive cells do not contribute to mature islets (). Instead, endocrine cells that will go on to contribute to the mature islets begin to differentiate at e13, a period known as the secondary transition ().Citation15 Some transcription factors critically involved in β cell differentiation include NK2 homeobox 2 (Nkx2.2), Nkx6.1, islet 1 (Isl-1), neuronal differentiation 1 (NeuroD1), motor neuron and pancreas homeobox 1 (Mnx1), paired box gene 4 (Pax4) and Pdx1.Citation12 The function of these factors in pancreatic endocrine differentiation has been extensively reviewed and will not be covered here.Citation13-Citation15 cells formed during the secondary transition, which extends into the early postnatal period (), will serve as the source of cells for replication at late gestation, postnatally and in adults. Strategies to reactivate neogenic pathways in adult pancreas are under active research and could aid in developing therapies to expand adult β cell mass.
Regulation of Embryonic and Neonatal β Cell Proliferation
Overall, less is known about regulation of embryonic β cell replication compared with adult β cell replication. Proliferation of existing cells can first be observed at e16.5 in the mouse ().Citation16 Studies in sheep have demonstrated that fetal over-nutrition, in which pregnant ewes are fed a high fat diet, increases prenatal β cell proliferation.Citation17 Thus, as discussed below in adults, physiological stimuli can enhance β cell proliferation during development. Although several factors have been identified that play a role in the regulation of embryonic and neonatal β cell proliferation (), most of these have no obvious role in regulation of the cell cycle. One cell cycle regulator that does play a role in embryonic β cell proliferation is the cell cycle inhibitor, p27Kip1. Inactivation of p27Kip1 during embryogenesis results in an increase in β cell proliferation and subsequently β cell mass.Citation18 There was no change, however, in early postnatal β cell proliferation, suggesting that p27Kip1 is not crucial to postnatal proliferation.
As mentioned above, Pdx1 is expressed in multipotent pancreatic progenitors in the early stages of pancreas development, but by e16.5, Pdx1 expression becomes enhanced in insulin-positive cells and is found at only low levels in exocrine cells.Citation19 This expression pattern is maintained into adulthood and Pdx1 plays a critical role in maintenance of the mature β cell phenotype.Citation20 Inactivation of Pdx1 in embryonic insulin-expressing cells results in a dramatic decrease in β cell proliferation at late gestation, leading to decreased β cell mass at birth and early onset diabetes.Citation21 Two large Maf (musculoaponeurotic fibrosarcoma oncogene homolog) transcription factors that are closely related to one another, MafA and MafB, are critical for β cell differentiation and embryonic Pdx1 expressionCitation22 and therefore may have an indirect effect on embryonic β cell replication.
Inactivation of the eIF2α endoplasmic reticulum resident kinase, PERK (protein kinase RNA-like endoplasmic reticulum kinase), specifically in embryonic β cells (PERKΔbeta) results in a 2-fold decrease in β cell proliferation at e16.5, which persists through postnatal day (P) 8.Citation16 No difference in the number of insulin-expressing cells is evident in PERKΔbeta mice compared with control mice at e16.5, suggesting that PERK deficiency does not affect β cell differentiation. Inactivation of PERK in adult β cells has no effect on β cell mass or function,Citation16 although its role in β cell mass expansion in response to stimuli such as pregnancy or obesity has not yet been examined.
CTGF, a member of the CCN (Cyr61, CTGF, Nov) family of secretory proteins, is involved in various cellular functions such as adhesion, migration, proliferation, extra-cellular matrix (ECM) remodeling and angiogenesis.Citation23 In the pancreas, CTGF is expressed in insulin-positive cells, blood vessel endothelium and ductal epithelium during embryogenesis in the mouse, but only in endothelium and ducts after P3.Citation3 We discovered that homozygous and heterozygous Ctgf-null mutant mice have a significant reduction in β cell proliferation at e18.5.Citation3 Hypothesizing that CTGF overexpression would increase β cell proliferation, we generated transgenic mice in which CTGF could be inducibly overexpressed.Citation4 Overexpression of CTGF in insulin-producing cells during embryogenesis induced proliferation of both α and β cells, resulting in increased α and β cell mass while maintaining the appropriate ratio of these two cell types. Interestingly, only MafA-negative insulin-positive cells were observed to proliferate in this model. Since MafA expression is associated with more mature cells,Citation24 these results suggest that only immature cells respond to CTGF. CTGF overexpression had no effect on the number of endocrine progenitors, indicating that CTGF influences endocrine cell proliferation and not neogenesis.
Regulation of Postnatal β Cell Proliferation
The perinatal period is characterized by heightened β cell proliferation and increased β cell mass.Citation25 At the time of weaning, there is considerable islet remodeling with significant β cell death but also a burst of β cell neogenesis; however, there is no significant contribution of neogenesis to β cell mass expansion post-weaning ().Citation25 Instead, adult β cell mass is mainly maintained and increased via proliferation.Citation26 In rats, by 30–40 d of age, β cell replication rates stabilize.Citation25 At this time, the α:β cell ratio is approximately 1:4 in mice.Citation27
Postnatal β cell proliferation is heavily regulated by specific cell cycle activators and inhibitors that, surprisingly, play no role in embryonic β cell replication (). Although loss of the cell cycle activator Cyclin D2 during embryogenesis has no effect on β cell mass, postnatally, its absence causes decreased β cell replication and a 4-fold reduction in β cell mass compared with controls.Citation28 Similarly, the absence of the cell cycle activator cyclin-dependent kinase 4 (Cdk4) has no effect on embryonic β cell mass but Cdk4-null mutants have a 3.5-fold reduction in β cell proliferation at P10.Citation29 β cells have been shown to have less redundancy in cell cycle activators. For example, β cells lack Cdk6, a close homolog of Cdk4 that is co-expressed with Cdk4 in other cell types.Citation29 Thus, loss of a single activator results in a dramatic phenotype, although why the effects are often restricted to the postnatal period is not clear.
p16Ink4a, a cell cycle inhibitor, increases in β cells with age and contributes to the normal age-related decline in β cell proliferation.Citation30 Germline deletion of p16Ink4a ameliorates this decline and β cells of aged mutant mice proliferate similarly to those of young control mice. Conversely, transgenic overexpression of p16Ink4a in β cells results in decreased β cell proliferation in young transgenic mice, similar to that seen in older control mice.Citation30 Bmi1 is an inhibitor of the Ink4a/Arf locus. At 2 weeks of age, Bmi1 null mice show a significant increase in p16Ink4a with a subsequent reduction in β cell proliferation, demonstrating that Bmi-1 is necessary for p16Ink4a suppression.Citation31 However, the role of Bmi-1 during embryogenesis is yet to be elucidated. Conditional deletion of Enhancer of zeste homolog 2 (Ezh2), another inhibitor of the Ink4a/Arf locus, resulted in a decrease in β cell proliferation and β cell mass by one month of age with a concomitant increase in p16Ink4a levels,Citation32 but the effect of Ezh2 loss during embryogenesis has not been reported.
The effects of Ezh2 on β cell replication and the fact that platelet-derived growth factor (PDGF) is known to regulate Ezh2 signaling in fibroblasts led Chen and colleagues to analyze the role of PDGF signaling in β cell replication. They discovered that there is an age-related decline in expression of PDGF receptors (PDGFR) in β cells, which is coincident with the decrease in Ezh2 and increase in p16Ink4a. Inactivation of the Pdgfr gene results in a decrease in Ezh2 mRNA levels and a subsequent decrease in β cell proliferation seen by 2–3 weeks of age. Conversely, transgenic overexpression of PDGF in β cells induces a 9-fold increase in β cell proliferation in 14-mo-old transgenic mice compared with control mice, suggesting that regulation of Ezh2 by the PDGF signaling pathway can improve the age-related decline in β cell proliferation.Citation33
A major regulator of genes involved in cell cycle progression in many cell types is the transcription factor forkhead box M1 (FoxM1). FoxM1 regulates genes involved in the G1-SCitation34 and G2-MCitation35 transitions of the cell cycle as well as karyokinesis and cytokinesis. Inactivation of Foxm1 throughout the pancreatic epithelium using a Cre-lox mouse model demonstrated that FoxM1 is critical to preventing nuclear accumulation of the cell cycle inhibitor p27Kip1. In the absence of pancreatic Foxm1, there is decreased β cell proliferation and β cell mass expansion postnatally. In addition, Foxm1 mutant β cells show a severe impairment in response to several proliferative stimuli, indicating that many different stimuli converge on FoxM1 to mediate increased proliferation.Citation36-Citation38 Interestingly, the requirement for FoxM1 in β cell replication is first seen around weaning; Foxm1 inactivation had no effect on β cell mass or proliferation during embryogenesis.Citation38 The reader is referred to other reviews of the role of cell cycle activators and inhibitors in β cell replication and aging.Citation39-Citation42
In addition to cell cycle activators and inhibitors regulating postnatal β cell replication, there are several known factors and signaling pathways that control β cell mass expansion in adults under normal conditions and in response to physiological stimuli (). For example, insulin resistance, which is often accompanied by increased insulin secretion, in the setting of either obesity or pregnancy is known to stimulate an increase in β cell mass via proliferation.Citation43,Citation44 Likewise, elevated glucose is a stimulus for β cell replication. Adult rats given a 96-h glucose infusion showed a transient increase in β cell replication accompanied by β cell hypertrophy, which led to an increase in β cell mass; replication declined within seven days after infusion.Citation45 Furthermore, with a six day glucose infusion, there is evidence of neogenesis in addition to an increase in β cell proliferation in adult rats.Citation46 The signaling mechanism(s) by which neogenesis or proliferation is stimulated by glucose has not yet been identified, although increased glucose metabolism increases secretion of insulin, which itself is a β cell growth factor.Citation47
Insulin/insulin-like growth factor (IGF) signaling is one of several growth factor signaling pathways that plays a role in postnatal β cell mass expansion. Binding of insulin or IGF to their cognate receptors results in activation of a well-studied second messenger cascade culminating in the phosphorylation and activation of protein kinase B (PKB/AKT) by phosphoinositide-dependent protein kinase-1 (PDK1) and mammalian target of rapamycin complex 2 (mTORC2) kinases.Citation48 Deletion of either PDK1 or Rictor, a key component of mTORC2, results in a decrease in β cell replication in part through upregulation of the p27Kip1 cell cycle inhibitor.Citation49 Activation of AKT leads to phosphorylation and inhibition of activity of the FoxO1 transcription factor, which then actively inhibits Pdx1 transcription by binding to the Pdx1 promoter.Citation50 Indeed, Pdx1 is essential for β cell mass expansion in response to insulin resistance.Citation51 These results highlight the importance of insulin/IGF induction of the AKT pathway to stimulate β cell proliferation through the inhibition of p27Kip1 and FoxO1.
The transcription factor nuclear factor of activated T cells (NFAT) and its regulator, calcineurin, have recently been found to be critical in adult β cell replication. Loss of calcineurin b1 in a β cell-specific manner results in decreased β cell replication in mice at four weeks of age with a significant loss in β cell mass by 12 weeks of age.Citation52 Both calcineurin and NFAT are activated in response to increased glucose metabolismCitation53 and elevated intracellular calcium.Citation54
Pregnancy is a time of metabolic stress in which β cell mass and function are enhanced to compensate for insulin resistance associated with weight gain, placental hormone production and fetal growth.Citation55 Circulating factors such as growth hormone, prolactin and placental lactogens (PL) are elevated during pregnancy and contribute to increased β cell mass, although PL is the major stimulus for maternal β cell proliferation during pregnancy.Citation44 PL-mediated β cell proliferation is FoxM1-dependent.Citation36 Transgenic overexpression of PL also stimulates β cell replication in non-pregnant mice.Citation56 Another secreted factor involved in maternal β cell replication during pregnancy is serotonin, although the activity of serotonin seems to be more of a local effect within the islet environment itself rather than systemic. The serotonin receptor subtype, Htr2b, is necessary for appropriate β cell proliferation and mass compensation during pregnancy.Citation57 It would be interesting to examine whether serotonin treatment in non-pregnant mice has an effect on β cell replication.
Other circulating factors that enhance adult β cell replication in vivo include hepatocyte growth factor (HGF),Citation58 Exendin-4/GLP-1 Citation59 and IGF-1.Citation60 Our laboratory has been examining the potential role of CTGF in adult β cell mass expansion. CTGF increases proliferation in many cell types, including pancreatic endocrine cells during development,Citation4 but the mechanism by which this occurs in the endocrine pancreas is yet to be elucidated. Studies in immortalized mammary epithelial cells suggest that IGF1 regulates Ctgf expression via the PI3/Akt pathway.Citation61 Since this pathway is also involved in β cell replication, future studies should examine whether CTGF interacts with the insulin signaling pathway during times of β cell mass expansion. Although CTGF is not normally expressed in adult β cells in human or mouse, we have observed re-expression of CTGF in maternal islets during pregnancy in mice,Citation3 suggesting that it plays a role in adaptive changes that enhance β cell mass and/or function.
Overexpression of CTGF in Adult β Cells
CTGF is required for embryonic α and β cell proliferation, but whether it plays a role in adult β cell replication has yet to be determined. In order to assess whether CTGF treatment can improve β cell proliferation and/or function, we generated transgenic mice in which CTGF could be conditionally overexpressed under control of the Tetracycline operator (TetO).Citation4 Cell type-specific expression is thus controlled by the choice of promoter driving the TetO binding protein, reverse tetracycline transactivator (rtTA). Here we used the rat insulin promoter (RIP) to drive expression in insulin-producing cells.Citation5 The timing of CTGF expression in insulin-producing cells was controlled by doxycycline (DOX) administration. We first determined the level of Ctgf overexpression in our system using real time quantitative PCR. DOX was administered for one week and islets isolated for total RNA. We estimate that there is a 400-fold increase in total Ctgf in the CTGF-overexpressing islets (Fig. S1). While this may seem extremely high, it should be kept in mind that several different methods have shown that CTGF expression is normally undetectable in adult mouse or human islets.Citation3 Studies in which CTGF expression was induced in embryonic β cells had previously revealed a 10-fold increase in total CTGF in whole pancreata.Citation4
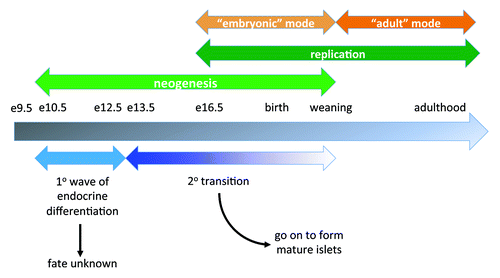
Using this transgenic mouse model, we first examined whether induction of CTGF in β cells of ~7-week-old mice could increase β cell proliferation and β cell mass. Previous studies have shown that mouse β cells at this age are still highly responsive to several different proliferative stimuli.Citation62 Ctgf expression was induced for one, three and five weeks to determine whether longer exposure of β cells to CTGF had a more beneficial effect. cell function was assessed using intraperitoneal glucose tolerance tests. Given its re-expression in maternal islets during pregnancy, we hypothesized that CTGF might improve β cell function and thus glucose homeostasis. Animals expressing CTGF in their β cells showed no improvements in glucose homeostasis at any time point examined ( and data not shown). We also examined glucose tolerance following longer periods of CTGF overexpression (up to 12 weeks), but no improvement was observed in glucose tolerance compared with littermate controls (data not shown). Importantly, there were no observed deleterious effects of CTGF overexpression on glucose homeostasis at any time point examined.
Figure 2. Effects of adult β cell CTGF overexpression on β cell function and mass. (A, B) Intraperitoneal glucose tolerance tests reveal no difference in glucose homeostasis between RIP-rtTA control mice (closed squares) and RIP-rtTA;TetO-CTGF mice (open triangles) treated with doxycycline for 1 week (A) or 5 weeks (B) beginning at ~7 weeks of age. (C, D) Immunolabeling of adult pancreata with antibodies against insulin (green) and Ki67 (red) to assess β cell proliferation (blue: DAPI, nuclei) (Magnification 200). (E) There is no difference in the percent of β cell proliferation in CTGF overexpressing mice compared with control mice after one week of doxycycline treatment (p = 0.61). (F) There is no statistically significant difference in β cell mass in CTGF overexpressing mice compared control mice after one week of doxycycline treatment (p = 0.24). In A, n = 8 for control, n = 4 for bigenic. In B, n = 2 for control, n = 2 for bigenic. In E, n = 3 for each genotype. In F, n = 3 for each genotype.
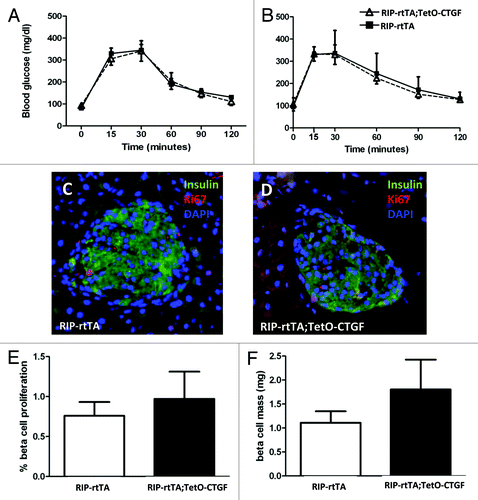
Cell replication and cell mass were also examined after one week of DOX treatment and were found to be unchanged in CTGF overexpressing mice (). Therefore, β cells of mice between 7–9 weeks of age do not seem to be responsive to CTGF, at least in this model. As noted previously, the ability of CTGF to induce β cell proliferation during embryogenesis was confined to MafA-negative, or immature, β cells. Thus, in retrospect, it may not be too surprising that adult β cells under non-stimulatory conditions do not respond to increased CTGF. However, at three weeks of age in the mouse, there remains a small population of immature MafA-negative β cells.Citation63 We therefore induced CTGF overexpression in animals right at weaning (three weeks of age) for one week and examined glucose homeostasis, β cell mass and β cell proliferation.
However, we observed no significant changes in any of these parameters (). Because pathological levels of CTGF are associated with fibrosis in models of pancreatitisCitation64 and pancreatic cancer,Citation65 pancreata from CTGF overexpressing and control mice were examined for collagen deposition using Direct Red 80/Fast Green staining. We were encouraged to observe no increase in peri-islet collagen was observed after one, three or five weeks of CTGF overexpression in β cells (Fig. S2 and data not shown). In addition, there was no increase in peri-ductal or peri-vascular collagen after one week of CTGF overexpression. However, increased collagen was apparent after 5 weeks of CTGF overexpression in peri-ductal and peri-vascular regions (Fig. S2). These results suggest caution when overexpressing CTGF, as there is a finite time period before fibrosis may develop.
Figure 3. Effects of β cell CTGF overexpression at weaning on β cell function and mass. (A) Intraperitoneal glucose tolerance tests reveal no difference in glucose homeostasis between RIP-rtTA control mice (closed squares) and RIP-rtTA;TetO-CTGF mice (open triangles) treated with doxycycline for one week, beginning at 3 weeks of age. (B, C) Immunolabeling of 4-week-old pancreata with antibodies against insulin (green) and Ki67 (red) to assess β cell proliferation after one week of CTGF overexpression (blue: DAPI, nuclei) (Magnification 200). (D) There is no difference in the percent of β cell proliferation in CTGF overexpressing mice compared with control mice after one week of doxycycline treatment (p = 0.16). (E) β cell mass is unchanged after one week of CTGF overexpression (p = 0.91). In A, n = 8 for control, n = 3 for bigenic. In D, n = 2 for each genotype. In E, n = 2 for each genotype.
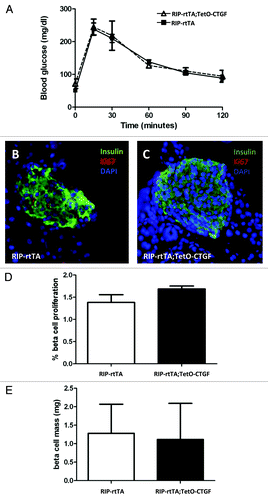
Since CTGF is an inducer of angiogenesis in some settings,Citation66,Citation67 we also examined whether CTGF overexpression in β cells would enhance islet vascularity. To this end, CTGF expression was induced in β cells for one or five weeks. There was no difference in islet vascular density as assessed by PECAM immunolabeling between control and CTGF overexpressing mice after one or five weeks of CTGF overexpression ().
Figure 4. Effects of adult β cell CTGF overexpression on islet vascularity. Immunolabeling of control (A, D) and CTGF-overexpressing (B, E) pancreata after one (A, B) or five (D, E) weeks of CTGF induction (insulin: red; PECAM: green; DAPI: blue, nuclei) (Magnification 200). (A-C) Preliminary data suggests that one week of CTGF overexpression may increase islet vascular density in ~7-week-old mice. (D-F) Islet density is not increased after five weeks of CTGF overexpression. In C, n = 1 for each genotype. In F, n = 2 for each genotype.
Experimental Procedures
Animals.
Generation of RIP-rtTACitation5 and TetO-CTGFCitation4 transgenic mice has been described previously. Mice were administered 2 mg/ml of doxycycline (DOX) in a 2% Splenda solution in drinking water at the time of weaning (for 3-week-old mice) or at 6.5–7.5 weeks of age. Mice were treated with DOX for one, three and five weeks. All animal experiments were approved by the Institutional Animal Care and Use Committee of Vanderbilt University Medical Center.
PCR genotyping.
Genotyping of RIP-rtTA;TetO-CTGF mice was performed using the following primers: forward 5'-GGA GGT CTA TAT AAG CAG AGC TCG-3' and reverse 5'-TTA AGT TAC GCC ATG TCT CCG TA-3' for TetO-CTGF; forward 5'-CGC TGT GGG GCA TTT TAC TTT AG-3' and reverse 5'-CAT GTC CAG ATC GAA ATC GTC for RIP-rtTa.
Real-time PCR for Ctgf expression.
Entire pancreata were dissected and islets were isolated by the Vanderbilt University Islet Procurement and Analysis Core and placed immediately into RNAlater (Ambion). Total RNA was extracted using the RNAqueous kit (Ambion) according to the manufacturer’s instructions. RNA concentration and integrity were assessed by the Vanderbilt University Genome Sciences Resource Core using the 2100 Electrophoresis Biolanalyzer (Agilent). cDNA was synthesized using the SuperScript III First-Strand synthesis system (Invitrogen). Reactions were performed in technical duplicate with iQ SYBR Green Supermix (Bio-Rad) according to the manufacturer’s instructions at an annealing temperature of 61.1°C. Data were collected using an iCycler iQ Real-time PCR Detection System (Bio-Rad) and software (Bio-Rad). Primers were optimized by melting curve and standard curve assays first before application. Expression levels were normalized against the levels of hypoxanthineguanine phosphoribosytransferase (HPRT). The following primers were used to detect total Ctgf expression: forward 5'-TTC TGC GAT TTC GGC TCC-3' and reverse 5'-ACC ATC TTT GGC AGT GCA CA-3'.
Tissue dissection, preparation and histology.
At the end of the DOX treatment period, pancreata were dissected into cold PBS and fixed immediately in 4% paraformaldehyde at 4°C for 4 h. Tissues were incubated in a 30% sucrose solution overnight and were embedded in Surgipath© HSC 22 Frozen Section compound and sectioned at 7 μm and stored at -80°C.
Immunolabeling.
Protein localization was assessed by incubation of tissue sections with various antibodies. Insulin and Ki67: labeling was performed on frozen sections and required antigen retrieval for Ki67 in 1 sodium citrate (10 mM) for 14 min at 40% power. Primary antibodies included guinea pig anti-insulin (1:1,000; Upstate) and mouse anti-Ki67 (1:500; BD PharMingen). Primary antibodies were detected by species-specific donkey secondary antibodies conjugated to either Cy2 or Cy3 flurophores (1:300; Jackson ImmunoResearch Laboratories, Inc.). PECAM: platelet endothelial β cell adhesion molecule (PECAM) labeling was performed on frozen sections after 3% hydrogen peroxide quenching using rat anti-PECAM (1:50; BD PharMingen) and the Vector M.O.M kit (Vector Labs). An anti-rat biotinylated secondary antibody (1:500; Vector Labs) applied with Vectastatin ABC kit (Vector Labs) was used for labeling followed by TSA amplification (Invitrogen).
All primary antibodies were incubated overnight in a humid chamber at 4°C. Fluorophores were excited and imaged using a ScanScope FL slide scanner (Aperio Technologies, Inc.). Images from each experiment were processed identically with ImageScope software (Aperio Technologies, Inc.).
Intraperitoneal glucose tolerance tests (IPGTT).
IPGTTs were performed on control (RIP-rtTA) and bigenic (RIP-rtTA;TetO-CTGF) mice treated with DOX as described previously in reference Citation3, using an Accuchek Aviva glucometer and test strips (Roche Pharmaceuticals).
β cell proliferation.
Entire pancreata were serially sectioned and slides were immunolabeled for insulin and Ki67. One section was analyzed every 250 μm (11–16 sections per animal). Every islet in that section was photographed and the total number of cells positive for insulin was counted, as were the number of proliferating cells (co-labeled for insulin and Ki67) using Metamorph 6.1 software (Molecular Devices). The percentage of proliferating cells was determined by dividing the number of Ki67/insulin double-positive cells by the total number of insulin-positive cells and multiplying by 100.
β cell mass.
Entire pancreata were removed, weighed, fixed and serially sectioned. One section per slide was analyzed every 250 μm (11–16 sections per animal) throughout the pancreas as previously described in reference Citation3 and Citation41. Briefly, sections were prepared for insulin immunoperoxidase labeling and eosin counterstaining. Vector DAB Peroxidase Substrate kit (Vector Labs) was used for immunohistochemical labeling of insulin for β cell mass measurement. Images of anti-insulin-labeled sections were scanned using a ScanScope CS slide scanner (Aperio Technologies, Inc.). Images from each experiment were processed identically with the ImageScope software (Aperio Technologies, Inc.). β cell mass was measured by obtaining the fraction of cross-sectional area of pancreatic tissue positive for insulin as calculated by modified Genie algorithm within the Spectrum software (Aperio Technologies, Inc.,) and multiplying this by the pancreatic wet weight.
Collagen assessment.
Entire pancreata were serially sectioned and three sections per animal, at least 250 μm apart, were randomly selected and analyzed. Sections were stained for collagen by incubating with Direct Red 80/Fast Green (Sigma-Aldrich Company) for one hour at room temperature in a humid chamber. The slides were washed in acidified water and dehydrated using an increasing ethanol series to xylene. Images of collagen-labeled sections were scanned using a ScanScope CS slide scanner (Aperio Technologies, Inc.).
Islet microvascular density.
The blood vessel endothelium density per islet was determined by immunolabeling for insulin and PECAM. Entire pancreata were serially sectioned and slides immunolabeled for insulin and PECAM. One section per slide was analyzed every 250 μm (11–16 sections per animal) throughout the pancreas and the PECAM-positive area of every islet (designated as containing 10 insulin-positive cells or more) per section was quantified using Metamorph software. To determine the percentage of blood vessel area, the total PECAM area was divided by the total insulin-positive area and multiplied by 100.
Conclusions and Future Perspectives
The data presented here suggest that CTGF has no influence on β cell replication or mass in the post-weaning β cell under normal circumstances and that it can only induce replication of embryonic β cells.Citation4 Much work is still needed to elucidate the mechanism(s) by which CTGF promotes β cell proliferation, but from what is known in mammary cell lines, this regulation may occur via the PI3/Akt signaling pathway. Adult β cells are in general less receptive to proliferative stimuli than are embryonic or neonatal β cells, although the β cells at the time point we studied have been responsive in other studies.Citation62 It is possible that CTGF plays a role only in embryonic β cell replication or alternatively, would affect adult β cell replication in the presence of a strong proliferative stimulus such as obesity-induced insulin resistance, response to injury or pregnancy. These possibilities will be examined in the future.
Current efforts in the field are focused in large part on directed differentiation of human embryonic stem cells or induced pluripotent stem cells toward a β cell phenotype by mimicking normal pancreatic development. The best protocols allow for production of glucose-responsive β cells, although this process requires an in vivo maturation step that remains ill-defined.Citation68 CTGF is necessary for adequate β cell mass at birth and is sufficient to induce embryonic β cell mass expansion. Thus, it could be useful in stem cell directed differentiation protocols or in the expansion of insulin-producing cells prior to their full maturation.
Clearly, expansion of immature insulin-positive cell populations is regulated by factors that differ from regulators of mature β cell mass expansion. In many cases, these changes in requirement from an “embryonic” form of regulation to an “adult” form of regulation occur around weaning (see model in ). While the mechanism(s) underlying this switch have yet to be elucidated, it is possible that metabolic and physiological cues associated with switching from breast milk to chow could influence the mode of β cell replication. Alternatively, some sort of clock mechanism might come into play where a β cell undergoes a certain number of “embryonic” cell divisions and then switches to the postnatal form of replication. A better understanding of β cell development and the identification of factors important for β cell maturation and replication would be useful to further improving strategies for replacing β cell mass in individuals with diabetes. Changes in β cell mass are the result of a balance between the negative effects of apoptosis and the positive effects of proliferation or neogenesis. Successful treatments to increase β cell mass will attempt to tip the scales in the direction of proliferation and/or neogenesis and CTGF could be such a factor.
Additional material
Download Zip (1.3 MB)Acknowledgments
We thank Dr. Maria Golson and Ms. Kathryn Henley for helpful discussions and critical reading of the manuscript and Mr. Amit Chakraborty for technical help. This research involved use of the Vanderbilt Transgenic Mouse/ES Cell Shared Resource, the Islet Procurement and Analysis Core and the Functional Genomics Shared Resource (supported in part by National Institutes of Health Grants CA68485 and DK20593) and was supported by 1F32DK092001–01 (U.G.) and Grant 1–2007–548 from the Juvenile Diabetes Research Foundation International (M.G.).
Note
Supplemental materials can be found at: www.landesbioscience.com/journals/cc/article/20545
Related Research Data
References
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003; 52:102 - 10; http://dx.doi.org/10.2337/diabetes.52.1.102; PMID: 12502499
- Diseases NIoDaDaK. . National Diabetes Fact Sheet. 2011.
- Crawford LA, Guney MA, Oh YA, Deyoung RA, Valenzuela DM, Murphy AJ, et al. Connective tissue growth factor (CTGF) inactivation leads to defects in islet cell lineage allocation and beta-cell proliferation during embryogenesis. Mol Endocrinol 2009; 23:324 - 36; http://dx.doi.org/10.1210/me.2008-0045; PMID: 19131512
- Guney MA, Petersen CP, Boustani A, Duncan MR, Gunasekaran U, Menon R, et al. Connective tissue growth factor acts within both endothelial cells and beta cells to promote proliferation of developing beta cells. Proc Natl Acad Sci USA 2011; 108:15242 - 7; http://dx.doi.org/10.1073/pnas.1100072108; PMID: 21876171
- Milo-Landesman D, Surana M, Berkovich I, Compagni A, Christofori G, Fleischer N, et al. Correction of hyperglycemia in diabetic mice transplanted with reversibly immortalized pancreatic beta cells controlled by the tet-on regulatory system. Cell Transplant 2001; 10:645 - 50; PMID: 11714200
- Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, et al. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development 1996; 122:983 - 95; PMID: 8631275
- Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet 2002; 32:128 - 34; http://dx.doi.org/10.1038/ng959; PMID: 12185368
- Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci USA 2000; 97:1607 - 11; http://dx.doi.org/10.1073/pnas.97.4.1607; PMID: 10677506
- Jacquemin P, Durviaux SM, Jensen J, Godfraind C, Gradwohl G, Guillemot F, et al. Transcription factor hepatocyte nuclear factor 6 regulates pancreatic endocrine cell differentiation and controls expression of the proendocrine gene ngn3. Mol Cell Biol 2000; 20:4445 - 54; http://dx.doi.org/10.1128/MCB.20.12.4445-4454.2000; PMID: 10825208
- Seymour PA, Freude KK, Dubois CL, Shih HP, Patel NA, Sander M. A dosage-dependent requirement for Sox9 in pancreatic endocrine cell formation. Dev Biol 2008; 323:19 - 30; http://dx.doi.org/10.1016/j.ydbio.2008.07.034; PMID: 18723011
- Oliver-Krasinski JM, Kasner MT, Yang J, Crutchlow MF, Rustgi AK, Kaestner KH, et al. The diabetes gene Pdx1 regulates the transcriptional network of pancreatic endocrine progenitor cells in mice. J Clin Invest 2009; 119:1888 - 98; http://dx.doi.org/10.1172/JCI37028; PMID: 19487809
- Sander M, Sussel L, Conners J, Scheel D, Kalamaras J, Dela Cruz F, et al. Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development 2000; 127:5533 - 40; PMID: 11076772
- Kim SK, MacDonald RJ. Signaling and transcriptional control of pancreatic organogenesis. Curr Opin Genet Dev 2002; 12:540 - 7; http://dx.doi.org/10.1016/S0959-437X(02)00338-6; PMID: 12200159
- Jensen J. Gene regulatory factors in pancreatic development. Dev Dyn 2004; 229:176 - 200; http://dx.doi.org/10.1002/dvdy.10460; PMID: 14699589
- Pan FC, Wright C. Pancreas organogenesis: from bud to plexus to gland. Dev Dyn 2011; 240:530 - 65; http://dx.doi.org/10.1002/dvdy.22584; PMID: 21337462
- Zhang W, Feng D, Li Y, Iida K, McGrath B, Cavener DR. PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab 2006; 4:491 - 7; http://dx.doi.org/10.1016/j.cmet.2006.11.002; PMID: 17141632
- Ford SP, Zhang L, Zhu M, Miller MM, Smith DT, Hess BW, et al. Maternal obesity accelerates fetal pancreatic beta-cell but not alpha-cell development in sheep: prenatal consequences. Am J Physiol Regul Integr Comp Physiol 2009; 297:R835 - 43; http://dx.doi.org/10.1152/ajpregu.00072.2009; PMID: 19605766
- Georgia S, Bhushan A. p27 Regulates the transition of beta-cells from quiescence to proliferation. Diabetes 2006; 55:2950 - 6; http://dx.doi.org/10.2337/db06-0249; PMID: 17065330
- Guz Y, Montminy MR, Stein R, Leonard J, Gamer LW, Wright CV, et al. Expression of murine STF-1, a putative insulin gene transcription factor, in beta cells of pancreas, duodenal epithelium and pancreatic exocrine and endocrine progenitors during ontogeny. Development 1995; 121:11 - 8; PMID: 7867492
- Holland AM, Hale MA, Kagami H, Hammer RE, MacDonald RJ. Experimental control of pancreatic development and maintenance. Proc Natl Acad Sci USA 2002; 99:12236 - 41; http://dx.doi.org/10.1073/pnas.192255099; PMID: 12221286
- Gannon M, Ables ET, Crawford L, Lowe D, Offield MF, Magnuson MA, et al. pdx-1 function is specifically required in embryonic beta cells to generate appropriate numbers of endocrine cell types and maintain glucose homeostasis. Dev Biol 2008; 314:406 - 17; http://dx.doi.org/10.1016/j.ydbio.2007.10.038; PMID: 18155690
- Artner I, Blanchi B, Raum JC, Guo M, Kaneko T, Cordes S, et al. MafB is required for islet beta cell maturation. Proc Natl Acad Sci USA 2007; 104:3853 - 8; http://dx.doi.org/10.1073/pnas.0700013104; PMID: 17360442
- Moussad EE, Brigstock DR. Connective tissue growth factor: what’s in a name?. Mol Genet Metab 2000; 71:276 - 92; http://dx.doi.org/10.1006/mgme.2000.3059; PMID: 11001822
- Zhang C, Moriguchi T, Kajihara M, Esaki R, Harada A, Shimohata H, et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol Cell Biol 2005; 25:4969 - 76; http://dx.doi.org/10.1128/MCB.25.12.4969-4976.2005; PMID: 15923615
- Finegood DT, Scaglia L, Bonner-Weir S. Dynamics of beta-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes 1995; 44:249 - 56; http://dx.doi.org/10.2337/diabetes.44.3.249; PMID: 7883109
- Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004; 429:41 - 6; http://dx.doi.org/10.1038/nature02520; PMID: 15129273
- Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H. beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev 1998; 12:1763 - 8; http://dx.doi.org/10.1101/gad.12.12.1763; PMID: 9637677
- Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest 2004; 114:963 - 8; PMID: 15467835
- Martín J, Hunt SL, Dubus P, Sotillo R, Néhmé-Pélluard F, Magnuson MA, et al. Genetic rescue of Cdk4 null mice restores pancreatic beta-cell proliferation but not homeostatic cell number. Oncogene 2003; 22:5261 - 9; http://dx.doi.org/10.1038/sj.onc.1206506; PMID: 12917627
- Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 2006; 443:453 - 7; http://dx.doi.org/10.1038/nature05092; PMID: 16957737
- Dhawan S, Tschen SI, Bhushan A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev 2009; 23:906 - 11; http://dx.doi.org/10.1101/gad.1742609; PMID: 19390085
- Chen H, Gu X, Su IH, Bottino R, Contreras JL, Tarakhovsky A, et al. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev 2009; 23:975 - 85; http://dx.doi.org/10.1101/gad.1742509; PMID: 19390090
- Chen H, Gu X, Liu Y, Wang J, Wirt SE, Bottino R, et al. PDGF signalling controls age-dependent proliferation in pancreatic β-cells. Nature 2011; 478:349 - 55; http://dx.doi.org/10.1038/nature10502; PMID: 21993628
- Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ, et al. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol Cell Biol 2005; 25:10875 - 94; http://dx.doi.org/10.1128/MCB.25.24.10875-10894.2005; PMID: 16314512
- Laoukili J, Kooistra MR, Brás A, Kauw J, Kerkhoven RM, Morrison A, et al. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol 2005; 7:126 - 36; http://dx.doi.org/10.1038/ncb1217; PMID: 15654331
- Zhang H, Zhang J, Pope CF, Crawford LA, Vasavada RC, Jagasia SM, et al. Gestational diabetes mellitus resulting from impaired beta-cell compensation in the absence of FoxM1, a novel downstream effector of placental lactogen. Diabetes 2010; 59:143 - 52; http://dx.doi.org/10.2337/db09-0050; PMID: 19833884
- Ackermann Misfeldt A, Costa RH, Gannon M. Beta-cell proliferation, but not neogenesis, following 60% partial pancreatectomy is impaired in the absence of FoxM1. Diabetes 2008; 57:3069 - 77; http://dx.doi.org/10.2337/db08-0878; PMID: 18728229
- Zhang H, Ackermann AM, Gusarova GA, Lowe D, Feng X, Kopsombut UG, et al. The FoxM1 transcription factor is required to maintain pancreatic beta-cell mass. Mol Endocrinol 2006; 20:1853 - 66; http://dx.doi.org/10.1210/me.2006-0056; PMID: 16556734
- Gunasekaran U, Gannon M. Type 2 diabetes and the aging pancreatic beta cell. Aging 2011; 3:565 - 75; PMID: 21765202
- Kushner JA. Beta-cell growth: an unusual paradigm of organogenesis that is cyclin D2/Cdk4 dependent. Cell Cycle 2006; 5:234 - 7; http://dx.doi.org/10.4161/cc.5.3.2399; PMID: 16410729
- Dhawan S, Georgia S, Bhushan A. Formation and regeneration of the endocrine pancreas. Curr Opin Cell Biol 2007; 19:634 - 45; http://dx.doi.org/10.1016/j.ceb.2007.09.015; PMID: 18061427
- Cozar-Castellano I, Fiaschi-Taesch N, Bigatel TA, Takane KK, Garcia-Ocaña A, Vasavada R, et al. Molecular control of cell cycle progression in the pancreatic beta-cell. Endocr Rev 2006; 27:356 - 70; http://dx.doi.org/10.1210/er.2006-0004; PMID: 16638909
- Pick A, Clark J, Kubstrup C, Levisetti M, Pugh W, Bonner-Weir S, et al. Role of apoptosis in failure of beta-cell mass compensation for insulin resistance and beta-cell defects in the male Zucker diabetic fatty rat. Diabetes 1998; 47:358 - 64; http://dx.doi.org/10.2337/diabetes.47.3.358; PMID: 9519740
- Sorenson RL, Brelje TC. Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res 1997; 29:301 - 7; http://dx.doi.org/10.1055/s-2007-979040; PMID: 9230352
- Bonner-Weir S, Deery D, Leahy JL, Weir GC. Compensatory growth of pancreatic beta-cells in adult rats after short-term glucose infusion. Diabetes 1989; 38:49 - 53; http://dx.doi.org/10.2337/diabetes.38.1.49; PMID: 2642434
- Topp BG, McArthur MD, Finegood DT. Metabolic adaptations to chronic glucose infusion in rats. Diabetologia 2004; 47:1602 - 10; http://dx.doi.org/10.1007/s00125-004-1493-5; PMID: 15349726
- Paris M, Bernard-Kargar C, Berthault MF, Bouwens L, Ktorza A. Specific and combined effects of insulin and glucose on functional pancreatic beta-cell mass in vivo in adult rats. Endocrinology 2003; 144:2717 - 27; http://dx.doi.org/10.1210/en.2002-221112; PMID: 12746336
- Kozma SC, Thomas G. Regulation of cell size in growth, development and human disease: PI3K, PKB and S6K. Bioessays 2002; 24:65 - 71; http://dx.doi.org/10.1002/bies.10031; PMID: 11782951
- Gu Y, Lindner J, Kumar A, Yuan W, Magnuson MA. Rictor/mTORC2 is essential for maintaining a balance between beta-cell proliferation and cell size. Diabetes 2011; 60:827 - 37; http://dx.doi.org/10.2337/db10-1194; PMID: 21266327
- Kitamura T, Nakae J, Kitamura Y, Kido Y, Biggs WH 3rd, Wright CV, et al. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest 2002; 110:1839 - 47; PMID: 12488434
- Kulkarni RN, Jhala US, Winnay JN, Krajewski S, Montminy M, Kahn CR. PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J Clin Invest 2004; 114:828 - 36; PMID: 15372107
- Heit JJ, Apelqvist AA, Gu X, Winslow MM, Neilson JR, Crabtree GR, et al. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature 2006; 443:345 - 9; http://dx.doi.org/10.1038/nature05097; PMID: 16988714
- Lawrence MC, Bhatt HS, Easom RA. NFAT regulates insulin gene promoter activity in response to synergistic pathways induced by glucose and glucagon-like peptide-1. Diabetes 2002; 51:691 - 8; http://dx.doi.org/10.2337/diabetes.51.3.691; PMID: 11872668
- Liu J, Farmer JD Jr., Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991; 66:807 - 15; http://dx.doi.org/10.1016/0092-8674(91)90124-H; PMID: 1715244
- Buchanan TA, Metzger BE, Freinkel N, Bergman RN. Insulin sensitivity and B-cell responsiveness to glucose during late pregnancy in lean and moderately obese women with normal glucose tolerance or mild gestational diabetes. Am J Obstet Gynecol 1990; 162:1008 - 14; PMID: 2183610
- Vasavada RC, Garcia-Ocaña A, Zawalich WS, Sorenson RL, Dann P, Syed M, et al. Targeted expression of placental lactogen in the beta cells of transgenic mice results in beta cell proliferation, islet mass augmentation, and hypoglycemia. J Biol Chem 2000; 275:15399 - 406; http://dx.doi.org/10.1074/jbc.275.20.15399; PMID: 10809775
- Kim H, Toyofuku Y, Lynn FC, Chak E, Uchida T, Mizukami H, et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med 2010; 16:804 - 8; http://dx.doi.org/10.1038/nm.2173; PMID: 20581837
- Otonkoski T, Beattie GM, Rubin JS, Lopez AD, Baird A, Hayek A. Hepatocyte growth factor/scatter factor has insulinotropic activity in human fetal pancreatic cells. Diabetes 1994; 43:947 - 53; http://dx.doi.org/10.2337/diabetes.43.7.947; PMID: 8013761
- Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes 1999; 48:2270 - 6; http://dx.doi.org/10.2337/diabetes.48.12.2270; PMID: 10580413
- Hügl SR, White MF, Rhodes CJ. Insulin-like growth factor I (IGF-I)-stimulated pancreatic beta-cell growth is glucose-dependent. Synergistic activation of insulin receptor substrate-mediated signal transduction pathways by glucose and IGF-I in INS-1 cells. J Biol Chem 1998; 273:17771 - 9; http://dx.doi.org/10.1074/jbc.273.28.17771; PMID: 9651378
- Zhou Y, Capuco AV, Jiang H. Involvement of connective tissue growth factor (CTGF) in insulin-like growth factor-I (IGF1) stimulation of proliferation of a bovine mammary epithelial cell line. Domest Anim Endocrinol 2008; 35:180 - 9; http://dx.doi.org/10.1016/j.domaniend.2008.05.003; PMID: 18586434
- Rankin MM, Kushner JA. Adaptive beta-cell proliferation is severely restricted with advanced age. Diabetes 2009; 58:1365 - 72; http://dx.doi.org/10.2337/db08-1198; PMID: 19265026
- Artner I, Hang Y, Mazur M, Yamamoto T, Guo M, Lindner J, et al. MafA and MafB regulate genes critical to beta-cells in a unique temporal manner. Diabetes 2010; 59:2530 - 9; http://dx.doi.org/10.2337/db10-0190; PMID: 20627934
- di Mola FF, Friess H, Martignoni ME, Di Sebastiano P, Zimmermann A, Innocenti P, et al. Connective tissue growth factor is a regulator for fibrosis in human chronic pancreatitis. Ann Surg 1999; 230:63 - 71; http://dx.doi.org/10.1097/00000658-199907000-00010; PMID: 10400038
- Hartel M, Di Mola FF, Gardini A, Zimmermann A, Di Sebastiano P, Guweidhi A, et al. Desmoplastic reaction influences pancreatic cancer growth behavior. World J Surg 2004; 28:818 - 25; http://dx.doi.org/10.1007/s00268-004-7147-4; PMID: 15457365
- Brigstock DR. Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61). Angiogenesis 2002; 5:153 - 65; http://dx.doi.org/10.1023/A:1023823803510; PMID: 12831056
- Yang H, Huang Y, Chen X, Liu J, Lu Y, Bu L, et al. The role of CTGF in the diabetic rat retina and its relationship with VEGF and TGF-beta, elucidated by treatment with CTGFsiRNA. Acta Ophthalmol 2009;
- Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S, et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol 2008; 26:443 - 52; http://dx.doi.org/10.1038/nbt1393; PMID: 18288110
- Dutta S, Bonner-Weir S, Montminy M, Wright C. Regulatory factor linked to late-onset diabetes?. Nature 1998; 392:560; http://dx.doi.org/10.1038/33311; PMID: 9560151
- Rose SD, Swift GH, Peyton MJ, Hammer RE, MacDonald RJ. The role of PTF1-P48 in pancreatic acinar gene expression. J Biol Chem 2001; 276:44018 - 26; http://dx.doi.org/10.1074/jbc.M106264200; PMID: 11562365
- Wang S, Jensen JN, Seymour PA, Hsu W, Dor Y, Sander M, et al. Sustained Neurog3 expression in hormone-expressing islet cells is required for endocrine maturation and function. Proc Natl Acad Sci USA 2009; 106:9715 - 20; http://dx.doi.org/10.1073/pnas.0904247106; PMID: 19487660
- Dubois CL, Shih HP, Seymour PA, Patel NA, Behrmann JM, Ngo V, et al. Sox9-haploinsufficiency causes glucose intolerance in mice. PLoS ONE 2011; 6:e23131; http://dx.doi.org/10.1371/journal.pone.0023131; PMID: 21829703
- Zhang H, Ables ET, Pope CF, Washington MK, Hipkens S, Means AL, et al. Multiple, temporal-specific roles for HNF6 in pancreatic endocrine and ductal differentiation. Mech Dev 2009; 126:958 - 73; http://dx.doi.org/10.1016/j.mod.2009.09.006; PMID: 19766716
- Artner I, Le Lay J, Hang Y, Elghazi L, Schisler JC, Henderson E, et al. MafB: an activator of the glucagon gene expressed in developing islet alpha- and beta-cells. Diabetes 2006; 55:297 - 304; http://dx.doi.org/10.2337/diabetes.55.02.06.db05-0946; PMID: 16443760
- Buteau J, Accili D. Regulation of pancreatic beta-cell function by the forkhead protein FoxO1. Diabetes Obes Metab 2007; 9:Suppl 2 140 - 6; http://dx.doi.org/10.1111/j.1463-1326.2007.00782.x; PMID: 17919188
- Garcia-Ocaña A, Takane KK, Syed MA, Philbrick WM, Vasavada RC, Stewart AF. Hepatocyte growth factor overexpression in the islet of transgenic mice increases beta cell proliferation, enhances islet mass, and induces mild hypoglycemia. J Biol Chem 2000; 275:1226 - 32; http://dx.doi.org/10.1074/jbc.275.2.1226; PMID: 10625667
- Cozar-Castellano I, Takane KK, Bottino R, Balamurugan AN, Stewart AF. Induction of beta-cell proliferation and retinoblastoma protein phosphorylation in rat and human islets using adenovirus-mediated transfer of cyclin-dependent kinase-4 and cyclin D1. Diabetes 2004; 53:149 - 59; http://dx.doi.org/10.2337/diabetes.53.1.149; PMID: 14693709
- Westmoreland JJ, Wang Q, Bouzaffour M, Baker SJ, Sosa-Pineda B. Pdk1 activity controls proliferation, survival, and growth of developing pancreatic cells. Dev Biol 2009; 334:285 - 98; http://dx.doi.org/10.1016/j.ydbio.2009.07.030; PMID: 19635472
- Zehri A. Differential effects of pulsatile vs. chronic hyperglycemia on fetal pancreatic beta cell population. Physiology. Tuscon: University of Arizona, 2011:22.