Abstract
Hyperglycemia during hyper-CVAD chemotherapy is associated with poor outcomes of acute lymphoblastic leukemia (ALL) (Cancer 2004; 100:1179–85). The optimal clinical strategy to manage hyperglycemia during hyper-CVAD is unclear. To examine whether anti-diabetic pharmacotherapy can influence chemosensitivity of ALL cells, we examined the impacts of different anti-diabetic agents on ALL cell lines and patient samples. Pharmacologically achievable concentrations of insulin, aspart and glargine significantly increased the number of ALL cells, and aspart and glargine did so at lower concentrations than human insulin. In contrast, metformin and rosiglitazone significantly decreased the cell number. Human insulin and analogs activated AKT/mTOR signaling and stimulated ALL cell proliferation (as measured by flow cytometric methods), but metformin and rosiglitazone blocked AKT/mTOR signaling and inhibited proliferation. Metformin 500 μM and rosiglitazone 10 μM were found to sensitize Reh cells to daunorubicin, while aspart, glargine and human insulin (all at 1.25 mIU/L) enhanced chemoresistance. Metformin and rosiglitazone enhanced daunorubicin-induced apoptosis, while insulin, aspart and glargine antagonized daunorubicin-induced apoptosis. In addition, metformin increased etoposide-induced and L-asparaginase-induced apoptosis; rosiglitazone increased etoposide-induced and vincristine-induced apoptosis. In conclusion, our results suggest that use of insulins to control hyperglycemia in ALL patients may contribute to anthracycline chemoresistance, while metformin and thiazolidinediones may improve chemosensitivity to anthracycline as well as other chemotherapy drugs through their different impacts on AKT/mTOR signaling in leukemic cells. Our data suggest that the choice of anti-diabetic pharmacotherapy during chemotherapy may influence clinical outcomes in ALL.
Introduction
Epidemiologic data suggest important roles of type 2 diabetes mellitus (DM2) in carcinogenesisCitation1-Citation4 and cancer prognosis.Citation5 The hyper-CVAD regimen (fractionated cyclophosphamide, vincristine, doxorubicin and dexamethasone alternating with methotrexate and high-dose cytarabine) is currently a standard treatment for acute lymphocytic leukemia (ALL), Burkitt lymphoma (BL) and lymphoblastic lymphoma (LL).Citation6 This regimen, which includes high-dose dexamethasone and methylprednisolone, frequently leads to hyperglycemia. Our retrospective study of 278 adult patients with previously untreated ALL who achieved a complete response with hyper-CVAD showed that hyperglycemia (glucose ≥ 200 mg/dL on ≥ 2 determinations) occurred in up to 37% of patients during induction chemotherapy.Citation7 Hyperglycemic patients had shorter median complete remission duration (CRD) (24 vs. 52 mo, p = 0.001) and a shorter median survival (29 vs. 88 mo, p < 0.001) than non-hyperglycemic (glucose ≥ 200 mg/dL on < 2 determinations) patients.Citation7 When controlled for predictors of ALL outcomes in multivariate analysis, hyperglycemia was an independent factor for early relapse and mortality; patients with hyperglycemia were 1.57 times more likely to relapse and 1.71 times more likely to die than those without hyperglycemia.Citation7
Mean fasting glucose > 6.25 mM (> 112.5 mg/dL) is associated with significant increase in cancer death.Citation8 There are multiple reasons or mechanisms that can lead to the association of hyperglycemia with increased cancer deaths. Deregulated energy metabolism is a hallmark of cancer,Citation9 and use of glycolysis to generate the cancer cell’s energy need from glucose is a major characteristic described by Warburg.Citation10 Cancer cells often have coordinated upregulation of hypoxia-induced factor-1 (HIF-1) and Myc and downregulation of tumor suppressor p53 to result in an increase in flux through the glycolytic pathway.Citation11 In the context of insulin resistance in obesity and DM2, elevated circulating insulin and insulin-like growth factors (IGF) will stimulate the AKT signaling pathway, which will lead to downstream upregulation of HIF-1α and Myc and downregulation of p53, and this coordinated downstream regulation will lead to increased glycolytic flux. This increased glycolytic flux confers the avidity of glucose uptake by malignant cells, and hyperglycemia will translate into an abundance of fuel for the malignant cells and the ability to tolerate hypoxia, radiation and chemotherapy.
Our recent experimental data show that high levels of insulin and glucose promote cancer cell growth in vitro.Citation12 The American Diabetic Association and American Cancer Society have issued a consensus statement that the possible mechanisms for a direct link between cancer and DM2 are hyperinsulinemia, hyperglycemia and inflammation.Citation13 The evidence for specific drugs affecting cancer risk is limited, and observed associations may have been confounded by indications for specific drugs, effects on other cancer risk factors, such as body weight and hyperinsulinemia, and the complex progressive nature of hyperglycemia and pharmacotherapy in DM2. The consensus states that cancer risk should not be a major factor when choosing between available diabetes therapies for the average patient.
A DM2 patient also diagnosed with cancer is not the “average” patient. In the context of the adverse impact of hyperglycemia on the clinical outcomes of ALL patients undergoing hyper-CVAD chemotherapy, the issue of the choice of pharmacotherapy for hyperglycemia in these patients already diagnosed with a malignant neoplastic disease is distinct from the issues addressed in the consensus statement.Citation13 Our recent investigation into the impact of anti-diabetic medications on various cancer cell lines found that biguanides and thiazolidinediones inhibited cancer cells.Citation12 We hypothesize that biguanides and thiazolidinediones can perturb the AKT/mTOR signaling pathway in different ways, resulting in differences in leukemic cell proliferation, apoptosis and chemosensitivity. Therefore, we conducted laboratory studies with ALL cell lines and primary leukemic cell samples of ALL patients to examine the impacts of different classes of anti-diabetic pharmacotherapy on ALL cells and their intracellular signaling mechanisms and to ascertain what class(es) of anti-diabetic drugs may be beneficial for the clinical outcomes of hyperglycemic ALL patients.
Results
Insulin stimulated, while metformin and rosiglitazone inhibited, ALL cells.
Using human ALL cell lines and primary patient samples (), we investigated the impact of anti-diabetic medications on leukemic cell proliferation. First, we examined the impact of human regular insulin, aspart and glargine on the number of live ALL cells in culture. Reh and Nalm-6 cells were incubated in control media without insulins, human regular insulin, aspart and glargine at 80 mIU/L for 72 h, and then the cells were dispersed by trypsin and EDTA treatment for counting the number of trypan blue dye-excluding cells using a hemocytometer. Glargine significantly (p = 0.010; one-way ANOVA, post-hoc Dunnett T3 test) increased the number of live Reh leukemic cells from (8.656 ± 1.313) x 105/ml (mean ± 95% confidence interval) to (10.688 ± 1.034) x 105/ml; aspart also significantly (p = 0.019) increased it to (10.500 ± 0.779) x 105/ml. There were (9.813 ± 1.475) x 105/ml live cells in the human regular insulin group, but the difference was not statistically significant (p = 0.177). Similarly, glargine significantly (p = 0.011) increased the number of live Nalm-6 leukemic cells from (15.625 ± 1.852) x 105/ ml (mean ±95% confidence interval) to (17.813 ± 1.107) x 105/ml; aspart also significantly (p = 0.050) increased it to (17.313 ± 0.526) x 105/ml. There were (16.781 ± 1.262) x 105/ ml live cells in the human regular insulin group, but the difference was not statistically significant (p = 0.228).
Table 1. Clinical characteristic of acute lymphoblastic leukemia patients who donated cell samples for in vitro analysis
Further investigation showed that human insulin, glargine and aspart dose-dependently increased the number of live ALL cells as measured by the MTS assayCitation14-Citation16 compared with ALL cells cultured without insulin and insulin analogs (). Four ALL cell lines (Reh, Nalm-6, Raji and CCRF-CEM) and seven consecutive primary leukemic cell samples of ALL patients (patients 1 to 7) were evaluated. Representative log-dose response curves (Reh, Nalm-6 and patient 1) are shown (), and the curves for Raji, CCRF-CEM and patients 2 to 7 are qualitatively similar (Fig. S1). Assuming that the burden of malignant cells can be estimated by compounding the percentage increase in cells every 3 d, chronic exposure for a month to a concentration of insulin or analogs that increases the cells by 5% over 3 d will result in a 62.9% increase in tumor burden [i.e., (1.05)Citation10 = 1.629]. Based on the sigmoidal log-dose response curve, the concentrations that increased leukemia cells by 5% over 3 d for regular insulin, aspart and glargine for ALL cell lines and patient samples tested are reported in . The mean concentrations that increased leukemic cells by 5% in 3 d were not significantly [p > 0.0167 (Bonferroni correction for multiple comparison)] different between cell lines and primary samples for regular insulin, aspart and glargine. Combining the data from cell lines and primary samples for analysis, the concentration of glargine is significantly lower than that of regular insulin (one-way ANOVA on ranks, post-hoc intergroup comparison using Dunnett T3 test; p = 0.015). A concentration of 50 mIU/L is two times the upper limit of normal fasting serum insulin level, and this concentration is reachable pharmacologicallyCitation17 in insulin-resistant patients as well. At 50 mIU/L, the mean relative increase of leukemic cells in 3 d was not significantly [p > 0.0167 (Bonferroni correction for multiple comparison)] different between cell lines and primary samples for regular insulin and aspart. Primary samples were increased by glargine to a higher proportion than cell lines (p = 0.006). Combining the data from cell lines and primary samples for analysis, the leukemic cells were increased by aspart and glargine to a significant degree (one-way ANOVA on ranks, post-hoc intergroup comparison using Dunnett T3 test; p < 0.001 and p < 0.001, respectively) higher proportion than that by regular insulin (). Therefore, insulin analogs aspart and glargine stimulated proliferation of leukemic cells to significantly higher degrees than human regular insulin at pharmacologically relevant concentrations.
Figure 1. Differential effects of diabetes medications on leukemic cell proliferation. (A) The cell viability was measured by the MTS assay. The cell viability relative to the control samples was plotted against the concentration of human insulin (red), glargine (green) or aspart (blue). Insulin and analogs increased the number of live ALL cells. Glargine and aspart stimulated ALL cells more than human insulin at equal IU concentrations (i.e., equi-hypoglycemic). (B) The viability relative to control was measured as in (A). Representative log-dose response curves are shown. Metformin inhibited ALL cell lines and primary patient samples (see color key). (C) Similar to (B), representative log-dose response curves demonstrating the inhibitory effect of rosiglitazone are shown (see color key for ALL cell lines and primary patient samples). (D) Reh and Nalm-6 cells were incubated for 2 and 4 d, respectively, with insulin (5 mIU/L, red bar), aspart (0.6 mIU/L, blue bar) or glargine (0.2 mIU/L, green bar), and a Coulter counter was used to count cells. Error bars represent 95% confidence intervals (n = 4). p-values (one-way ANOVA, post-hoc intergroup comparisons) indicate significant increases in the cell number relative to control (white bar). (E) Reh cells were treated for 4 d with metformin (71 µM, blue bar) or rosiglitazone (11 µM, yellow bar). Nalm-6 cells were treated for 6 d with metformin (32 µM, blue bar) or rosiglitazone (4 µM, yellow bar). Cell numbers were counted using a Coulter counter. Error bars represent 95% confidence intervals (n = 4). P-values (one-way ANOVA, post-hoc intergroup comparisons) indicate significant decreases in the cell number relative to control (white bar).
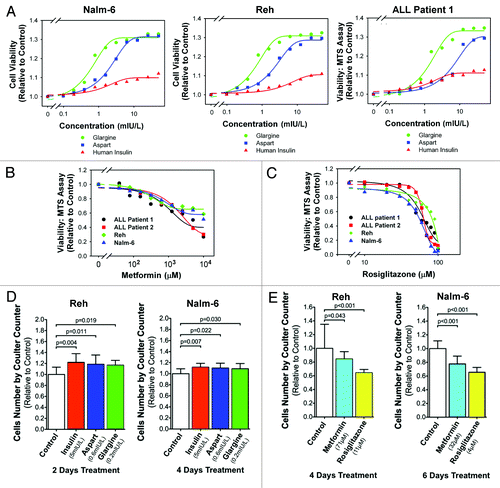
Table 2. Concentrations of insulin and insulin analogs that can increase leukemic cells by 5% in 3 d
Table 3. Mean ratio of viable cells treated with insulin and insulin analogs at 50 mIU/L (2 × upper limit of normal fasting serum insulin level for most laboratories) for 72 h relative to control
In contrast, exposure to metformin () or rosiglitazone () decreased the number of live ALL cells in a dose-dependent manner in the ALL cells (data for Reh, Nalm-6, patient 1 and patient 2 are shown in ; data for Raji, CCRF-CEM, patient 4, patient 8 and patient 9 are shown in Fig. S2A and B). Assuming that the burden of malignant cells can be estimated by compounding the percentage decrease in cells every 3 d, chronic exposure for a month to a concentration of insulin or analogs that increases the cells by 5% over 3 d will result in a 40% decrease in tumor burden [i.e., (0.95)10 = 0.599]. Based on the sigmoidal log-dose response curve, the concentration that decreases the leukemia cells by 5% over 3 d for metformin and rosiglitazone for the four cell lines as above and patients1, 2, 4, 8 and 9 are reported in . These patient samples were used, because cells of less than six passages from patients 3, 5, 6 and 7 were used up, and patients 8 and 9 were recruited. The median concentration of metformin that decreased leukemic cells by 5% in 3 d was 32.1 ∝M (range: 10.8–70.6); for rosiglitazone, it was 16.2 ∝M (range: 2.68–40.1). Therefore, metformin and rosiglitazone inhibited proliferation of leukemic cells at pharmacologically relevant concentrations.Citation18,Citation19
Table 4. Concentrations of metformin and rosiglitazone that can decrease leukemic cells by 5% in 3 d
We next experimentally evaluated and confirmed that the concentrations of insulin and insulin analogs estimated in significantly increased the number of cultured ALL cells. Based on , Reh cells were incubated with control culture medium, insulin (5 mIU/L), aspart (0.6 mIU/L), glargine (0.2 mIU/L) for 2 d and Nalm-6 cells for 4 d. The respective media were changed and refreshed every 2 d. The number of cells in each sample was measured by a Coulter counter. Relative to control culture medium, 2-d treatments with insulin, aspart or glargine significantly (one-way ANOVA, post-hoc intergroup comparisons with p-values as labeled in , left part) increased the cell number by averages of 22.1%, 18.6% or 16.9%, respectively. Nalm-6 cells also proliferate significantly (, right part) faster, by 11.7%, 10.2% or 8.9%, in response to 4-d treatments with insulin, aspart or glargine, respectively. Therefore, pharmacologically achievable concentrations of insulin, aspart and glargine significantly increased the number of ALL cells, and aspart and glargine increased ALL cells to the same degree at lower concentrations than human insulin.
We also experimentally evaluated and confirmed that the concentrations of metformin and rosiglitazone estimated in significantly decreased the number of cultured ALL cells measured using a Coulter counter. The number of Reh cells decreased significantly (, left part) by 15.4% and 35.6% compared with control after incubation with metformin (71 ∝M) and rosiglitazone (11 ∝M), respectively, for 4 d. Similarly, the number of Nalm-6 cells decreased significantly (, right part) by 22.3% and 34.5% compared with control after incubation with metformin (32 ∝M) and rosiglitazone (4 ∝M), respectively, for 6 d. Therefore, pharmacologically achievable concentrations of metformin and rosiglitazone significantly decreased the cell number.
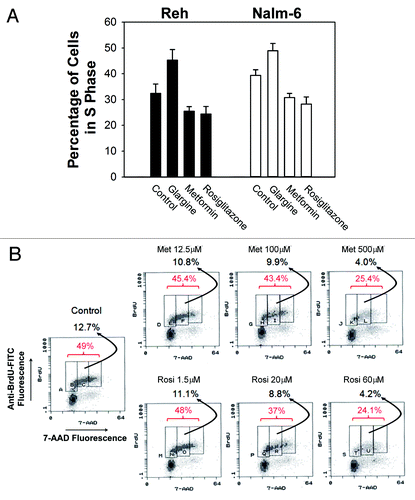
The impact of these drugs on cell proliferation was investigated by measuring the percentage of cells in S phase of the cell cycle. First, we used flow cytometry after PI staining. After incubation for 48 h, glargine 16 mIU/L significantly (p < 0.001, one-way ANOVA, post-hoc Dunnett T3 test) increased, while metformin 100 ∝M and rosiglitazone 20 ∝M significantly (p < 0.001 and p < 0.001 respectively) decreased the number of cells in S phase of the cell cycle compared with controls in Reh cells (). Similar results were obtained in Nalm-6 cells (). Then, we used the more sophisticated technique of 2-dimensional flow cytometry measuring BrdU incorporation and DNA content, The percentage of replicating cells in S phase was measured by dual-staining with anti-BrdU-FITC and 7-AAD. Experimentally treated cells and sham-treated control cells were incubated with BrdU (100 ∝M) for 1 h prior to processing for flow cytometry. Both metformin and rosiglitazone dose-dependently decreased the percentage of Reh cells in the S phase of the cell cycle (). Therefore, glargine stimulated while metformin and rosiglitazone inhibited proliferation of ALL cells.
Figure 2. Inhibitory effects of metformin and rosiglitazone on leukemic cell DNA replication. (A) The percentage of cells in S phase as determined by PI flow cytometry measuring DNA content were plotted on the bar chart with treatments labeled on the horizontal axis. The error bars represent 95% confidence intervals. The black bars are data from Reh cells, and white bars, Nalm-6 cells. (B) The percentage of replicating cells in S phase was determined by 2-dimensional flow cytometry measuring BrdU incorporation and DNA content Both metformin and rosiglitazone, at labeled concentrations, decreased the percentage of Reh cells in the S-phase of the cell cycle (red numbers) and the percentage of BrdU-labeled cells with DNA content between 2n and 4n (black numbers) .
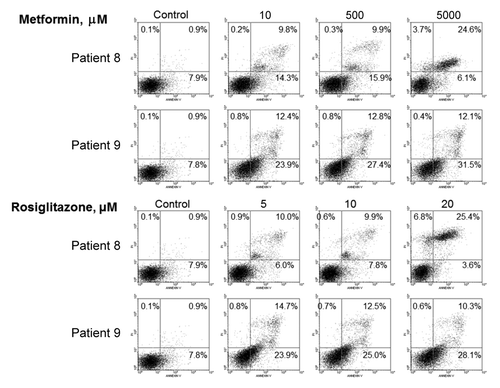
To examine whether apoptosis was involved in the inhibition of ALL cells by metformin and rosiglitazone, we measured apoptotic cells by annexin V binding followed by flow cytometry. Metformin and rosiglitazone both dose-dependently increased the percentage of apoptotic (annexin V-binding) cells in samples from patients 8 and 9 (). Therefore, induction of apoptosis might also play a role in the inhibition of ALL cells by metformin and rosiglitazone.
Figure 3. Induction of apoptosis by metformin and rosiglitazone in leukemic cells. The percentage of apoptotic cells was measured by dual-staining flow cytometry with annexin V-FITC and propidium iodide. The sum of the percentages indicated in the right upper and right lower quadrants represent the percentage of annexin V-binding apoptotic cells. Metformin and rosiglitazone both dose-dependently increased the percentage of apoptotic (annexin V-binding) cells in primary malignant cell sample from patients 8 and 9.
Taking all the above data into consideration, insulin and analogs had stimulatory effects on ALL cells opposite to the inhibitory effects of metformin and rosiglitazone. Therefore, different anti-diabetic pharmacotherapeutic agents had different (positive or negative) direct impact on the proliferation of ALL cells.
Insulin stimulated, while metformin and rosiglitazone inhibited, Akt/mTOR signaling in ALL cells.
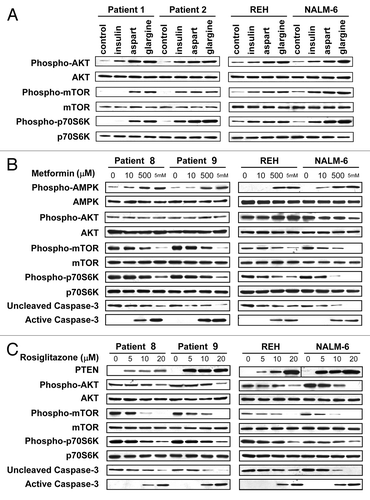
Insulin, insulin analogs, metformin and thiazolidinediones can regulate cell proliferation and cell death through the AKT/mTOR signaling pathway.Citation12 Reh and Nalm-6 and primary samples from patients 1 and 2 were incubated with insulin, aspart and glargine; insulin and analogs activated this pathway, and equi-hypoglycemic concentrations of aspart and glargine activated this pathway to a higher degree than human insulin (). Reh and Nalm-6 and primary samples from patients 8 and 9 were incubated with metformin (); metformin dose-dependently increased activation of AMPK, inhibition of phosphorylation/activation of mTOR and p70S6K and activation of caspase-3 (a marker of apoptosis) while having no effect on the phosphorylation of AKT. The same cells were incubated with rosiglitazone (); rosiglitazone dose-dependently increased expression of PTEN, accompanied by inhibition of phosphorylation/activation of AKT, mTOR and p70S6K and activation of caspase-3. Therefore, insulin and insulin analogs had effects opposite to those of metformin and rosiglitazone on the AKT/mTOR signaling pathway. Our results are consistent with results by metforminCitation20,Citation21 and rosiglitazoneCitation22 in ALL and other types of malignancies.
Figure 4. Differential effects of diabetes medications on AKT/mTOR signaling in leukemic cells. Immunoblots are shown with the antigens labeled to the left and the cell source and treatment labeled above. (A) 24 h treatments of human insulin, aspart and glargine at equi-hypoglycemic concentration (8 mIU/L) activated the AKT/mTOR signaling pathway, and aspart and glargine activated this pathway to a higher degree than human insulin. (B) Metformin at increasing concentrations for 48 h dose-dependently increased activation of AMPK, which inhibited phosphorylation/activation of mTOR and p70S6K, and activated caspase-3 (a marker of apoptosis) while having no effect on the phosphorylation of AKT. (C) Rosiglitazone at increasing concentrations for 48 h dose-dependently increased expression of PTEN, which inhibited phosphorylation/ activation of AKT, mTOR and p70S6K, and activated caspase-3.
Insulin promoted, while metformin and rosiglitazone antagonized, chemoresistance in ALL cells.
The impact of anti-diabetic medications on chemoresistance of ALL cells was first investigated using an anthracycline daunorubicin. The log-dose-response curves of daunorubicin (48-h incubations) were defined using the MTS assay ALL cells. In combination with daunorubicin, pharmacokinetically relevant concentrations of metformin (500 ∝M) and rosiglitazone (10 ∝M) were found to shift the log-dose-response curve of daunorubicin to the left, indicating chemosensitization, while pharmacokinetically relevant concentrations of aspart, glargine and human insulin (1.25 mIU/L) shifts the dose-response curve to the right, indicating chemoresistance in Reh cells (). Similar data were obtained using the leukemic cells from patient 9; metformin and rosiglitazone shift the log-dose-response curve of daunorubicin to the left, while aspart, glargine and human insulin shift the curve to the right also in cells from patient 9. To investigate the impact on daunorubicin-induced apoptosis, Reh cells treated with metformin 500 ∝M, rosiglitazone 10 ∝M, human insulin 8 mIU/L, aspart 8 mIU/L or glargine 8 mIU/L were found to have about 6% apoptotic cells (, top parts). Daunorubicin at 8 nM (IC25 for Reh) alone induced apoptosis in 15.5% of cells, while the combination with metformin increased the apoptotic cells to 31.8%, and the combination with rosiglitazone increased the apoptotic cells to 36.6% (, middle parts). Daunorubicin at 32 nM (IC75 for Reh) induced apoptosis in 29.8% of cells, while the combination with insulin, aspart or glargine decreased the apoptotic cells to < 10% (, bottom parts). Similar data were also obtained with sample from patient 9. ALL cells from patient 9 treated with metformin, rosiglitazone, human insulin, aspart or glargine had 6%, 12.5%, 14.1%, 13.2%, 16% and 14.5% apoptotic cells, respectively. Daunorubicin at 7 nM (IC25 for patient 9) alone induced apoptosis in 11.8% of cells, while the combination with metformin increased the apoptotic cells to 28.7%, and the combination with rosiglitazone increased the apoptotic cells to 37.5%. Daunorubicin at 20 nM (IC75 for patient 9) induced apoptosis in 34.4% of cells, while the combination with insulin, aspart or glargine decreased the apoptotic cells to 18%, 18.4% and 16.5%, respectively.
Figure 5. Differential effects of anti-diabetic medications on chemoresistance. (A) Data-fitted sigmoidal dose-response curves of daunorubicin (DNR) are shown. Cell viability relative to control was measured by MTS assay in Reh cells. The control (black) dose response curve was from cells treated with daunorubicin only. Metformin 500 μM (cyan) and rosiglitazone 10 μM (white with dash line) shifts the dose-response curve of daunorubicin to the left, indicating chemosensitization. Aspart (blue), glargine (red) and human insulin (green) (all at 1.25 mIU/L) shifts the dose-response curves to the right, indicating chemoresistance. (B) Reh cells were treated with metformin 500 μM, rosiglitazone 10 μM, human insulin 8 mIU/L, aspart 8 mIU/L or glargine 8 mIU/L in the absence or presence of daunorubicin 8 nM (IC25) or 32 nM (IC75) for 48 h. The percentage of apoptotic cells were measured using dual-staining flow cytometry with propidium iodide and annexin V-FITC. Representative scattergrams are shown as labeled. Metformin and rosiglitazone enhanced daunorubicin-induced apoptosis while insulin, aspart and glargine antagonized daunorubicin-induced apoptosis. (C) Reh cells were treated with control culture medium, metformin 500 μM or rosiglitazone 10 μM in the absence or presence of etoposide 0.1 μM for 72 h. Both metformin and rosiglitazone significantly (one-way ANOVA) increased the percentage of apoptotic cells in etoposide-treated cells. (D) Reh cells were treated with control culture medium, metformin 500 μM or rosiglitazone 10 μM in the absence or presence of vincristine 0.5 ng/ml for 72 h. Rosiglitazone significantly (one-way ANOVA) increased the percentage of apoptotic cells in vincristine-treated cells. (E) Reh cells were treated with control culture medium, metformin 500 μM or rosiglitazone 10 μM in the absence or presence of L-asparaginase 5 units/ml for 72 h. Metformin significantly (one-way ANOVA) increased the percentage of apoptotic cells in L-asparaginase-treated cells. (F) Reh cells were treated with control culture medium, metformin 500 μM or rosiglitazone 10 μM in the absence or presence of methotrexate 3 ng/ml for 72 h. Neither metformin nor rosiglitazone significantly (one-way ANOVA) increased the percentage of apoptotic cells in methotrexate-treated cells.
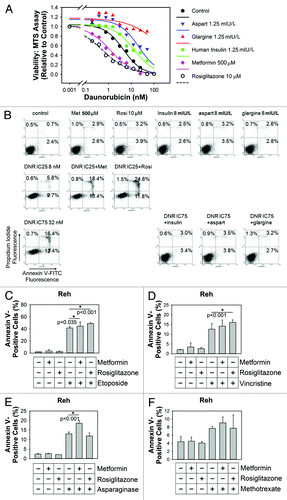
The impact of anti-diabetic medications on chemoresistance of ALL cells was also investigated using etoposide, vincristine, L-asparaginase and methotrexate. Using annexin V-binding for measuring the percentage of apoptotic Reh cells in flow cytometry analysis, human insulin, aspart and glargine (all at 8 mIU/L) did not significantly change the percentage of apoptotic cells treated with etoposide 0.21 ∝M, vincristine 15 ng/ ml, L-asparaginase 105 units/ml or methotrexate 53 ng/ml. In contrast, both metformin 500 ∝M and rosiglitazone 10 ∝M significantly (p = 0.035 and p < 0.001, respectively) increased the percentage of apoptotic cells when treated with etoposide 0.1 ∝M for 72 h (). In similar experiments using vincristine 0.5 ng/ml, rosiglitazone significantly (p < 0.001) increased the percentage of apoptotic cells in vincristine-treated cells (). In similar experiments using L-asparaginase 5 units/ml, metformin significantly (p < 0.001) increased the percentage of apoptotic cells in L-asparaginase-treated cells (). In similar experiments using methotrexate 3 ng/ml, although there is a trend that metformin may increase the percentage of apoptotic cells in methotrexate-treated cells, the statistical analysis did not reach the confidence level of 95% (p = 0.147) ().
In summary, insulin, aspart and glargine antagonized daunorubicin-induced apoptosis and cytotoxicity, but no changes in chemosensitivity to etoposide, vincristine, L-asparaginase and methotrexate were detected at the concentrations examined. In contrast, metformin increased daunorubicin-induced cytotoxicity and apoptosis, etoposide-induced apoptosis and L-asparaginase-induced apoptosis; rosiglitazone increased daunorubicin-induced cytotoxicity and apoptosis, etoposide-induced apoptosis and vincristine-induced apoptosis.
Discussion
The association of hyperglycemia with outcomes in ALL patients is multifactorial. Factors such as an altered metabolism that supports the proliferative state of leukemic cellsCitation23 and impaired immune functionCitation24 may contribute to early relapse. Hyperinsulinemia and increased levels of IGF-1 have also been shown to promote tumor growth in solid tumors.Citation25-Citation27 Recently we demonstrated that different types of anti-diabetic pharmacotherapy had a differential direct impact on breast and pancreatic cancer cells.Citation28 Here, we have found evidence that the engineered insulin analogs (aspart and glargine) may stimulate proliferation of ALL cells to a greater degree than regular human insulin.
The impact of glargine on cancer is highly controversial. A cohort study raised the concern for a higher than expected incidence of cancer in patients on glargine compared with human insulin,Citation29 but other epidemiologic studies did not corroborate the potential link to increased cancer incidence.Citation30-Citation33 The potential difference between glargine and human insulin on carcinogenesis and cancer incidence may need long-term (> 5 y) studies to detect convincingly.Citation34 Some publications support that glargine stimulates malignant cell growth more than insulin,Citation35,Citation36 while others do not.Citation37-Citation40 Our data about the higher stimulatory of glargine and aspart than human insulin in ALL cells and data by others in other cancer cellsCitation35,Citation36 would suggest that one area of future research should focus on whether cancer patients on glargine and/or aspart have worse oncologic outcomes than those on human insulin.
In addition to an indirect mechanism through the systemic effect of metformin on insulin level, metformin may suppress malignancy by direct mechanisms involving stem cell-like cancer cells, epithelial-to-mesenchymal transition (EMT) and cellular senescence.Citation41 Metformin also suppresses malignant cells through activation of AMPK, leading to inhibition of mTOR,Citation20,Citation21,Citation28,Citation42-Citation44 and the regulation of the FoxO3A transcriptional program by AMPK is also relevant in autophagic cell death of persistently stressed cells.Citation45 Thiazolidinedione PPARγ ligands inhibit proliferation and induce apoptosis of cancer cells,Citation28,Citation46,Citation47 and inhibit tumor angiogenesis and invasion.Citation46,Citation48 They attenuate signaling through the IGF-1R signaling pathwayCitation49,Citation50 as PPARγ activation upregulates the expression of PTEN, an inhibitor of signaling through AKT/mTOR.Citation51 Studies on aging and caner across the animal kingdom (nematodes, fruit flies and mice) suggest that elevation in glucose, insulin and IGF-1 are important factors linked to shorter lifespans and anti-insulin resistance drugs and interventions that interfere with insulin/IGF-1 signaling postpone or inhibit carcinogenesis.Citation52 Here, in terms of malignant cell proliferation, we have demonstrated that insulin and analogs stimulate, while metformin and thiazolidinediones inhibit, ALL cells using both cell lines and primary patient samples (–). Modeling of log dose responses predicted that pharmacologically achievable concentrations of insulin and analogs (), and metformin and rosiglitazone () might have significant impact on the number live ALL cells. We have experimentally confirmed that these predicted concentrations of the anti-diabetic medications do cause statistically significant changes in the number of ALL cells. Insulin and analogs activate the AKT/mTOR signaling pathway, while metformin and rosiglitazone inhibit mTOR signaling (). Insulin and analogs induced chemoresistance to daunorubicin, while metformin and thiazolidinediones improved chemosensitivity to daunorubicin and other agents (). The chemosensitivity to anthracyclines may be regulated by the Raf/ MEK/ERK and PI3K/PTEN/Akt/mTOR pathways,Citation53 and inhibition of the PI3K/PTEN/Akt/mTOR pathway can enhance chemosensitivity. Chemosensitivity to other agents (etoposide, vincristine, L-asparaginase and methotrexate) showed varied responses, which suggests that the pathways or mechanisms regulating chemosensitivty to these other agents are different from those for anthracyclines. Taken together, all these data support the notion that insulin and analogs are detrimental, while metformin and thiazolidinediones are beneficial, to the clinical outcomes of hyperglycemic ALL patients undergoing hyper-CVAD chemotherapy.
In contrast to cycled/pulsed treatment with classical cytotoxic chemotherapy that results in high percentages of malignant cells being killed, chronic inhibitory/suppressive therapy, which may have some similarity to metronomic therapy,Citation54 will only need a very modest impact to translate into clinically meaningful benefits over the long-term. For example, a mere 2.5% decrease in the number of cancer cells by a drug compared with control observed in 3 d may become a 53.2% decrease in tumor burden in 3 mo (i.e., 0.975Citation30 = 0.468). Antidiabetic pharmacotherapy involves chronic administration; modest short-term impact on malignant cells at normal human doses will become clinically significant over time. The effects of insulin or analogs compared with the opposite effects of metformin or thiazolidinediones on malignant cells will magnify the difference. A caveat for this argument is the possibility of tachyphylaxis (i.e., the antidiabetic drugs may stop having an effect on malignant cells after many days of exposure). However, epidemiologic evidence does not support this caveat, and diabetic patients with various types of cancer have different prognoses associated with the anti-diabetic drugs they use.Citation55-Citation57 An epidemiologic study revealed an increased risk of cancer-related death in diabetic patients treated with sulfonylureas and insulin preparations than in those treated with metformin,Citation58 and based on our previous results in reference Citation12 and results reported in this paper, this difference is very likely due to both detrimental effects of insulin preparations and the beneficial effect of metformin on cancer cell growth and chemoresistance. Diabetic patients with breast cancer receiving metformin and neoadjuvant chemotherapy have a higher complete response rate than do diabetics not receiving metformin.Citation59 This epidemiologic evidence and our finding that metformin adds to the cytotoxic effect of daunorubicin corroborate each other. Specifically for hyperglycemic ALL patients undergoing hyper-CVAD chemotherapy, a randomized clinical trial comparing intensive insulin therapy using glargine and aspart with conventional therapy (control) was terminated early because of a trend that survival in the intensive insulin therapy arm was worse than the control arm and a probability of 0.0001 to conclude in favor of the intensive insulin therapy arm.Citation60 Secondary analysis of the data in this clinical trial suggests that exogenous insulin may be associated with poor outcomes, while metformin and thiazolidinediones may be associated with improved outcomes.Citation60 The results in this paper and the results of our clinical trial corroborate one another.
To our knowledge, this is the first study that includes evidence from primary leukemic patient samples to demonstrate that insulin and insulin analogs are associated with a high proliferation rate and chemoresistance, while metformin and thiazolidinediones are associated with low proliferation rate, apoptosis and chemosensitivity of ALL cells. While we need to learn more about the interactions of anti-diabetic pharmacotherapy with malignancies and their response to treatments, future research should also seek confirmation of beneficial effects of biguanides and thiazolidinediones to the clinical outcomes of malignant diseases in prospective trials. Our results open the avenue to potentially influence the clinical outcomes and survival of hyperglycemic ALL patients simply by making an informed choice of anti-diabetic pharmacotherapy. The implications of our data may reach beyond the context of hyperglycemic ALL patients to normoglycemic patients. Currently, a phase III adjuvant trial (NCIC MA.32) is investigating the addition of metformin to chemotherapy for breast cancer.Citation61 In low glucose culture conditions, a combination of rapamycin and metformin kills cancer cells without killing normal cells;Citation62 these results, taken together with our findings, suggest that the combination of metformin and an mTOR inhibitor may have a wide therapeutic window. We are currently evaluating this idea of combining metformin with an mTOR inhibitor (everolimus) in a clinical trial of obese and overweight postmenopausal patients with metastatic hormone-responsive breast cancer on exemestane therapy. The safety of metformin and thiazolidinediones in normoglycemic patients and their chemosensitizing effects may justify clinical trials to evaluate the addition of metformin and/or thiazolidinediones to chemotherapy for ALL.
Methods
Cell lines and primary patient samples.
Lymphoblast cell lines Reh (non-T non-B ALL), Nalm-6 (pre-B ALL), Raji (Burkitt’s lymphoma) and CCRF-CEM (T-cell ALL) were all obtained from ATCC and were grown in RPMI 1640 medium (Invitrogen) supplemented with 1% heat-inactivated fetal calf serum (FCS), as described previously in reference Citation16. The glucose concentration in standard RPMI 1640 medium is 2 g/L. Media are replaced after 48 h, at which point glucose in the media has not decreased by more than 10%. Cells in logarithmic phase were used in all experiments starting with 2.0 x 105/mL. Nine patients () were recruited from the Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, People’s Republic of China to donate primary leukemic cell samples for in vitro studies. All the patient samples were obtained after written informed consent under a clinical research protocol approved by the Institutional Review Board at the Sun Yat-sen University in accordance with the precepts established by the Helsinki Declaration. The primary leukemic cells were cultured in the medium as above. Only cells less than six in vitro passages were used in experiments; for this reason, samples from seven consecutive patients were used in earlier experiments; in later experiments, samples from patient 1, 2 and 4 were used with new recruits of patients 8 and 9, as no more cells from patient 3, 5, 6 and 7 were available.
Reagents.
Metformin was dissolved in water, and rosiglitazone was dissolved in DMSO (Sigma) at a stock concentration of 40 mM and 5 mM, respectively, and stored at -20°C. Mouse antibodies against procaspase-3 and active caspase-3 (CM1) were from BD Biosciences. Antibodies against AMPKα, phospho-AMPKα (Thr172), AKT, phospho-AKT (Ser473), mTOR, phospho-mTOR (Ser2448), p70 S6 kinase and phospho-p70 S6 kinase (Thr389) were from Cell Signaling Technology; mouse monoclonal antibody against actin and Annexin V-FITC Kit were from Sigma-Aldrich. Anti-mouse immunoglobulin G and anti-rabbit immunoglobulin G horse radish peroxidase-conjugated antibodies were from Pierce Biotechnology.
Cell viability assay.
Cell viability was determined by MTS assay (CellTiter 96 Aqueous One Solution Cell Proliferation assay; Promega).Citation15,Citation16,Citation63 100 ∝l cells (2.0 x 105/mL) were seeded in 96-well plates, incubated with various concentrations of drugs for 72 h. Four hours prior to culture termination, 20 ∝L of MTS solution was added to each well of culture. Absorbance was read on a 96-well plate reader at a wavelength of 490 nm. Control cells received DMSO (< 0.1%) containing medium.
The numbers of live cells were also counted after dispersion by EDTA treatment using trypan blue dye exclusion with a hemocytometer. Alternatively, the total number of cells was counted using a Coulter counter, and the percentage of live cells in the cell population was determined by trypan blue dye exclusion.
Western blotting analysis.
Control or drug-treated cells were pelleted by centrifugation and rinsed with PBS. The cell pellets were then lysed in a total of 300 ∝l RIPA buffer [1x PBS, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 0.1 mg/ml PMSF and Complete Protease Inhibitor Mix (Roche), one tablet per 50 ml]. The DNA in the lysate was sheared by a sonicator. All western blot analysis was performed using whole-cell lysates prepared as described above. SDS-PAGE was performed using standard methods. The protein concentrations of samples were measured using a modified Lowry method (protein assay, Bio-Rad Laboratories, Inc.). Equal amounts of total protein from each sample were loaded onto the SDS polyacrylamide gel. Immunoblotting was performed using nitrocellulose membranes (Bio-Rad Laboratories, Inc.). Kodak X-AR film was used to record the image generated by enhanced chemiluminescence using the ECL kit (Pierce Chemical Co.).
Flow cytometry analysis.
Apoptosis measurement. Apoptosis was measured by flow cytometry using annexin V-FITC/Propidium iodide (PI) or 7-AAD/Cy5AnnV double staining. Cells were cultured in the presence of indicated concentrations of metformin, rosiglitazone, insulin, aspart, glargine and daunorubicin then harvested and washed and incubated in binding buffer (Annexin V Binding Buffer, Sigma-Aldrich) with 0.3% annexin-FITC for 15 min at room temperature. The cells were washed and resuspended in binding buffer. Propidium iodide or 7-AAD was added just before flow cytometric analysis.Citation15,Citation16
Cell cycle phase measurement.
Cells were cultured in the presence of indicated concentrations of metformin, rosiglitazone, glargine or control medium then harvested, washed, fixed and stained with PI according to standard protocols. The DNA contents of the cells were measured by PI fluorescence. The percentages of cells in different phases of the cell cycle were determined using the computer program Flowjo Version 7.6.4 (www.treestar.com).
Proliferating cells detection.
Cells treated and control were incubated with bromodeoxyuridine (BrdU) and harvested, washed with PBS and fixed with 66% ethanol overnight. Cells were centrifuged and washed with PBS, then stained with 7-amino-actinomycin D (7-AAD) and 2.5 ∝g/mL RNase and anti-BrdU-FITC in PBS solution for 30 min at room temperature. S-phase cell percentage was analyzed by dual-fluorescence flow cytometry according to the instruction of BrdU flow cytometry assay kit (BD Biosciences).
Statistics.
Log-dose response curves were obtained by fitting all the data points to a 4-parameter sigmoid curve model using the dynamic fit wizard of SigmaPlot version 12 (Systat Software Inc.). The mean response of at least three measurements for each tested concentration was plotted along with the fitted curve. Inhibitory concentrations that inhibit the cancer cells by 25%, 50% and 75% (IC25, IC50 and IC75, respectively) were determined from the fitted sigmoid curve. Comparison between two groups was evaluated by t-test or rank sum test where appropriate. Bonferroni correction for multiple comparisons was applied where appropriate. Comparisons involving more than two groups were performed using one-way analysis of variance (ANOVA) and Kruskal-Wallis one-way ANOVA on Ranks (for non-normally distributed data). Post-hoc intergroup comparisons were performed with Dunnett T3 or Holm-Sidak tests. p < 0.05 was considered significant.
Additional material
Download Zip (396.4 KB)Acknowledgments
This paper is dedicated to our beloved deceased colleague Dr. Mary Ann Weiser. The University of Texas MD Anderson Cancer Center, Division of Internal Medicine Multidisciplinary Research Program (PI: M. Weiser, succeeded by S.C. Yeung; co-PI: M. Andreeff). This study was also supported by grants from the following agencies: U.S. National Institute of Health (NIHRO1CA 089266, PI: M.H. Lee), Department of Defense, Breast Cancer Research Program (BCRP) of the Office of the Congressionally Directed Medical Research Programs (CDMRP) (Synergistic Idea Development Award BC062166, PI: S.C. Yeung and M.H. Lee), Susan G. Komen Foundation for Breast Cancer Research (Promise grant KG081048, PI: S.C. Yeung), the National Natural Science Fund of China (no. 90713036, PI: J. Pan; no. 30772367, PI: C. Chen), the Research Foundation of Education Bureau of Guangdong Province, China (Grant cxzd1103) to J. Pan, the Research Foundation of Guangzhou Bureau of Science and Technology, China (Grant to J. Pan), National Natural Science Funds of China for Distinguished Young Scholars (grant 81025021, PI: J. Pan), National Basic Research Program of China (973 Program grant 2009CB825506, PI: J. Pan), National High Technology Research and Development Program of China (863 Program grant 2008AA02Z420, PI: J. Pan), the Major Research Plan of the National Natural Science Fund of China (grant 90713036, PI: J. Pan), and the Fundamental Research Funds for Central Universities (PI: J. Pan). Enrique Fuentes-Mattei is supported by the MD Anderson Cancer Center Training Grant Program in Molecular Genetics (T32CA009299, PI: G. Lozano), and Guermarie Velazquez-Torres is supported by a National Cancer Institute grant minority supplement (parent grant R01CA089266, PI: M.H. Lee).
Financial Support
No funding bodies had any role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The University of Texas MD Anderson Cancer Center, Division of Internal Medicine Multidisciplinary Research Program; This study was also supported by grants from the following agencies: US. National Institute of Health (NIHRO1CA 089266, PI: M.H. Lee), Department of Defense, Breast Cancer Research Program (BCRP) of the Office of the Congressionally Directed Medical Research Programs (CDMRP) (Synergistic Idea Development Award BC062166, PI: S.C. Yeung and M.H. Lee), Susan G. Komen Foundation for Breast Cancer Research (Promise grant KG081048, PI: S.C. Yeung), National Natural Science Funds of China for Distinguished Young Scholars (grant 81025021, PI: J. Pan), National Basic Research Program of China (973 Program grant 2009CB825506, PI: J. Pan), the National Natural Science Fund of China (no. 90713036, PI: J. Pan; no. 30772367, PI: C. Chen), the Research Foundation of Education Bureau of Guangdong Province, China (Grant cxzd1103) to J. Pan, the Research Foundation of Guangzhou Bureau of Science and Technology, China (Grant to J. Pan), National High Technology Research and Development Program of China (863 Program grant 2008AA02Z420, PI: J. Pan), and the Fundamental Research Funds for Central Universities (PI: J. Pan). Enrique Fuentes-Mattei is supported by the MD Anderson Cancer Center Training Grant Program in Molecular Genetics (T32CA009299, PI: G. Lozano) and Guermarie Velazquez-Torres is supported by a National Cancer Institute grant minority supplement (parent grant R01CA089266, PI: M.H. Lee).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Note
Supplemental materials can be found at: www.landesbioscience.com/journals/cc/article/20770
Related Research Data
References
- Nilsen TI, Vatten LJ. Prospective study of colorectal cancer risk and physical activity, diabetes, blood glucose and BMI: exploring the hyperinsulinaemia hypothesis. Br J Cancer 2001; 84:417 - 22; http://dx.doi.org/10.1054/bjoc.2000.1582; PMID: 11161410
- Verlato G, Zoppini G, Bonora E, Muggeo M. Mortality from site-specific malignancies in type 2 diabetic patients from Verona. Diabetes Care 2003; 26:1047 - 51; http://dx.doi.org/10.2337/diacare.26.4.1047; PMID: 12663571
- Richardson LC, Pollack LA. Therapy insight: Influence of type 2 diabetes on the development, treatment and outcomes of cancer. Nat Clin Pract Oncol 2005; 2:48 - 53; http://dx.doi.org/10.1038/ncponc0062; PMID: 16264856
- Coughlin SS, Calle EE, Teras LR, Petrelli J, Thun MJ. Diabetes mellitus as a predictor of cancer mortality in a large cohort of US adults. Am J Epidemiol 2004; 159:1160 - 7; http://dx.doi.org/10.1093/aje/kwh161; PMID: 15191933
- van de Poll-Franse LV, Houterman S, Janssen-Heijnen ML, Dercksen MW, Coebergh JW, Haak HR. Less aggressive treatment and worse overall survival in cancer patients with diabetes: a large population based analysis. Int J Cancer 2007; 120:1986 - 92; http://dx.doi.org/10.1002/ijc.22532; PMID: 17230509
- Kantarjian HM, O’Brien S, Smith TL, Cortes J, Giles FJ, Beran M, et al. Results of treatment with hyper-CVAD, a dose-intensive regimen, in adult acute lymphocytic leukemia. J Clin Oncol 2000; 18:547 - 61; PMID: 10653870
- Weiser MA, Cabanillas ME, Konopleva M, Thomas DA, Pierce SA, Escalante CP, et al. Relation between the duration of remission and hyperglycemia during induction chemotherapy for acute lymphocytic leukemia with a hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone/methotrexate-cytarabine regimen. Cancer 2004; 100:1179 - 85; http://dx.doi.org/10.1002/cncr.20071; PMID: 15022284
- Seshasai SR, Kaptoge S, Thompson A, Di Angelantonio E, Gao P, Sarwar N, et al, Emerging Risk Factors Collaboration. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N Engl J Med 2011; 364:829 - 41; http://dx.doi.org/10.1056/NEJMoa1008862; PMID: 21366474
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646 - 74; http://dx.doi.org/10.1016/j.cell.2011.02.013; PMID: 21376230
- Warburg O. On the origin of cancer cells. Science 1956; 123:309 - 14; http://dx.doi.org/10.1126/science.123.3191.309; PMID: 13298683
- Yeung SJ, Pan J, Lee MH. Roles of p53, MYC and HIF-1 in regulating glycolysis - the seventh hallmark of cancer. Cell Mol Life Sci 2008; 65:3981 - 99; http://dx.doi.org/10.1007/s00018-008-8224-x; PMID: 18766298
- Feng YH, Velazquez-Torres G, Gully C, Chen J, Lee MH, Yeung SC. The impact of type 2 diabetes and antidiabetic drugs on cancer cell growth. J Cell Mol Med 2011; 15:825 - 36; http://dx.doi.org/10.1111/j.1582-4934.2010.01083.x; PMID: 20455996
- Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, et al. Diabetes and cancer: a consensus report. Diabetes Care 2010; 33:1674 - 85; http://dx.doi.org/10.2337/dc10-0666; PMID: 20587728
- Jin Y, Lu Z, Ding K, Li J, Du X, Chen C, et al. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: inactivation of the NF-kappaB pathway and generation of reactive oxygen species. Cancer Res 2010; 70:2516 - 27; http://dx.doi.org/10.1158/0008-5472.CAN-09-3950; PMID: 20215516
- Pan J, Quintás-Cardama A, Kantarjian HM, Akin C, Manshouri T, Lamb P, et al. EXEL-0862, a novel tyrosine kinase inhibitor, induces apoptosis in vitro and ex vivo in human mast cells expressing the KIT D816V mutation. Blood 2007; 109:315 - 22; http://dx.doi.org/10.1182/blood-2006-04-013805; PMID: 16912224
- Shi X, Jin Y, Cheng C, Zhang H, Zou W, Zheng Q, et al. Triptolide inhibits Bcr-Abl transcription and induces apoptosis in STI571-resistant chronic myelogenous leukemia cells harboring T315I mutation. Clin Cancer Res 2009; 15:1686 - 97; http://dx.doi.org/10.1158/1078-0432.CCR-08-2141; PMID: 19240172
- Malone JI, Lowitt S, Grove NP, Shah SC. Comparison of insulin levels after injection by jet stream and disposable insulin syringe. Diabetes Care 1986; 9:637 - 40; http://dx.doi.org/10.2337/diacare.9.6.637; PMID: 3542456
- van Berlo-van de Laar IR, Vermeij CG, Doorenbos CJ. Metformin associated lactic acidosis: incidence and clinical correlation with metformin serum concentration measurements. J Clin Pharm Ther 2011; 36:376 - 82; http://dx.doi.org/10.1111/j.1365-2710.2010.01192.x; PMID: 21545617
- Niemi M, Backman JT, Granfors M, Laitila J, Neuvonen M, Neuvonen PJ. Gemfibrozil considerably increases the plasma concentrations of rosiglitazone. Diabetologia 2003; 46:1319 - 23; http://dx.doi.org/10.1007/s00125-003-1181-x; PMID: 12898007
- Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res 2007; 67:10804 - 12; http://dx.doi.org/10.1158/0008-5472.CAN-07-2310; PMID: 18006825
- Grimaldi C, Chiarini F, Tabellini G, Ricci F, Tazzari PL, Battistelli M, et al. AMP-dependent kinase/mammalian target of rapamycin complex 1 signaling in T-cell acute lymphoblastic leukemia: therapeutic implications. Leukemia 2012; 26:91 - 100; http://dx.doi.org/10.1038/leu.2011.269; PMID: 21968881
- Farrow B, Evers BM. Activation of PPARgamma increases PTEN expression in pancreatic cancer cells. Biochem Biophys Res Commun 2003; 301:50 - 3; http://dx.doi.org/10.1016/S0006-291X(02)02983-2; PMID: 12535639
- Boren J, Cascante M, Marin S, Comín-Anduix B, Centelles JJ, Lim S, et al. Gleevec (STI571) influences metabolic enzyme activities and glucose carbon flow toward nucleic acid and fatty acid synthesis in myeloid tumor cells. J Biol Chem 2001; 276:37747 - 53; PMID: 11489902
- Bagdade JD, Root RK, Bulger RJ. Impaired leukocyte function in patients with poorly controlled diabetes. Diabetes 1974; 23:9 - 15; PMID: 4809622
- Gapstur SM, Gann PH, Colangelo LA, Barron-Simpson R, Kopp P, Dyer A, et al. Postload plasma glucose concentration and 27-year prostate cancer mortality (United States). Cancer Causes Control 2001; 12:763 - 72; http://dx.doi.org/10.1023/A:1011279907108; PMID: 11562117
- Goodwin PJ, Ennis M, Pritchard KI, Trudeau ME, Koo J, Madarnas Y, et al. Fasting insulin and outcome in early-stage breast cancer: results of a prospective cohort study. J Clin Oncol 2002; 20:42 - 51; http://dx.doi.org/10.1200/JCO.20.1.42; PMID: 11773152
- Meyerhardt JA, Catalano PJ, Haller DG, Mayer RJ, Macdonald JS, Benson AB 3rd, et al. Impact of diabetes mellitus on outcomes in patients with colon cancer. J Clin Oncol 2003; 21:433 - 40; http://dx.doi.org/10.1200/JCO.2003.07.125; PMID: 12560431
- Feng YH, Velazquez-Torres G, Gully C, Chen J, Lee MH, Yeung SC. The impact of type 2 diabetes and antidiabetic drugs on cancer cell growth. J Cell Mol Med 2011; 15:825 - 36; http://dx.doi.org/10.1111/j.1582-4934.2010.01083.x; PMID: 20477906
- Hemkens LG, Grouven U, Bender R, Günster C, Gutschmidt S, Selke GW, et al. Risk of malignancies in patients with diabetes treated with human insulin or insulin analogues: a cohort study. Diabetologia 2009; 52:1732 - 44; http://dx.doi.org/10.1007/s00125-009-1418-4; PMID: 19565214
- Colhoun HM, SDRN Epidemiology Group. Use of insulin glargine and cancer incidence in Scotland: a study from the Scottish Diabetes Research Network Epidemiology Group. Diabetologia 2009; 52:1755 - 65; http://dx.doi.org/10.1007/s00125-009-1453-1; PMID: 19603149
- Currie CJ, Poole CD, Gale EA. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia 2009; 52:1766 - 77; http://dx.doi.org/10.1007/s00125-009-1440-6; PMID: 19572116
- Home PD, Lagarenne P. Combined randomised controlled trial experience of malignancies in studies using insulin glargine. Diabetologia 2009; 52:2499 - 506; http://dx.doi.org/10.1007/s00125-009-1530-5; PMID: 19756478
- Jonasson JM, Ljung R, Talbäck M, Haglund B, Gudbjörnsdòttir S, Steineck G. Insulin glargine use and short-term incidence of malignancies-a population-based follow-up study in Sweden. Diabetologia 2009; 52:1745 - 54; http://dx.doi.org/10.1007/s00125-009-1444-2; PMID: 19588120
- Wilson C. Diabetes: long-term use of insulin glargine might increase the risk of breast cancer. Nat Rev Endocrinol 2011; 7:499; http://dx.doi.org/10.1038/nrendo.2011.112; PMID: 21709708
- Mayer D, Shukla A, Enzmann H. Proliferative effects of insulin analogues on mammary epithelial cells. Arch Physiol Biochem 2008; 114:38 - 44; http://dx.doi.org/10.1080/13813450801900645; PMID: 18465357
- Mayer D, Chantelau E. Treatment with insulin glargine (Lantus) increases the proliferative potency of the serum of patients with type-1 diabetes: a pilot study on MCF-7 breast cancer cells. Arch Physiol Biochem 2010; 116:73 - 8; http://dx.doi.org/10.3109/13813451003631439; PMID: 20199195
- Liefvendahl E, Arnqvist HJ. Mitogenic effect of the insulin analogue glargine in malignant cells in comparison with insulin and IGF-I. Horm Metab Res 2008; 40:369 - 74; http://dx.doi.org/10.1055/s-2008-1062739; PMID: 18393172
- Erbel S, Reers C, Eckstein VW, Kleeff J, Büchler MW, Nawroth PP, et al. Proliferation of colo-357 pancreatic carcinoma cells and survival of patients with pancreatic carcinoma are not altered by insulin glargine. Diabetes Care 2008; 31:1105 - 11; http://dx.doi.org/10.2337/dc07-2015; PMID: 18316392
- Müller K, Weidinger C, Führer D. Insulin glargine and insulin have identical effects on proliferation and phosphatidylinositol 3-kinase/AKT signalling in rat thyrocytes and human follicular thyroid cancer cells. Diabetologia 2010; 53:1001 - 3; http://dx.doi.org/10.1007/s00125-010-1674-3; PMID: 20179899
- Nagel JM, Staffa J, Renner-Müller I, Horst D, Vogeser M, Langkamp M, et al. Insulin glargine and NPH insulin increase to a similar degree epithelial cell proliferation and aberrant crypt foci formation in colons of diabetic mice. Horm Cancer 2010; 1:320 - 30; http://dx.doi.org/10.1007/s12672-010-0020-z; PMID: 21761363
- Del Barco S, Vazquez-Martin A, Cufí S, Oliveras-Ferraros C, Bosch-Barrera J, Joven J, et al. Metformin: multi-faceted protection against cancer. Oncotarget 2011; 2:896 - 917; PMID: 22203527
- Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res 2007; 67:6745 - 52; http://dx.doi.org/10.1158/0008-5472.CAN-06-4447; PMID: 17638885
- Isakovic A, Harhaji L, Stevanovic D, Markovic Z, Sumarac-Dumanovic M, Starcevic V, et al. Dual antiglioma action of metformin: cell cycle arrest and mitochondria-dependent apoptosis. Cell Mol Life Sci 2007; 64:1290 - 302; http://dx.doi.org/10.1007/s00018-007-7080-4; PMID: 17447005
- Wang LW, Li ZS, Zou DW, Jin ZD, Gao J, Xu GM. Metformin induces apoptosis of pancreatic cancer cells. World J Gastroenterol 2008; 14:7192 - 8; http://dx.doi.org/10.3748/wjg.14.7192; PMID: 19084933
- Chiacchiera F, Simone C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle 2010; 9:1091 - 6; http://dx.doi.org/10.4161/cc.9.6.11035; PMID: 20190568
- Fenner MH, Elstner E. Peroxisome proliferator-activated receptor-gamma ligands for the treatment of breast cancer. Expert Opin Investig Drugs 2005; 14:557 - 68; http://dx.doi.org/10.1517/13543784.14.6.557; PMID: 16004588
- Lyles BE, Akinyeke TO, Moss PE, Stewart LV. Thiazolidinediones regulate expression of cell cycle proteins in human prostate cancer cells via PPARgamma-dependent and PPARgamma-independent pathways. Cell Cycle 2009; 8:268 - 77; http://dx.doi.org/10.4161/cc.8.2.7584; PMID: 19164938
- Dong YW, Wang XP, Wu K. Suppression of pancreatic carcinoma growth by activating peroxisome proliferator-activated receptor gamma involves angiogenesis inhibition. World J Gastroenterol 2009; 15:441 - 8; http://dx.doi.org/10.3748/wjg.15.441; PMID: 19152448
- Papageorgiou E, Pitulis N, Manoussakis M, Lembessis P, Koutsilieris M. Rosiglitazone attenuates insulin-like growth factor 1 receptor survival signaling in PC-3 cells. Mol Med 2008; 14:403 - 11; http://dx.doi.org/10.2119/2008-00021.Papageorgiou; PMID: 18475308
- Cantini G, Lombardi A, Piscitelli E, Poli G, Ceni E, Marchiani S, et al. Rosiglitazone inhibits adrenocortical cancer cell proliferation by interfering with the IGF-IR intracellular signaling. PPAR Res 2008; 2008:904041; http://dx.doi.org/10.1155/2008/904041; PMID: 18670617
- Teresi RE, Shaiu CW, Chen CS, Chatterjee VK, Waite KA, Eng C. Increased PTEN expression due to transcriptional activation of PPARgamma by Lovastatin and Rosiglitazone. Int J Cancer 2006; 118:2390 - 8; http://dx.doi.org/10.1002/ijc.21799; PMID: 16425225
- Anisimov VN. Metformin for aging and cancer prevention. Aging (Albany NY) 2010; 2:760 - 74; PMID: 21084729
- Abrams SL, Steelman LS, Shelton JG, Wong EW, Chappell WH, Bäsecke J, et al. The Raf/MEK/ERK pathway can govern drug resistance, apoptosis and sensitivity to targeted therapy. Cell Cycle 2010; 9:1781 - 91; http://dx.doi.org/10.4161/cc.9.9.11483; PMID: 20436278
- Pasquier E, Kavallaris M, André N. Metronomic chemotherapy: new rationale for new directions. Nat Rev Clin Oncol 2010; 7:455 - 65; http://dx.doi.org/10.1038/nrclinonc.2010.82; PMID: 20531380
- He X, Esteva FJ, Ensor J, Hortobagyi GN, Lee MH, Yeung SC. Metformin and thiazolidinediones are associated with improved breast cancer-specific survival of diabetic women with HER2+ breast cancer. Ann Oncol 2011; http://dx.doi.org/10.1093/annonc/mdr534; PMID: 22112968
- He XX, Tu SM, Lee MH, Yeung SC. Thiazolidinediones and metformin associated with improved survival of diabetic prostate cancer patients. Ann Oncol 2011; 22:2640 - 5; http://dx.doi.org/10.1093/annonc/mdr020; PMID: 21415239
- Sadeghi N, Abbruzzese JL, Yeung SC, Hassan M, Li D. Metformin use is associated with better survival of diabetic patients with pancreatic cancer. Clin Cancer Res 2012; 18:2905 - 12; http://dx.doi.org/10.1158/1078-0432.CCR-11-2994; PMID: 22465831
- Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 2006; 29:254 - 8; http://dx.doi.org/10.2337/diacare.29.02.06.dc05-1558; PMID: 16443869
- Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, et al. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol 2009; 27:3297 - 302; http://dx.doi.org/10.1200/JCO.2009.19.6410; PMID: 19487376
- Vu K, Busaidy N, Cabanillas ME, Konopleva M, Faderl S, Thomas DA, et al. A randomized controlled trial of an intensive insulin regimen in hyperglycemic acute lymphoblastic leukemia patients. Clin Lymphoma Myeloma Leuk 2012; In press
- Goodwin PJ, Stambolic V, Lemieux J, Chen BE, Parulekar WR, Gelmon KA, et al. Evaluation of metformin in early breast cancer: a modification of the traditional paradigm for clinical testing of anti-cancer agents. Breast Cancer Res Treat 2011; 126:215 - 20; http://dx.doi.org/10.1007/s10549-010-1224-1; PMID: 20976543
- Apontes P, Leontieva OV, Demidenko ZN, Li F, Blagosklonny MV. Exploring long-term protection of normal human fibroblasts and epithelial cells from chemotherapy in cell culture. Oncotarget 2011; 2:222 - 33; PMID: 21447859
- Jin Y, Chen Q, Shi X, Lu Z, Cheng C, Lai Y, et al. Activity of triptolide against human mast cells harboring the kinase domain mutant KIT. Cancer Sci 2009; 100:1335 - 43; http://dx.doi.org/10.1111/j.1349-7006.2009.01159.x; PMID: 19383029