Abstract
The aberrantly increased lipogenesis is a universal metabolic feature of proliferating tumor cells. Although most normal cells acquire the bulk of their fatty acids from circulation, tumor cells synthesize more than 90% of required lipids de novo. The sterol regulatory element-binding protein 1 (SREBP1), encoded by SREBF1 gene, is a master regulator of lipogenic gene expression. SREBP1 and its target genes are overexpressed in a variety of cancers; however, the role of SREBP1 in endometrial cancer is largely unknown. We have screened a cohort of endometrial cancer (EC) specimen for their lipogenic gene expression and observed a significant increase of SREBP1 target gene expression in cancer cells compared with normal endometrium. By using immunohistochemical staining, we confirmed SREBP1 protein overexpression and demonstrated increased nuclear distribution of SREBP1 in EC. In addition, we found that knockdown of SREBP1 expression in EC cells suppressed cell growth, reduced colonigenic capacity and slowed tumor growth in vivo. Furthermore, we observed that knockdown of SREBP1 induced significant cell death in cultured EC cells. Taken together, our results show that SREBP1 is essential for EC cell growth both in vitro and in vivo, suggesting that SREBP1 activity may be a novel therapeutic target for endometrial cancers.
Introduction
The number of women with newly diagnosed endometrial cancer (EC) has increased 20% since 1987, yet the number of deaths from EC posted a 168% increase during the same time period. Estimated new cases and deaths from EC in the United States in 2011 are 46,470 and 8,120, respectively (www.cancer.gov/cancertopics/types/endometrial). Although well-differentiated endometrial cancers have a favorable outcome, the poorly differentiated form has a worse prognosis, largely due to lack of biomarkers and therapeutic targets to predict and to restrict cancer progression. Therefore, identifying new bio-markers and therapeutic targets may provide novel approaches that can improve the clinical outcome of endometrial cancer.
Sterol regulatory element binding proteins (SREBPs) are a family of transcription factors that regulate lipid homeostasis by controlling the expression of the key and rate-limiting enzymes required for cholesterol and fatty acid (FA) synthesis. In mammals, SREBPs are composed of three isoforms: SREBP1a, SREBP1c and SRBEP2. SREBP1a and SREBP1c, encoded by a single gene SREBF1 with alternative promoter usage, mainly control lipogenic gene expression, while SREBP2, encoded by a separate gene SREBF2, predominantly regulates cholesterogenic gene expression.Citation1,Citation2 As transcription factors, SREBPs are unique in that they are synthesized as inactive precursors bound to the endoplasmic reticulum (ER) membranes, and their processing is mainly controlled by the intracellular sterol content. When sterol level decreases, the SREBP precursors are transported to Golgi apparatus, where they undergo a sequential two-step cleavage process. As a result, the NH2-terminal active domain, also known as the mature SREBP, is released and translocated into the nucleus (designated nSREBP), which then activates SREBP target genes to increase the biosynthesis of cholesterol and FAs. In contrast, when the intracellular sterol level is high, the SREBP precursors will remain in the ER. This model provides an elegant explanation of how intracellular sterol and FA homeostasis is maintained.Citation3-Citation9 The unique regulation and activation properties of each SREBP isoform facilitate the coordinated regulation of lipid metabolism.
Lipogenesis is almost universally upregulated in human cancers.Citation10 Consistent with an essential role of SREBP1 in sensing and regulating intracellular lipid homeostasis, increased expression of SREBP1 has been detected in colorectal carcinoma, breast and prostate cancer and hepatocarcinoma.Citation11-Citation14 Moreover, elevated expression of SREBP1 is closely correlated with malignant transformation, cancer progression and metastasis for several cancer types, particularly hormone-responsive tissue-derived cancers, such as breast and prostate cancers.Citation11,Citation13,Citation15,Citation16 For example, SREBP1 expression correlates with FASN and Ki-67 expression in colorectal cancer, indicating a role for SREBP1 in supporting rapid cellular proliferation.Citation16 SREBP1 is elevated in clinical prostate cancer samples compared with benign prostatic hypertrophy.Citation12 Gene expression profiling of hepatocellular carcinoma (HCC) tissue and non-cancerous liver tissue showed increased lipogenic signaling in HCC. Elevated SREBP1 expression in hepatocellular carcinoma is a known predictor of increased mortality.Citation13,Citation15 Overexpression of SREBP1 in human hepatoma HuH7 and Hep3B cells enhanced cellular proliferation and foci formation, while knockdown of SREBP1 in these cells reduced cell replication and anchorage-independent cell growth.Citation15 A dramatic increase of SREBP1 has been correlated with the progression of prostate cancer toward androgen-independence.Citation12 Oncogenic transformation of normal breast epithelial cells was accompanied by increased SREBP1 and FASN expression, consistent with the observation of increased SREBP1 abundance in human breast cancers.Citation17-Citation19 Previous studies have established that SREBP1, through induction of FASN and subsequent fatty acids production, regulates PPARγ transactivation.Citation20,Citation21
Although lipogenesis is known to be elevated in EC cells, the role of SREBP1 in endometrial cancer remains largely unclear. Herein, we report that the nuclear SREBP1 is significantly increased in endometrial cancer, and the expression levels are positively correlated with cancer progression. Importantly, knockdown of endogenous SREBP1 not only reduced SREBP1 target gene expression, but also suppressed cellular proliferation and colony formation of endometrial cancer cells. Furthermore, a xenograft tumor model clearly demonstrated that reduction of SREBP1 expression reduced tumor growth rate. These observations indicate that SREBP1 is required for endometrial cancer cell proliferation and cancer progression, supporting the model that enhanced SREBP1 transactivation may contribute to endometrial cancer progression through induction of lipogenic gene expression and lipogenesis.
Results
Elevated expression of SREBP1 in endometrial carcinoma compared with the adjacent normal endometrium.
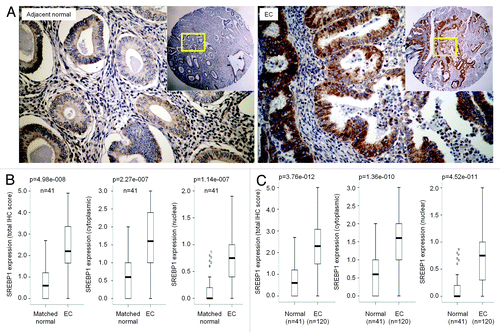
It has been reported that FASN, the lipogenic gene encoding fatty acid synthase, is overexpressed in endometrial cancers;Citation22,Citation23 however, the underlying mechanisms are still unknown.Citation24 SREBP1 is the major transcription factor that directly activates the expression of FASN and other lipogenic genes.Citation21,Citation25 Therefore, we asked whether increased SREBP1 expression and/or activity contribute to the elevated FASN expression in EC. To determine the levels of SREBP1 expression, we performed immunohistochemical staining on formalin-fixed, paraffin-embedded sections using anti-SREBP1 antibody. The levels of nuclear and cytoplasmic SREBP1 were scored for both mature and precursor forms, respectively. As shown in , SREBP1 is expressed in both normal and cancerous tissues, but its levels were significantly increased in poorly differentiated EC. The majority of SREBP1 was found in the cytoplasm of epithelial cells of normal endometrium and well-differentiated tumors, while nuclear SREBP1 was mainly detected in high-grade tumors, including moderately differentiated to poorly differentiated tumors (; Fig. S1). These observations indicate that SREBP1 expression and activation positively correlates with the progression of endometrial cancer.
Figure 1. SREBP1 expression in endometrial cancer (EC) determined by IHC. (A) Immunohistochemistry (IHC) staining of endometrial cancer specimens and matched adjacent non-cancerous tissues for SREBP1 protein expression and subcellular localization. (B) Boxplot of IHC staining score for SREBP1 in whole cell, cytoplasm and nucleus in cancer and matched non-cancerous tissues. (C) Boxplot of IHC staining score for SREBP1 in whole cell, cytoplasm and nucleus in all cancer specimens recruited to this study and non-cancerous endometrial tissues.
SREBP1 status in endometrium during the menstrual cycle and post-menopause.
Because endometrial cancer progression correlates with menopausal status, we characterized the expression of SREBP1 in endometrium during the menstrual cycle and in post-menopausal endometrium. As a result of cyclic steroid hormone levels throughout the menstrual cycle, the endometrium undergoes characteristic proliferative, secretory and menstrual phases and displays a wide spectrum of normal and pathological appearances. As shown in , SREBP1 protein was markedly increased in glandular epithelial cells during proliferative phase, and SREBP1 protein was located in both nucleus and cytoplasm. In contrast, at the secretory phase, the epithelial cells in endometrium lost the expression of SREBP1 protein, while the nuclear distribution of SREBP1 became negligible (). The observation of increased SREBP1 expression and activation (nuclear translocation) in proliferative phase is consistent with the notion that the proliferating cells in endometrium require de novo lipogenesis. SREBP1 protein was not detectable in the majority of post-menopausal endometrium nor in the stromal cells, regardless of the endometrial phases ( and data not shown).
Figure 2. Increased SREBP1 expression in atypical hyperplasia. (A) IHC staining was conducted with anti-SREBP1 antibody on endometrial tissues derived from normal, hyperplasia without atypia and atypical hyperplasia. Secretory, proliferative and post-menopausal normal endometrial tissues were stained. (B) Boxplot of IHC staining score for SREBP1 in whole cell, cytoplasm and nucleus in normal hyperplasic, and cancerous tissues in all specimens recruited to this study as indicated. Statistical analysis of SREBP1 expression was performed showing the p-value for the difference among the experimental groups (bottom panels).
Enhanced SREBP1 expression and nuclear translocation in atypical hyperplasia.
Atypical endometrial hyperplasia (AH) is a precancerous condition. Patients with AH have increased risk of developing invasive endometrial cancer compared to those patients who have hyperplasia without atypia. To test whether the expression and distribution of SREBP1 correlates with the AH status in endometrial hyperplasia, we performed immunohistostaining of SREBP1 in hyperplastic endometrial samples. As shown in , we observed significant differences in both the expression levels and the distribution of SREBP1 in hyperplastic tissues without atypia compared with normal tissues. SREBP1 expression in atypical hyperplasia, when scored at whole-cell level, was significantly higher than that in normal and non-atypical hyperplasia (). In addition, cytoplasmic SREBP1 was increased in AH when compared with normal tissue (). Elevated nuclear SREBP1 in AH was observed but did not reach significant levels when compared with normal (p = 0.180), non-AH (p = 0.173) and EC (p = 0.958), which could be due to the limited number of patient samples (Fig. S2). These results indicate that SREBP1 activation occurs in AH, the precancerous condition, which may contribute to cancer progression.
Figure 3. SREBP1 is required for lipogenic genes and cell proliferation in endometrial cancer cells. (A) Quantitative RT-PCR analysis of mRNA abundance of SREBP1a and SREBP1c in endometrial cancer cells. RNA abundance was shown as fold change relative to that in ECC-1 cells. (B) Western blotting (WB) analysis of lipogenic gene expression in commonly used endometrial cancer cell lines. Actin serves as protein loading control. (C) AN3CA cells were transduced with a set of lentiviral vector expressing shRNA targeting SREBF1. Western blot analysis of SREBP1 and SCD1 were performed showing a successful knockdown of SREBP1 and reduced expression of SCD1 as a transcriptional target of SREBP1. (D) Quantitative RT-PCR analysis of mRNA abundance of SREBP1, SREBP2 and their targets including FASN and SCD1 in #2 and #4 transduced cells.
SREBP1 is responsible for lipogenic gene expression in endometrial cancer cells.
The levels of SREBP1 can be regulated by multiple mechanisms, including transcriptional or posttranslational regulation.Citation26,Citation27 To examine whether increased SREBP1 protein in EC patient samples was due to enhanced gene expression, we performed quantitative RT-PCR (qRT-PCR) analysis on cDNA samples prepared from surgically resected samples of normal and cancerous tissues and compared the mRNA levels of SREBF1a, SREBF1c, SREBF2 and SCD (Stearoyl CoA desaturase), transcriptional targets of SREBPs. As shown in , SREBP1c expression was significantly increased, while the levels of SREBP1a and SREBP2 transcripts were markedly reduced in cancer tissues. Similar to SREBF1c, the mRNA levels of SCD were significantly increased in cancer, which is consistent to increased SREBP1 protein expression and activation as evidenced by nuclear translocation.
Having established the correlation between SREBP1 expression and endometrial cancer progression ( and ), we next determined whether elevated levels of SREBP1 was required for expression of the lipogenic genes in EC cells. To answer this question, we analyzed SREBP1 expression in five commonly used endometrial cancer cell lines, including ECC, HEC-1-A, RL95-2, KLE and AN3 CA. As shown in , SREBP1 expression was undetectable in well-differentiated ECC-1 cells, but it was highly expressed in medium and poorly differentiated RL95-2 and AN3 CA cells. Since the SREBP1 antibody cannot distinguish SREBP1a from SREBP1c proteins, we performed qRT-PCR to determine which SREBP1 isoform was predominantly expressed. RL95-2 cells express both isoforms at relatively high levels; in contrast, AN3 CA expresses SREBP1a but not SREBP1c (data not shown). Next, we determined the expression of SREBP target genes, such as FASN, SCD and ACLY (ATP citrate lyase). Relatively high expression of FASN, SCD and ACLY were detected in AN3 CA cells (data not shown). To test whether the endogenous SREBP1 was required for lipogenic gene expression, we chose AN3 CA cells and depleted SREBP1 by using an shRNA approach. We used five different shRNAs targeting mRNA of SREBF1. As shown in , shRNA#2 and shRNA#4 exhibited high knockdown efficiency as confirmed at both protein and mRNA levels. The expression of SREBP1 target genes was also significantly reduced (), supporting that the expression of these genes is at least in part dependent on SREBP1 function.
SREBP1 is required for cell proliferation.
Cell proliferation is not only tightly controlled by mitogenic signaling but also requires activation of biosynthetic pathways for the generation of macromolecules, including proteins and lipids. Because SREBP1 regulates lipogenesis, the metabolic process that supplies cells with phospholipids, we expected that the knockdown of endogenous SREBP1 would suppress cell proliferation and growth. To determine the functional consequences of SREBP1 inactivation in cultured EC cells, we analyzed the effect of knockdown of SREBP1 on cell growth. As shown in , we observed reduced cell number in shSREBF1-expressing cells compared with the vector control, suggesting that SREBP1 is required for cell proliferation. Next, to determine whether SREBP1 is required for the progression of any specific cell cycle phases, we conducted FACs analysis. We found that depletion of SREBP1 had no significant effect on cell cycle distribution (data not shown). These observations suggest that instead of affecting certain specific cell cycle phases, depletion of SREBP1 may increase the total cell cycle duration.
Figure 4. Knockdown of SREBP1 expression represses colony formation of endometrial cancer cells. (A) Two x 105 of AN3 CA cells were seeded per well in a 6-well cell culture device. Cellular growth was determined by counting the cells at different time points as indicated. Cells with knockdown of endogenous SREBP1 were partially defective for cell growth. (B) Colony formation experiments were conducted in 10-cm cell culture dish. 2,000 cells were seeded and allowed to grow for 2 weeks. Medium was replaced every three days. Cells were stained with crystal violet. (C) The number and size of colonies were calculated using software Gel-Pro Analyzer. (D) Western blot was conducted on #2 and #4 cells, which were used for in vivo tumor growth experiment. (E–F) AN3 CA cells (#2, #4 and vector control) were implanted into SCID mice by injecting 2 x 106 cells subcutaneously. Tumor growth was measured every four days by using a digital caliper and tumor volume was calculated.
Knockdown of SREBP1 impairs EC cell colony-forming capacity and tumor growth in vivo.
To determine the role of SREBP1 in tumor colony formation, we performed clonogenic assays. As shown in , knockdown of SREBF1 by shRNA significantly reduced the number of colonies. The shRNA#4 of SREBF1 achieved nearly 100% knockdown efficiency at protein levels, which reduced the number of large colonies (size > 15) by 90%, while the shRNA#2, by reducing SREBP1 protein expression by 80% (), inhibited the colony-forming capacity of AN3 CA cells by 25%. These results suggest that the colony numbers positively correlate with the levels of the SREBP1 expression. Next, we examined the contribution of SREBP1 to tumor growth in vivo by using a xenograft SCID mouse model. We implanted 2 x 106 of SREBP1 knockdown cells (shRNA#2 and shRNA#4 lines) subcutaneously and monitored tumor growth by measuring tumor size every 4 days. As shown in , the AN3 CA cells with depletion of SREBP1 by the shRNA#2 significantly reduced the tumor size compared with the vector control cells. Fewer tumors and slower growth rate were found in shRNA#4 AN3 CA cells implanted mice, consistent with the high efficiency of SREBP1 depletion by shRNA#4 (). These observations are also in line with the observation that SREBP1-knockdown cells had decreased growth rates compared with the control cohort (). Collectively, these studies demonstrate that SREBP1 is required for tumor growth in vivo.
Knockdown of SREBP1 induces apoptotic cell death.
The tumor growth is resulted from the net gain of cell number, which is determined by cell proliferation and death. It has been previously shown that inhibition of SREBP1 sensitizes tumor cells to death ligand,Citation28 suggesting that SREBP1-dependent evasion of cell death could contribute to the net cell growth. To examine whether SREBP1 is required for cell survival, we depleted SREBP1 and analyzed the sub-G1 population, which represents apoptotic cells. We observed that knockdown of SREBP1 increased the number of cells in sub-G1 cell fraction from 1.7% to 3.3% (data not shown), suggesting that reduction of SREBP1 leads to apoptotic cell death. Annexin V staining precedes the loss of membrane integrity of cells undergoing either apoptotic or necrotic cell death.Citation29 We stained the cells with Annexin V and 7-AAD. As shown in , the early cell death marked by Annexin V-positive and 7-AAD-negative increased from 6.3% to 9% (shRNA#2) and 18.6% (shRNA#4), respectively, in cells with reduced SREBP1. Cells in late apoptosis (both Annexin V and 7-AAD positive) were low in all three cell lines but showed an increase from 1.1% in vector control cell line to 2.1% in SREBP1 knockdown cells (#4) (), which is consistent with the sub-G1 fraction of these two cell lines.
Figure 5. Knockdown of SREBP1 promotes cell death. (A) AN3 CA cells transduced with shSREBF1 and control vector were subjected to Annexin V analysis for apoptotic cell death. (B) Late and early apoptotic cell death were shown as percentage to the total cells counted. Data were presented as mean ± SEM from triplicates.
Discussion
SREBP1, also known as adipocyte determination- and differentiation-dependent factor 1 (ADD1),Citation30 has been shown to be involved in tumorigenesis. Increased expression of SREBP1 has been reported in colorectal carcinoma, breast and prostate cancer and hepatocarcinoma. The levels of SREBP1 positively correlated with the severity of endometrial hyperplasia and cancer, which further supports the notion that SREBP1 is oncogenic. Similarly, elevated expression of SREBP1 was reported in coinciding with malignant transformation, cancer progression and metastasis for several cancer types, particularly hormone-responsive tissues, including breast and prostate cancers.Citation11,Citation13,Citation15,Citation16 Despite this progress, the role of SREBP1 in endometrial tumorigenesis remains unexplored.
We have shown that SREBP1 is required for the tumorigenesis of endometrial cancers. We found that SREBP1 is overexpressed in EC patients, and its activation, as indicated by nuclear translocation, correlates with high-grade, poor differentiation of endometrial cancer. The lowest expression of SREBP1 was seen in post-menopausal tissues. Similar to EC tissue, atypical hyperplastic tissues exhibit an elevated SREBP1 expression. The observation of high SREBP1 expression in proliferative, hyperplastic and cancerous tissues further supports the notion that de novo lipid synthesis is enhanced in cancers. To our knowledge, this is the first report that has systematically examined the role of SREBP1 in endometrial cancer cell proliferation and tumor growth. To directly test whether SREBP1 functions as transforming oncogene, we overexpressed SREBP1 in well-differentiated ECC-1 and immortalized NIH 3T3 cells. Overexpression of SREBP1 did not show tumorigenic capability (unpublished observations), suggesting that gain of SREBP1 alone is not sufficient for tumorigenesis. Collectively, these results suggest that SREBP1 is required for promoting endometrial cancer progression rather than oncogenic transformation. We note that this observation was only made in lipid-depleted serum culture condition, where cells were completely dependent on the de novo lipogenesis to support the demand for phospholipids and cholesterol in rapid proliferating cells.
Several key enzymes involved in lipogenesis have been investigated for their potential to be targeted in cancer therapy. This rapidly growing list includes fatty acid synthase (encoded by FASN gene), acetyl-CoA carboxylase (ACACA), Stearoyl-CoA desaturase (SCD), phospholipase PLA2G7 and ATP citrate lyase (ACLY). Suppression of the protein expression and activity levels with either interfering RNA or small-molecule inhibitors impairs tumor cell proliferation in cell culture and tumor growth in vivo.Citation31-Citation40 In particular, targeting FASN with chemical inhibitors has been proved to be effective in repressing tumor growth.Citation41 Inhibition of PLA2G7 by gene silencing together with lipid-lowering statins impaired prostate cancer cell proliferation and migration and induced apoptotic cell death.Citation42 However, the efficacy of the available agents in cancer therapy needs to be investigated thoroughly. As a proof of concept, our results demonstrated that SREBP1 is required for supporting tumor cell growth and survival. As a master regulator of lipogenic gene transcription, knockdown of SREBP1 efficiently reduced the expression of FASN and SCD (), which may represent a novel interesting approach to block tumor progression. The present study revealed that the expression of SREBP1 was increased in endometrial carcinomas and the expression of SREBP1 protein was increased in poorly differentiated (histologically higher-grade) tumors. Importantly, knockdown of SREBP1 efficiently repressed colonigenic capacity of endometrial cancer cells and in vivo tumor growth, indicating that SREBP1 may serve as a potential therapeutic target.
SREBP1 is synthesized as a 125-kDa precursor located in the membranes of the endoplasmic reticulum and nuclear localization is required for its activation. Besides targeting SREBP expression, strategies targeting SREBP synthesis and maturation may also provide therapeutic values. Fatostatin, a small molecule inhibitor of fatty acid synthesis by directly targeting the SREBP cleavage-activating protein (SCAP), was found to inhibit prostate cancer growth. Our current studies demonstrated the nuclear localization of SREBP1 in endometrial cancer, especially in poorly differentiated carcinoma, including type II cancers (data not shown). The underlying mechanisms through which SREBP1 is processed into matured nuclear form in endometrial cancer cells are still poorly understood and warrant further studies in the future.
It is important to note that epidemiological studies have identified obesity as one of the major risk factors for endometrial cancer, where obese women have a 2–4-fold greater risk of developing endometrial cancer compared with women of normal weight, regardless of menopausal status.Citation43-Citation47 As population size affected by this disease is expected to grow rapidly, particularly in developing countries, endometrial cancer is expected to be a serious global health problem. This study, by establishing the role of SREBP1, the master regulator of lipogenesis, in endometrial cancer growth, may have important implications in understanding how obesity contributes to the increased risk in EC tumorigenesis.
In summary, we have demonstrated that SREBP1 is significantly upregulated and functionally activated in endometrial cancer, and that SREBP1 plays important roles in endometrial cancer progression. Experimental approaches in cell culture revealed that expression of the endogenous SREBP1 is required for cancer cell growth and colony-forming capacity. Although further studies are necessary to clarify the molecular mechanisms underlying the overexpression and functional activation of SREBP1 in cancers, our results suggest that SREBP1 may represent a novel therapeutic target in treating endometrial cancer.
Materials and Methods
Endometrial cancer specimens and immunohistochemistry (IHC) staining.
Formalin-fixed and paraffin-embedded tumor specimens used in this study were from commercial source (Creative Biolabs) and from the tissue bank of the 90th Hospital of Jinan. All tumors were primary and untreated before surgery with complete clinicopathological information. Tumor size was defined as the maximum tumor diameter measured on the tumor specimens at the time of operation. H&E-stained sections of specimens were reviewed and the diagnosis confirmed by an expert gynecologic pathologist. All of the specimens were anonymous and tissues were collected in compliance with institutional review board regulations. Patients with endometrial cancer and tumor characteristics for this study population are is summarized in . Endometrial cancer tissues from the 90th Hospital of Jinan were built into a 60-core array with 2 mm diameter of the core size. Adjacent normal tissues were included for some cancer tissues. Endometrial Cancer cDNA Arrays (TissueScanTM cDNA Arrays) were purchased from OriGene Technologies, Inc.
Table 1. Clinicopathologic characteristics of the patient cohort with endometrial cancer
IHC staining for SREBP1 was performed on the paraffin-embedded tissue blocks in the Kimmel Cancer Center Pathology Core Facility at Thomas Jefferson University. Hematoxylin and eosin (H&E) stainings were reviewed to ensure the cancer tissue and normal epitheliums. IHC staining for these markers was performed on 5 ∝m thick sections. Briefly, tissue slides were deparaffinized with xylene and rehydrated through a graded alcohol series. The endogenous peroxidase activity was blocked by incubation in a 3% hydrogen peroxide solution for 15 min. Antigen retrieval was performed by immersing the slides in 10 mM sodium citrate buffer (pH 6.0) and maintained at a sub-boiling temperature for 5 min. The slides were rinsed in phosphate-buffered saline and incubated with 10% normal serum to block non-specific staining. The slides were then incubated with the primary antibody (anti-SREBP1, K-10) overnight at 4°C in a humidified chamber.
All of staining was assessed by pathologists blinded to the origination of the samples using a semi-quantitative method. Each specimen was assigned a score according to the intensity of the nucleic and cytoplasmic staining. Tissue was scored (H-score) based on the total percentage of positive cells and the intensity of the staining (1+, 2+ or 3+), where H = (% “1+” x 1) + (% “2+” x 2) + (% “3+” x 3). A minimum of 100 cells was evaluated in calculating the H-score.
Statistical analysis.
Means of continuous variables for SERBP-1 staining intensity between endometrial cancer and each normal endometrial phase or within normal endometrial phases were compared by one-way analysis of variance (multiple comparisons). The comparison between the clinicopathologic characteristics of endometrial cancer and SERBP1 staining intensity was evaluated with the Mann-Whitney U test. All statistical tests were two-sided, and p-values less than 0.05 were considered as statistically significant. The statistical analyses were performed using SPSS 13.0 software (SPSS Inc.).
Plasmids, antibodies and reagents.
A set of five of shRNAs targeting human SREBF1 were purchased from Open Biosystems (Cat.#: RHS4533). The 1,000-bp promoter of FASN was amplified from genomic DNA of AN3 CA cells and subcloned into KpnI/BglII of pGL3-basic vector (Promega). Mouse Scd1 promoter-driven luciferase reporter was a gift from Dr. Ntambi.Citation48 pcDNA4-FLAG-SREBP1a was obtained from Addgene.Citation49 All the mutant constructs for SREBF1 promoter-driven luciferase reporter were kindly provided by Dr. Yamada?Citation50 The retroviral vector (MSCV-IRES-GFP) was described in our prior publication.Citation51 Anti-SREBP1 (K-10 and H-160), anti-FASN (H300), anti-actin (C4) antibodies were purchased from Santa Cruz Biotechnology, anti-SCD (MC38) and anti-LC3A/B antibodies were purchased from Cell Signaling Technology and anti-ACLY antibody was from Abcam (#ab40993).
Cell culture.
The human embryonic kidney 293T cells (HEK 293T) were maintained in DMEM containing 1% penicillin/streptomycin and supplemented with 10% fetal bovine serum (FBS). Endometrial cancer cell lines, including ECC-1, HEC-1A, RL95-2, KLE and AN3 CA, were purchased from American Type Culture Collection (ATCC). The basal culture medium are RPMI-1640 (ECC-1), McCoy’s 5a (HEC-1A), DMEM: F12 (RL95-2, KLE) and Eagle’s Minimum Essential Medium (AN3 CA) according to ATCC. For cell maintenance, the basal medium was supplemented with 10% fetal bovine serum (FBS) except ECC-1 with 5% FBS. Under lipid-free culture condition as indicated, the basal medium was supplemented with 5% lipid-depleted FBS purchased from Cocalico Biologicals (#55-0116).
Cell transfection and transduction.
For transient transfection, Superfect Transfection Reagent (Qiagen) was used following manufacturer’s protocol. For cell transduction, lenti-viruses were prepared using Trans-Lentiviral shRNA Packaging Kit following manufacturer’s instruction (Open Biosystems) with modifications. Briefly, lenti-viral vector expressing shRNA will be introduced into HEK 293T cells by transient co-transfection with helper virus with calcium phosphate precipitation. After 6 h, cell culture medium was replaced and cells were allowed to grow for 36 h to produce viruses. The supernatant was then collected and filtered through a 0.45-∝m filter. Cells were infected at approximately 70% confluence in DMEM supplemented with 8 ∝g/ml of polybrene. The following day, the medium was changed to basal medium supplemented with 10% FBS and cultured for further assays. Cells were stably selected by supplementing the medium with 2 ∝g/ml puromycin for two weeks. The knockdown efficiency was determined by either western blot or qRT-PCR assays.
RNA isolation and quantitative RT-PCR.
Total RNA was prepared using TRIzol Reagent (Invitrogen) following manufacturer’s instructions. Five ∝g of total RNA was subjected to reverse transcription to synthesize cDNA using the SuperScriptTM II Reverse Transcriptase Kit (Invitrogen). For qRT-PCR, each reaction (25 ∝l) consisted 1 ∝l reverse transcription cDNA product and 100 nM of each primer. The primers used for qRT-PCR are listed as below:
Gene symbol Forward primer sequence Reverse primer sequence Amplicon sizeSREBP1a 5'-CGG CGC TGC TGA CCG ACA TC 5'-CCC TGC CCC ACT CCC AGC AT 104 bpSREBP1c 5'-GCG CAG ATC GCG GAG CCA T 5'-CCC TGC CCC ACT CCC AGC AT 116 bpSREBP2 5'-CAA GCT TCT AAA GGG CAT CG 5'-AGT AGG GAG AGA AGC CAG CC 140 bpFANS 5'-CAC AGG GAC AAC CTG GAG TT 5'-ACT CCA CAG GTG GGA ACA AG 97 bpSCD1 5'-CGA CGT GGC TTT TTC TTC TC 5'-CCT TCT CTT TGA CAG CTG GG 70 bpACLY 5'-GCC CAT CCC CAA CCA GCC AC 5'-TTG CAG GCG CCA CCT CAT CG 137 bpADIPOQ 5'-TCC TGC CAG TAA CAG GGA AG 5'-AGG GGA AGT GTC AGT ACC CG 168 bpaP2 5'-CTC TCC GTT CAG ATT GAA GGG G 5'-AAT CCC GCC TCC ATC CTA ACT 122 bpGAPDH 5'-GAG TCA ACG GAT TTG GTC GT 5'-TTG AGG TCA ATG AAG GGG TC 103 bp
Cellular proliferation and apoptosis assays.
For cell proliferation assays, cells were stably transfected with shRNA targeting SREBF1 and control were seeded at a density of 2 x 105 cells per well in 6-well culture device in DMEM containing 10% FBS. The total number of cells was counted for 5 d. Data are from at least three experiments done in triplicate (mean ± SEM).
Cell death was determined by PE Annexin V Apoptosis Detection Kit (BD Biosciences) following manufacturer’s instructions.
Colony formation assays.
A total of 2,000 cells were seeded in 100-mm plates and allowed to grow for two weeks. The culture medium was replaced every three days with fresh MEM supplemented with either regular FBS or lipid-depleted FBS. The number of colonies formed per plate was stained with crystal violet and determined quantitatively by Gel-Pro Analyzer (Media Cybernetics, Inc.).
Subcutaneous tumor implantation.
All animal experiments were performed following animal protocols that have been approved by the Institutional Animal Care and Use Committee (IACUC) at Thomas Jefferson University. AN3 CA cells (2 x 106) with either knockdown of SREBP1 or vector control were implanted by subcutaneous injection in the flank of male 6- to 8-week old SCID mice. Comparisons were made for 10 animals in each group between AN3 CA/vector, AN3 CA/shSREBF1#2 and AN3 CA/shSREBF1#4. The tumor growth rates were examined using serial caliper measurements. The tumor volume were calculated using the equation (a x b2)/2 where “a” and “b” are length and width of the tumor, respectively. At the completion of the experiments, tumors were excised, weighed and statistical significance of differences in tumor volume were logarithm transformed and analyzed using a linear mixed model. Separate slope and intercepts were computed for each group (Vector, #2, #4), then we compared across groups using a global test followed by pair-wise comparisons via linear contrasts. Data prior to day 12 were ignored due to zeroes at day 0 (inability to take logarithms) and an initial nonlinearity/change in some of the animals growth patterns prior to day 12. Thus, the intercept at day 12 is interpretable as initiation of growth, and the slope is interpretable as rate of growth. For the models, we report coefficients, confidence intervals of coefficients and two-sided p-values.
| Abbreviations: | ||
| EC | = | endometrial cancer |
| SREBP1 | = | sterol regulatory element-binding protein 1 |
| SREBF1 | = | sterol regulatory element-binding transcription factor 1 |
| SCD | = | stearoyl CoA desaturase |
| FASN | = | fatty acid synthase |
Additional material
Download Zip (348 KB)Acknowledgments
Work conducted at the Kimmel Cancer Center was supported by the NIH Cancer Center Core grant (P30CA56036). This project is funded in part by the Pennsylvania Department of Health grant (C.W.), and the National Natural Science Foundation of China [81102006 (J.Z.), 81173614 (J.J.), and 81172008 (T.Z)] and the Shandong Natural Science Foundation (Z2008C13). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Note
Supplemental materials can be found at: http://www.landesbioscience.com/journals/cc/article/20811
Related Research Data
References
- Hua X, Wu J, Goldstein JL, Brown MS, Hobbs HH. Structure of the human gene encoding sterol regulatory element binding protein-1 (SREBF1) and localization of SREBF1 and SREBF2 to chromosomes 17p11.2 and 22q13. Genomics 1995; 25:667 - 73; http://dx.doi.org/10.1016/0888-7543(95)80009-B; PMID: 7759101
- Edwards PA, Tabor D, Kast HR, Venkateswaran A. Regulation of gene expression by SREBP and SCAP. Biochim Biophys Acta 2000; 1529:103 - 13; PMID: 11111080
- Wang X, Sato R, Brown MS, Hua X, Goldstein JL. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell 1994; 77:53 - 62; http://dx.doi.org/10.1016/0092-8674(94)90234-8; PMID: 8156598
- Hua X, Yokoyama C, Wu J, Briggs MR, Brown MS, Goldstein JL, et al. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc Natl Acad Sci USA 1993; 90:11603 - 7; http://dx.doi.org/10.1073/pnas.90.24.11603; PMID: 7903453
- Yokoyama C, Wang X, Briggs MR, Admon A, Wu J, Hua X, et al. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell 1993; 75:187 - 97; PMID: 8402897
- Briggs MR, Yokoyama C, Wang X, Brown MS, Goldstein JL. Nuclear protein that binds sterol regulatory element of low density lipoprotein receptor promoter. I. Identification of the protein and delineation of its target nucleotide sequence. J Biol Chem 1993; 268:14490 - 6; PMID: 8390995
- Wang X, Briggs MR, Hua X, Yokoyama C, Goldstein JL, Brown MS. Nuclear protein that binds sterol regulatory element of low density lipoprotein receptor promoter. II. Purification and characterization. J Biol Chem 1993; 268:14497 - 504; PMID: 8314806
- Shimano H. Sterol regulatory element-binding proteins (SREBPs): transcriptional regulators of lipid synthetic genes. Prog Lipid Res 2001; 40:439 - 52; http://dx.doi.org/10.1016/S0163-7827(01)00010-8; PMID: 11591434
- Horton JD, Shimomura I. Sterol regulatory element-binding proteins: activators of cholesterol and fatty acid biosynthesis. Curr Opin Lipidol 1999; 10:143 - 50; http://dx.doi.org/10.1097/00041433-199904000-00008; PMID: 10327282
- Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care 2006; 9:358 - 65; http://dx.doi.org/10.1097/01.mco.0000232894.28674.30; PMID: 16778563
- Yang YA, Morin PJ, Han WF, Chen T, Bornman DM, Gabrielson EW, et al. Regulation of fatty acid synthase expression in breast cancer by sterol regulatory element binding protein-1c. Exp Cell Res 2003; 282:132 - 7; http://dx.doi.org/10.1016/S0014-4827(02)00023-X; PMID: 12531699
- Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, Gleave ME, et al. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res 2004; 64:2212 - 21; http://dx.doi.org/10.1158/0008-5472.CAN-2148-2; PMID: 15026365
- Yahagi N, Shimano H, Hasegawa K, Ohashi K, Matsuzaka T, Najima Y, et al. Co-ordinate activation of lipogenic enzymes in hepatocellular carcinoma. Eur J Cancer 2005; 41:1316 - 22; http://dx.doi.org/10.1016/j.ejca.2004.12.037; PMID: 15869874
- Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011; 140:1071 - 83; http://dx.doi.org/10.1053/j.gastro.2010.12.006; PMID: 21147110
- Yamashita T, Honda M, Takatori H, Nishino R, Minato H, Takamura H, et al. Activation of lipogenic pathway correlates with cell proliferation and poor prognosis in hepatocellular carcinoma. J Hepatol 2009; 50:100 - 10; http://dx.doi.org/10.1016/j.jhep.2008.07.036; PMID: 19008011
- Li JN, Mahmoud MA, Han WF, Ripple M, Pizer ES. Sterol regulatory element-binding protein-1 participates in the regulation of fatty acid synthase expression in colorectal neoplasia. Exp Cell Res 2000; 261:159 - 65; http://dx.doi.org/10.1006/excr.2000.5054; PMID: 11082286
- Furuta E, Pai SK, Zhan R, Bandyopadhyay S, Watabe M, Mo YY, et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res 2008; 68:1003 - 11; http://dx.doi.org/10.1158/0008-5472.CAN-07-2489; PMID: 18281474
- Yoon S, Lee MY, Park SW, Moon JS, Koh YK, Ahn YH, et al. Up-regulation of acetyl-CoA carboxylase alpha and fatty acid synthase by human epidermal growth factor receptor 2 at the translational level in breast cancer cells. J Biol Chem 2007; 282:26122 - 31; http://dx.doi.org/10.1074/jbc.M702854200; PMID: 17631500
- Yang YA, Han WF, Morin PJ, Chrest FJ, Pizer ES. Activation of fatty acid synthesis during neoplastic transformation: role of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Exp Cell Res 2002; 279:80 - 90; http://dx.doi.org/10.1006/excr.2002.5600; PMID: 12213216
- Kim JB, Wright HM, Wright M, Spiegelman BM. ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc Natl Acad Sci USA 1998; 95:4333 - 7; http://dx.doi.org/10.1073/pnas.95.8.4333; PMID: 9539737
- Kim JB, Spiegelman BM. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev 1996; 10:1096 - 107; http://dx.doi.org/10.1101/gad.10.9.1096; PMID: 8654925
- Lupu R, Menendez JA. Targeting fatty acid synthase in breast and endometrial cancer: An alternative to selective estrogen receptor modulators?. Endocrinology 2006; 147:4056 - 66; http://dx.doi.org/10.1210/en.2006-0486; PMID: 16809439
- Tsuji T, Yoshinaga M, Togami S, Douchi T, Nagata Y. Fatty acid synthase expression and clinicopathological findings in endometrial cancer. Acta Obstet Gynecol Scand 2004; 83:586 - 90; PMID: 15144342
- Yin C, Qie S, Sang N. Carbon source metabolism and its regulation in cancer cells. Crit Rev Eukaryot Gene Expr 2012; 22:17 - 35; PMID: 22339657
- Kim JB, Sarraf P, Wright M, Yao KM, Mueller E, Solanes G, et al. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J Clin Invest 1998; 101:1 - 9; http://dx.doi.org/10.1172/JCI1411; PMID: 9421459
- Chen G, Liang G, Ou J, Goldstein JL, Brown MS. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc Natl Acad Sci USA 2004; 101:11245 - 50; http://dx.doi.org/10.1073/pnas.0404297101; PMID: 15266058
- Walker AK, Yang F, Jiang K, Ji JY, Watts JL, Purushotham A, et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev 2010; 24:1403 - 17; http://dx.doi.org/10.1101/gad.1901210; PMID: 20595232
- Eberhard Y, Gronda M, Hurren R, Datti A, MacLean N, Ketela T, et al. Inhibition of SREBP1 sensitizes cells to death ligands. Oncotarget 2011; 2:186 - 96; PMID: 21406729
- Lao T, Chen S, Sang N. Two mutations impair the stability and function of ubiquitin-activating enzyme (E1). J Cell Physiol 2012; 227:1561 - 8; http://dx.doi.org/10.1002/jcp.22870; PMID: 21678405
- Tontonoz P, Kim JB, Graves RA, Spiegelman BM. ADD1: a novel helix-loop-helix transcription factor associated with adipocyte determination and differentiation. Mol Cell Biol 1993; 13:4753 - 9; PMID: 8336713
- Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura M, et al. ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Res 2008; 68:8547 - 54; http://dx.doi.org/10.1158/0008-5472.CAN-08-1235; PMID: 18922930
- Zu XY, Zhang QH, Liu JH, Cao RX, Zhong J, Yi GH, et al. ATP citrate lyase inhibitors as novel cancer therapeutic agents. Recent Pat Anticancer Drug Discov 2012; 7:154 - 67; http://dx.doi.org/10.2174/157489212799972954; PMID: 22339355
- Luo DX, Tong DJ, Rajput S, Wang C, Liao DF, Cao D, et al. Targeting acetyl-CoA carboxylases: small molecular inhibitors and their therapeutic potential. Recent Pat Anticancer Drug Discov 2012; 7:168 - 84; http://dx.doi.org/10.2174/157489212799972918; PMID: 22339356
- Wang C, Rajput S, Watabe K, Liao DF, Cao D. Acetyl-CoA carboxylase-a as a novel target for cancer therapy. [Schol Ed] Front Biosci (Schol Ed) 2010; 2:515 - 26; http://dx.doi.org/10.2741/s82; PMID: 20036965
- Tong L, Harwood HJ Jr.. Acetyl-coenzyme A carboxylases: versatile targets for drug discovery. J Cell Biochem 2006; 99:1476 - 88; http://dx.doi.org/10.1002/jcb.21077; PMID: 16983687
- Roongta UV, Pabalan JG, Wang X, Ryseck RP, Fargnoli J, Henley BJ, et al. Cancer cell dependence on unsaturated fatty acids implicates stearoyl-CoA desaturase as a target for cancer therapy. Mol Cancer Res 2011; 9:1551 - 61; http://dx.doi.org/10.1158/1541-7786.MCR-11-0126; PMID: 21954435
- Kim SJ, Choi H, Park SS, Chang C, Kim E. Stearoyl CoA desaturase (SCD) facilitates proliferation of prostate cancer cells through enhancement of androgen receptor transactivation. Mol Cells 2011; 31:371 - 7; http://dx.doi.org/10.1007/s10059-011-0043-5; PMID: 21331774
- Minville-Walz M, Pierre AS, Pichon L, Bellenger S, Fèvre C, Bellenger J, et al. Inhibition of stearoyl-CoA desaturase 1 expression induces CHOP-dependent cell death in human cancer cells. PLoS One 2010; 5:e14363; http://dx.doi.org/10.1371/journal.pone.0014363; PMID: 21179554
- Igal RA. Stearoyl-CoA desaturase-1: a novel key player in the mechanisms of cell proliferation, programmed cell death and transformation to cancer. Carcinogenesis 2010; 31:1509 - 15; http://dx.doi.org/10.1093/carcin/bgq131; PMID: 20595235
- Fritz V, Benfodda Z, Rodier G, Henriquet C, Iborra F, Avancès C, et al. Abrogation of de novo lipogenesis by stearoyl-CoA desaturase 1 inhibition interferes with oncogenic signaling and blocks prostate cancer progression in mice. Mol Cancer Ther 2010; 9:1740 - 54; http://dx.doi.org/10.1158/1535-7163.MCT-09-1064; PMID: 20530718
- Liu H, Liu JY, Wu X, Zhang JT. Biochemistry, molecular biology, and pharmacology of fatty acid synthase, an emerging therapeutic target and diagnosis/prognosis marker. Int J Biochem Mol Biol 2010; 1:69 - 89; PMID: 20706604
- Vainio P, Lehtinen L, Mirtti T, Hilvo M, Seppänen-Laakso T, Virtanen J, et al. Phospholipase PLA2G7, associated with aggressive prostate cancer, promotes prostate cancer cell migration and invasion and is inhibited by statins. Oncotarget 2011; 2:1176 - 90; PMID: 22202492
- Bratos K, Roszak A, Cikowska-Woźniak E, Niecewicz P. [Analysis of epidemiologic risk factors for endometrial cancer]. Ginekol Pol 2002; 73:945 - 50; PMID: 12722379
- Gull B, Karlsson B, Milsom I, Granberg S. Factors associated with endometrial thickness and uterine size in a random sample of postmenopausal women. Am J Obstet Gynecol 2001; 185:386 - 91; http://dx.doi.org/10.1067/mob.2001.115869; PMID: 11518897
- Weiderpass E, Persson I, Adami HO, Magnusson C, Lindgren A, Baron JA. Body size in different periods of life, diabetes mellitus, hypertension, and risk of postmenopausal endometrial cancer (Sweden). Cancer Causes Control 2000; 11:185 - 92; http://dx.doi.org/10.1023/A:1008946825313; PMID: 10710204
- Shoff SM, Newcomb PA. Diabetes, body size, and risk of endometrial cancer. Am J Epidemiol 1998; 148:234 - 40; http://dx.doi.org/10.1093/oxfordjournals.aje.a009630; PMID: 9690359
- Goodman MT, Hankin JH, Wilkens LR, Lyu LC, McDuffie K, Liu LQ, et al. Diet, body size, physical activity, and the risk of endometrial cancer. Cancer Res 1997; 57:5077 - 85; PMID: 9371506
- Chu K, Miyazaki M, Man WC, Ntambi JM. Stearoyl-coenzyme A desaturase 1 deficiency protects against hypertriglyceridemia and increases plasma high-density lipoprotein cholesterol induced by liver X receptor activation. Mol Cell Biol 2006; 26:6786 - 98; http://dx.doi.org/10.1128/MCB.00077-06; PMID: 16943421
- Toth JI, Datta S, Athanikar JN, Freedman LP, Osborne TF. Selective coactivator interactions in gene activation by SREBP-1a and -1c. Mol Cell Biol 2004; 24:8288 - 300; http://dx.doi.org/10.1128/MCB.24.18.8288-8300.2004; PMID: 15340088
- Amemiya-Kudo M, Shimano H, Yoshikawa T, Yahagi N, Hasty AH, Okazaki H, et al. Promoter analysis of the mouse sterol regulatory element-binding protein-1c gene. J Biol Chem 2000; 275:31078 - 85; http://dx.doi.org/10.1074/jbc.M005353200; PMID: 10918064
- Zhou J, Zhang W, Liang B, Casimiro MC, Whitaker-Menezes D, Wang M, et al. PPARgamma activation induces autophagy in breast cancer cells. Int J Biochem Cell Biol 2009; 41:2334 - 42; http://dx.doi.org/10.1016/j.biocel.2009.06.007; PMID: 19563910