Abstract
The molecular mechanisms underlying mutant p53 (mutp53) “gain-of-function” (GOF) are still insufficiently understood, but there is evidence that mutp53 is a transcriptional regulator that is recruited by specialized transcription factors. Here we analyzed the binding sites of mutp53 and the epigenetic status of mutp53-regulated genes that had been identified by global expression profiling upon depletion of endogenous mutp53 (R273H) expression in U251 glioblastoma cells. We found that mutp53 preferentially and autonomously binds to G/C-rich DNA around transcription start sites (TSS) of many genes characterized by active chromatin marks (H3K4me3) and frequently associated with transcription-competent RNA polymerase II. Mutp53-bound regions overlap predominantly with CpG islands and are enriched in G4-motifs that are prone to form G-quadruplex structures. In line, mutp53 binds and stabilizes a well-characterized G-quadruplex structure in vitro. Hence, we assume that binding of mutp53 to G/C-rich DNA regions associated with a large set of cancer-relevant genes is an initial step in their regulation by mutp53. Using GAS1 and HTR2A as model genes, we show that mutp53 affects several parameters of active transcription. Finally, we discuss a dual mode model of mutp53 GOF, which includes both stochastic and deterministic components.
Introduction
The TP53 gene encodes a versatile cellular regulator, p53,Citation1 involved in transcriptional control of a large set of target genes associated with cell cycle arrest, apoptosis, metabolism and differentiation.Citation2 Loss of normal p53 function in various cellular contexts is both a prerequisite for, and a path leading to, tumor outgrowth. In tumors, p53 function is inactivated by mutations in the TP53 gene with a frequency varying between 5% and 50%, depending on tissue origin.Citation3 About 74% of TP53-mutated tumors harbor missense-point mutations that result in a high-level expression of mutant p53 (mutp53), which is usually localized in the nucleus.Citation3 Missense mutations are most frequently located in the DNA-binding domain (e.g., at the “hotspot” amino acid positions R175, G245, R248, R249, R273 and R282) that is responsible for sequence-specific binding of p53 to regulatory regions of target genes. Hence, mutp53 has lost the specificity of wild-type p53 (wtp53) and thus its tumor-suppressive functions. Mutp53, however, has retained the capacity of wtp53 to interact with DNA in a structure-specific fashion.Citation4 It by now is widely accepted that at least some mutp53 proteins in tumor cells are functional and exert an oncogenic potential termed “gain-of-function” (GOF).Citation5,Citation6 Expression of mutp53 increases drug resistanceCitation7 and enhances the potential for the outgrowth of singularized cells.Citation8 Furthermore, mutp53 promotes the transition from an epithelial to a mesenchymal differentiation state,Citation9 a process that is functionally linked to invasive growth and metastatic dissemination,Citation10 and the generation of a cellular subset that behaves like cancer stem cells (CSCs) within epithelial tumors.Citation11 These data provide a link between stem-like properties of tumor cells and mutp53 function. Similar to normal stem/progenitor cells, CSCs are able to self-renew, and although genetically and epigenetically flawed, they retain the potency to repopulate the distinct cellular phenotypes within the tumor. This property of CSCs requires transcriptional plasticity, reminiscent of the transcriptional competence of differentiation-related genes in embryonic stem cells.Citation12 Considering the diverse effects of mutp53 on transcriptionCitation5 and the diverse phenotypic consequences of mutp53 expression, promoting transcriptional plasticity in tumor cells emerges as a major feature of mutp53 GOF. Consequently, mutp53 is involved in the regulation of a large number of genes, thereby creating the need for genome-wide analyses to investigate mutp53 functions as a transcriptional regulator.
Despite recent efforts to identify transcriptional targets of mutp53 and to explore the mechanism underlying gene regulation by mutp53, either expressed endogenously or ectopically in p53-null cells, the emerging picture is still blurry. The mechanism of mutp53 activity appears to be very heterogeneous and strongly dependent on the biological context.Citation5,Citation6 This can be attributed to the diversity of mutant p53 species and to heterogeneity of the genetic background, but can also be ascribed to the interdependence of transcriptional functions of mutp53 with various, context-dependent signaling pathways. Currently a scenario is favored in which mutp53 takes advantage of the diverse protein-protein interactions inherent to the wtp53 protein and regulates transcription of genes in an indirect fashion. Via interactions with other TFs, mutp53 exerts either cooperative or counteracting effects on their target genes in a stimulus-dependent or -independent manner. This scenario is illustrated, e.g., by the cooperation of mutp53 with NF-Y in positive regulation of CCAT-box containing cell cycle genes upon DNA damage,Citation13 the functional interaction of mutp53 with the vitamin D receptor (VDR) on its target genes after exposure of cells to vitamin D,Citation14 the SMAD-mediated antagonism between mutp53 and p63 activity in regulation of metastasis-related, TGF-β-induced genes,Citation15 the co-stimulation of the pro-angiogenic ID4 gene via mutp53-E2F1 protein complexes,Citation16 the binding of mutp53 to promoters of mevalonate pathway genes at least partly by interaction with SREBP transcription factorsCitation17 and the promotion of etoposide resistance via ETS2-dependent co-regulation of the DNA repair related TDP2 gene.Citation18 A second scenario is based on the capacity of mutp53 to interact with structured DNA, like stem-loop structuresCitation19 and non-B DNA structures formed by trinucleotide [(CAG)·(CTG)]n repeats.Citation20 Along with the binding of mutp53 to intronic and intergenic sequences enriched in repetitive DNA and prone to form non-B DNA structures, these data led to a model that attributes mutp53 GOF to its ability to modulate transcription on a global level via binding to structured DNA.Citation21 Both scenarios are complementary rather than mutually exclusive; however, data that incorporate both aspects of transcriptional regulation by mutp53 into one model are still insufficient.
Here we extended our previous observations by a large-scale analysis of mutp53 binding sites coupled with global expression profiling and analysis of the epigenetic status of mutp53 regulated promoters. Our data led us to propose a role for mutp53 as a co-factor that is able to modulate transcription rates of a multitude of active genes, thereby contributing to their transcriptional plasticity. This mode of mutp53 action primarily is based on DNA binding to G-rich sequences at active promoters.
Results
Mutp53 binds to G/C rich regulatory regions around promoters of many genes
On the basis of a small set of mutp53 binding sites, we previously proposed that mutp53 modulates transcription on a global scale via direct binding to intronic and intergenic sequences prone to form non-B DNA structures.Citation21 To explore binding of mutp53 to genomic DNA in more detail, we performed ChIP-chip experiments using a custom tiling array that covers a total of 902 genes including putative mutp53 target genes derived from knockdown and overexpression studies in our lab (unpublished data) as well as several wtp53 target genes (Table S1). The probes on the tiling array were designed to cover whole genes including 1,500 nt upstream and downstream of transcriptional start and stop sites. As we have previously observed a significant binding of mutp53 to repetitive DNA,Citation21 we anticipated a relatively low sequence specificity and, consequently, high variability of mutp53 binding to gene regions. Surprisingly, however, we observed significant reproducibility of mutp53 binding in four biological replicates, e.g., in the mutp53 target gene EGR1Citation22 ().
Figure 1. Array-wide analysis of mutp53 binding sites. (A) Consensus signals for the whole genome and for chromosome 5 are shown. The EGR1 gene is shown as an example of a known mutp53 target gene. The lower panel displays the detected peaks in the EGR1 gene dependent on the selected p value threshold during peak calling. CpG-islands, DNaseI-HS regions and G4 motif locations extracted from the public databases are plotted. (B) Distribution of mutp53-BR around TSS. The median length of mutp53-BR varied from 1270 bp to 990 bp. (C) Motifs overrepresented in mutp53 binding regions (p value 0.001) identified by MEME-ChIP analysis.
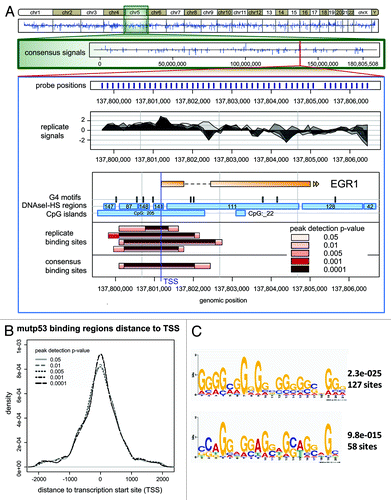
For analysis of mutp53-DNA binding sites, we used the p value-based peak calling function from CARPETCitation23 and observed significant enrichment of mutp53-bound regions (mutp53-BR) within ± 1 kb around transcription start sites (TSS) (; Table S2), demonstrating selectivity of mutp53 binding toward TSS regions. The frequent association of mutp53-BR with TSS coincides with a significant overlap with CpG islands (p value raw = 8.46e-129) (Fig. S1A), e.g., 90% mutp53-BR (at p value 0.001) intersect with CpG islands (Table S2). In comparison, 3,540 out of 5,387 (ca. 66%) of Ensembl TSS that map to the tiling array intersect with CpG islands in a window of ± 496 bp (corresponding to the median length of mutp53-BR at a p value of 0.001). Thus, mutp53-BR are remarkably overrepresented at CpG-associated TSS. Consequently, mutp53-BR feature, as determined by EpiGRAPH analysis,Citation24 a higher content of CpG dinucleotides and significant enrichment for a “CCCC” pattern compared with random regions on the tiling array (Fig. S1A). Accordingly, using 30 nt as maximum motif width, we identified by MEME-ChIP analysis of mutp53-BR predominant, 20–21 nt long G-rich motifs (), which are prone to form non-B DNA conformations. In support, calculation of base-pair step parameters by EpiGRAPH revealed that mutp53-BR exhibit higher Roll and lower Twist values and higher values (p value raw = 0) for the solvent accessible surface area (Fig. S1B), indicating significant structural differences between mutp53-bound and random regions. Furthermore, comparison of mutp53-BR with the coordinates of predicted G-quadruplex forming repeats from non-B DBCitation25 revealed that 75% of mutp53-BR (at p value 0.001) comprise G-quadruplex motifs (Table S2). In summary, by ChIP-chip analysis on a tiling array we observed a high predisposition of mutp53 for binding to TSS-associated G/C-rich regions, which are prone to exist in a non-B DNA conformation, particularly a G-quadruplex structure.
Mutp53 stabilizes G-quadruplex folding
To test directly the binding of mutp53 to G-rich DNA in non-B DNA conformation, we performed EMSA using a 52-mer oligonucleotide (Pu52) (), which contains the purine-rich nuclease-hypersensitive element (NHE) III1, located upstream of the P1-promoter of the human MYC gene.Citation26 Upon addition of potassium ions, this sequence forms an intramolecular G-quadruplex structure, which runs faster than double-stranded (ds) Py52/Pu52 DNA in polyacrylamide gel (Fig. S2A). In accordance with our ChIP-chip and in silico data, we observed by EMSA a preferential binding of mutp53 proteins (G245S, R248W and R273H mutations) as well as of wtp53 to the quadruplex structure of the Pu52 oligonucleotide (), but not to dsDNA composed of annealed Pu52 and Py52 oligonucleotides (), thereby supporting the specificity of the interaction of mutp53 with DNA folded into a quadruplex structure. In addition, significant binding of mutant p53 (R248W and R273H mutations) was detected by EMSA using another G-rich oligonucleotide (TERT-Pu61) derived from the human TERT promoterCitation27 (Fig. S2B). Compared with wtp53, mutp53 proteins bind either weakly (G245S and R273H) or not (R248W), to a p53 response element within a ds oligonucleotide (P53CON)Citation28 (). Hence, specific binding to structured DNA, particularly to G-quadruplex DNA, is an inherent feature of the p53 protein that is preserved in p53 mutants.
Figure 2. Analysis of the interaction of mutant p53 proteins and of wtp53 with the MYC-PU52 quadruplex structure by EMSA and CD spectroscopy. (A-C) Comparison of the binding affinity of mutant p53 and of wtp53 for the MYC-Pu52 intramolecular quadruplex structure (A), double-stranded (ds) oligonucleotide (Py52/Pu52) (B) and p53CON ds sequence (C). Twenty-five, 50 and 100 ng of wtp53, G245S, R248W and R273H mutant p53 were incubated under binding conditions (see M&M) with 32P-radiolabeled DNA oligonucleotides and 30 ng competitor DNA. (D) CD spectra of the MYC-Pu52 oligonucleotide (0.5 µM) in 5 mM TRIS-HCl, supplemented with 24 mM KCl, supplemented with mutp53R273H (3 µM) and 24 mM KCl. CD spectra were recorded after 24 h when equilibrium was attained. The dotted line corresponds to the CD spectrum of mutp53 alone (0.5 µM). Strong absorption of protein shifts measurement to shorter wavelength.
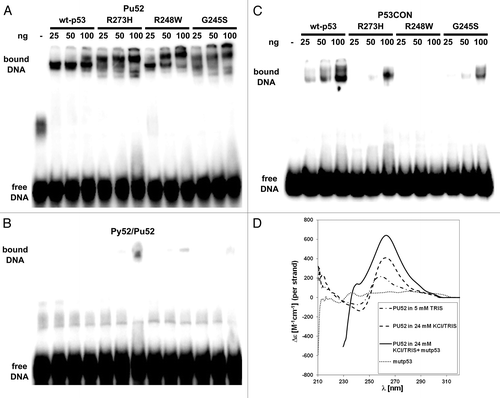
Next, we tested the influence of mutp53 on G-quadruplex structure stability, which could be either promoted or destabilized by interacting mutp53 protein. shows a CD spectrum of the MYC-Pu52, measured in Tris buffer and supplemented with potassium ions, which stabilize quadruplex formation. The presence of a quadruplex structure is reflected in the peak around 260 nm, where a parallel quadruplex displays a characteristic positive peak.Citation29 A distinct increase of this peak is observed upon addition of mutp53 (R273H) protein to the solution. Binding of mutp53 (R273H) to the MYC-Pu52 was verified by EMSA and western blotting directly after CD measurement (Fig. S2C). Similarly to MYC-Pu52, a less prominent, but reproducible increase in the CD signal around 260 nm was detected for the TERT-Pu61 incubated with mutp53 (R273H) protein (Fig. S2D), which forms slow-migrating complexes with TERT-Pu61, detectable by EMSA (Fig. S2E). In summary, the results indicate that mutp53 not only binds to G-quadruplex DNA, but also stabilizes quadruplex folding.
Autonomous binding of mutp53 to G/C-rich genomic DNA
The EMSA data support the notion that mutp53 can bind autonomously to structured DNA within gene regulatory regions. However, we also considered that mutp53 in U251 cells might interact with SP1 and ETS1, i.e., transcription factors that have been reported to interact with mutp53 and also bind to G/C-rich DNA motifs.Citation30,Citation31 Both SP1 and ETS1 are located in the nuclei of U251 cells, where they are associated with chromatin (Fig. S3A and B). We performed reciprocal co-IPs under conditions optimized for chromatin-associated proteins, and found that despite high expression of mutp53 and SP1, the fraction of both proteins forming a complex is very low (Fig. S4). Similarly, ETS1 protein was not, or only hardly, detectable after pull-down with a p53-specific antibody (Fig. S4).
As mutp53 might nevertheless cooperate with SP1 and ETS1 in a chromatin context, we analyzed by ChIP-chip binding of SP1 and ETS1 within genes covered by our tiling array. Applying the peak calling procedure used before for mutp53 ChIP-chip samples, we identified ETS1 and SP1 binding regions, which, compared with mutp53-BR, infrequently overlap with CpG islands and DNaseI-HS regions (; Table S2). Considering that the tiling array was not designed for ETS1 and SP1 target genes, it is not surprising that ETS1- and SP1-binding regions are less often located at TSS of genes covered by the tiling array (Fig. S5A and Table S2). The specificity of ETS1 and SP1 ChIP is supported by motif analysis of ETS1- and SP1-binding regions located at TSS. Using 10 nt as maximum motif length, we, by using the MEME-ChIP sequence analysis tool,Citation32 identified the GC-rich motifs typical for these TFs (Fig. S5B). Direct comparison of binding regions identified for mutp53, ETS1 and SP1 by our ChIP-chip studies revealed that only a small fraction of mutp53-BR may also serve as binding sites for either ETS1 or SP1 or both TFs (). Furthermore, the longer median length of mutp53-BR compared with that of ETS1 and SP1 binding regions also argues for an independent interaction of mutp53 with DNA.
Figure 3. Array-wide analysis of mutp53, SP1 and ETS1 binding sites. (A) Distribution of the scores calculated for the overlap of mutp53, ETS1 and SP1 binding regions with CpG-islands and DNaseI-HS regions. The distribution was used to identify a significance threshold, where peaks with a score > = 20 are assumed to overlap significantly. (B) Venn-diagram displaying the number of overlapping mutp53, ETS1 and SP1 binding regions.
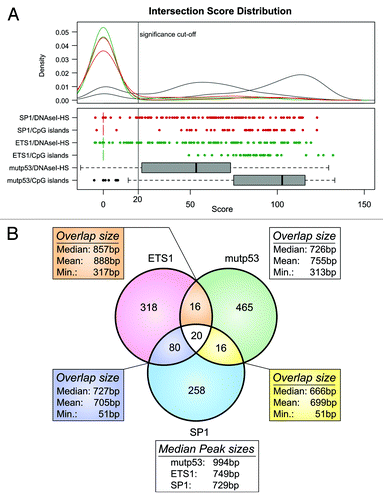
Taking into account that a recent study has shown significant overlap of SP1 binding sites with G-quadruplex motifs and in vitro binding to such a structure,Citation33 we compared the occurrence of G4 motifs in SP1, ETS1 and mutp53 binding regions identified in our experiments and in ChIP-Seq studies from the ENCODE project.Citation34,Citation35 To adjust for potential experimental bias that might obscure a binding preference to G4-motifs around TSSs, we restricted the ETS1, SP1 and mutp53 binding peaks to those located in a window of ± 1 kb around TSSs and calculated the enrichment z-scores to quantify the overlap between binding peaks and G4 motifs. In our data set we observed higher scores for mutp53 binding peaks in comparison to the ETS1 and SP1 binding peaks, suggesting a more frequent and specific binding of mutp53 in G4-rich regions (Fig. S5C). To emphasize this observation, the enrichment z-scores were also calculated for ETS1 and SP1 ChIP-Seq data derived from the ENCODE TF Binding Supertrack (contains computed peaks from different cell lines) and ENCODE/HAIB track (contains peaks from individual cell lines and replicates). This analysis revealed an even less frequent binding of ETS1 and SP1 to G4-motifs in three and five cell lines, respectively, analyzed in the ENCODE/HAIB project (Fig. S5C).
Altogether, our data strongly support autonomous binding of mutp53 to regulatory DNA sequences with high G/C content and provide evidence for preferential binding of mutp53 to G4-motif-enriched regions around TSSs. Owing to such DNA binding properties, we assume that mutp53 has the potential to influence transcription of a large number of genes.
Transient depletion of mutp53 influences transcription of multiple genes
To elucidate the role of mutp53 in the regulation of gene transcription, we transiently depleted mutp53 via siRNA. We observed a high metabolic stability of the mutp53 protein in U251 cells, as the amount of mRNA is already reduced ~10-fold 48 h after transfection (Fig. S6A), while the protein level starts to decrease only 48 h post-transfection, reaching a > 10-fold reduction at 72−96 h after transfection (Fig. S6B, upper panel). The mutp53 level is completely restored around 216 h post-transfection (Fig. S6B, lower panel). Transient depletion of mutp53 resulted in a slightly decelerated growth of p53-siRNA transfected cells as compared with control cells treated with scrambled siRNA (Fig. S6C). The cell growth advantage conferred by mutp53 is even more pronounced upon stable depletion. On an example of a cell clone derived from the U251 cells stably transfected with p53-specific shRNA,Citation21 we observed significant decrease in soft agar cloning efficiency (Fig. S6D) and propagation of neurospheres (Fig. S6E). As both assays are widely used to assess the cancer stem-like properties of tumor cells, we conclude that the long-term effect of mutp53 depletion is a reduction of the tumorigenic properties in U251 cell line.
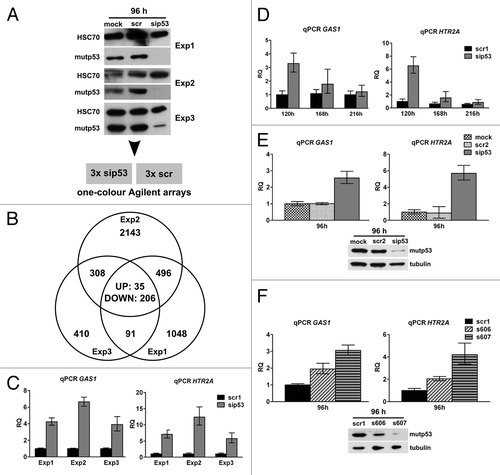
For microarray gene expression analysis, U251 cells were transfected either with p53-specific (sip53) or control siRNA (scr) and harvested 96 h post-transfection, where mutp53 depletion was at its maximum and had been effective for at least 24 h (). Bioinformatic analysis of microarray data from three independent transfection experiments with a cut-off of SLR (signal-log-ratio) = 0.8 yielded 4,737 probes corresponding to 3,679 differentially regulated genes (“compound list,” Table S3) upon mutp53 depletion, with an overlap of 241 (35 UP and 206 DOWN) probes representing 209 genes regulated in common (). The total number of regulated genes per experiment correlated with the effectiveness of mutp53 depletion; the degree of regulation was, with a few exceptions, usually moderate. Furthermore, only active genes were regulated, as these genes were transcribed before and after mutp53 depletion. Interestingly, PPARGC1A and FRMD5, two genes we have previously described as mutp53 target genes in U251-derived cell clones with stable mutp53 reductionCitation21 were only slightly regulated 96 h after transient mutp53 depletion (1.92- and 1.55-fold respectively, Fig. S6F), indicating varying kinetics of transcriptional regulation after mutp53 depletion.
Figure 4. Expression analysis in U251 cells after mutp53 depletion. (A) U251 cells were transfected with p53- and scr-siRNA in three biological replicates and mutp53 protein was detected 96 h post-transfection by WB. HSC70 was used as a loading control. (B) Total RNA from three replicate transfections with p53- and scr-siRNA was subjected to microarray gene expression analysis. The Venn diagram shows the number of probes regulated in three experiments. (C) Transcript levels of GAS1 and HTR2A genes after mutp53-depletion were measured relative to the scr-control (scr1) in the same samples that had been subjected to microarray analysis. (D) Transcript levels of GAS1 and HTR2A genes were measured in a time-course experiment after mutp53-depletion until re-expression of mutp53 occurred (for WB, see Fig. S6B). (E and F) Transcript levels of GAS1 and HTR2A genes were measured 96 h after mutp53 depletion in two additional experiments including mock transfections and a different scr-siRNA (scr2; E) as well as two different p53-siRNAs (s606, s607) (F). WB analysis of mutp53 expression in transfected cells is shown below the graphs. Tubulin was used as a loading control.
To correlate changes in gene expression with mutp53 binding, we compared the list of genes regulated after mutp53-depletion with mutp53-BR (p value 0.001) derived from ChIP-chip experiments. Out of 634 genes containing a mutp53-BR close to their TSS, only 165 genes were differentially regulated in at least one siRNA experiment (“compound list”). The finding that a large number of genes with mutp53-BR close to their TSS are not regulated upon mutp53-depletion, as well as the small fraction of genes commonly regulated in three siRNA experiments (n = 209) and varying kinetics for different genes, indicate that the transcriptional response after mutp53 depletion displays an inherent plasticity. We conclude from these results that mutp53 is not the sole determinant of transcriptional changes, but rather acts as a co-factor in a framework determined by additional stimuli and the physiological context.
Validation of putative mutp53 target genes
In contrast to deterministic regulation of a defined set of target genes by classical transcription factors, data on the transcriptional regulation by mutp53 point toward a cell, and context-specific impact of mutp53 on transcription.Citation5 Therefore, mutp53-regulated candidate genes must be unambiguously validated in the experimental system analyzed before addressing the mechanism(s) underlying transcriptional regulation by mutp53. We performed a series of rigorous validation experiments, including mutp53 knockdown with three different p53-specific siRNAs, two scrambled controls as well as mock transfected samples and selected GAS1 and HTR2A as models genes that were consistently and most strongly regulated in all validation experiments (−F).
In a time-course experiment, where siRNA-transfected cells were kept in a culture until mutp53 was re-expressed, regulation of GAS1 and HTR2A genes strictly correlated with mutp53 protein levels (Fig. S6B). One hundred and twenty hours post-transfection, when mutp53 protein levels are strongly reduced, the genes were still significantly upregulated. But around 216 h post-transfection, when mutp53 protein reached its normal level again, transcription of GAS1 and HTR2A returned to the levels observed in control siRNA transfected cells (). Upregulation of GAS1 and HTR2A could also be observed upon transfection with a different control siRNA () and two commercially available, validated siRNAs (), and again correlated with the reduction efficiency of mutp53 as observed by western blotting (). As outlined above, cell context plays an important role in the regulation of genes by mutp53, as genes consistently regulated in U251 cells (e.g., GAS1, HTR2A, PMEPA1 and BDKRB1) showed no mutp53-dependent changes in transcription in two other tumor cell lines, HT29 and MDA-MB-468, that express mutp53 (R273H) (Fig. S7).
Taken together, we identified GAS1 and HTR2A as genes regulated concomitantly with mutp53 levels in U251 cells, thereby qualifying these genes for further analysis regarding the underlying mechanism(s) of transcriptional regulation by mutp53.
Mutp53 regulates genes at the transcriptional level
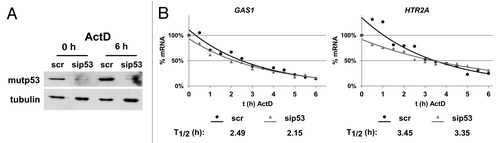
The transcript levels of genes can either be regulated by modulating their transcription or by changes in mRNA stability. We first checked whether upregulation of GAS1 and HTR2A genes upon mutp53 depletion results from altered mRNA stability. U251 cells were transfected with control and p53-specific siRNAs, and mRNA stability was assessed after actinomycin D (ActD) treatment at the time of maximal mutp53 depletion (96 h after transfection) (). Transcript levels of GAS1 and HTR2A genes were measured every 30 min for 6 h after ActD treatment and the half-life of the transcripts was calculated (). In case of the GAS1 gene, the observed half-life of 2.49 h matches a half-life of ~2 h that has been measured in mouse cells.Citation36 No significant increase in mRNA stability that could account for a 4–12-fold upregulation was observed for GAS1 and HTR2A genes, strongly arguing for transcriptional regulation of both genes.
Figure 5. Stability of GAS1 and HTR2A transcripts after mutp53-depletion. (A) Mutp53 protein levels at the start and at the end of ActD treatment. U251 cells were transfected with p53- and scr-siRNA. At the time of maximal mutp53 depletion (96 h), transcription was stopped by addition of ActD (1 µg/ml final concentration) and RNA was isolated every 30 min for 6 h. Tubulin was used as a loading control. (B) GAS1 and HTR2A transcript levels in scr- and sip53-transfected U251 cells were measured by qPCR for every time point after ActD treatment, normalized to GAPDH and calculated as %-mRNA remaining. The mRNA half-life was calculated using an exponential regression curve derived from the data points.
Mutp53 regulates active genes
Transcription of genes can be controlled by selective binding of transcription factors, recruitment of the basal transcription machinery and co-factors, regulation of RNA polymerase II (Pol II) initiation and elongation as well as by epigenetic mechanisms like histone modifications and chromatin accessibility. In selecting the levels of transcriptional control to be tested, we considered that the transcripts of most genes regulated by p53 siRNA in U251 cells, including GAS1 and HTR2A genes, were readily detectable in mutp53-depleted and control-transfected U251 cells, indicating that their promoters were already active before depletion of mutp53. Therefore, reversion from a stably silenced gene status is rather unlikely to play a role in the upregulation of GAS1 and HTR2A genes by mutp53 depletion, and we thus excluded an analysis of Polycomb complex binding, heterochromatin marks and DNA methylation. Instead we focused on changes in active histone marks, H3K4me3 and H3K9ac, and Pol II recruitment and processivity. H3K4me3 and H3K9ac are found to be enriched in active promoter regions,Citation37,Citation38 and the phosphorylation of serine 5 (S5-P) and serine 2 (S2-P) in the CTD of Pol II has been described to define initiated and elongating Pol II complexes, respectively.Citation39,Citation40
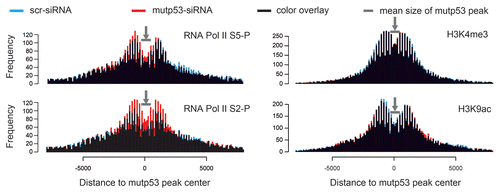
U251 cells were transfected with control and p53-siRNAs, and 96 h later, cells were subjected to ChIP and analyzed on our custom tiling array to obtain a map of histone marks and Pol II S5-P/S2-P distribution (see data in Table S4). In total, 69% and 65% of all genes covered by the array, and 83% and 76% TSS-associated mutp53-BR, carry H3K4me3 and H3K9ac around TSSs (± 2 kb) in U251 cells. In general, comparing control and p53 siRNA-treated cells, we observed slight differences in the occurrence of H3K4me3 and H3K9ac in the vicinity of mutp53-BR after mutp53 depletion across the array (): a slight decrease of H3K4me3 at the flanks of mutp53-BR and a slight decrease of H3K9ac within mutp53-BRs. These global changes correlate with the prevailing downregulation of genes as a consequence of mutp53 depletion. On the other hand, an increase in Pol II S5-P/S2-P within mutp53-BRs can be observed in mutp53-depleted cells in comparison to control cells (), which correlates with increased transcription of some genes including GAS1 and HTR2A. The signal drop in the center of the bimodal-like distribution patterns for the histone modifications, and Pol II S5-P/S2-P () indicates that a significant portion of mutp53-BR only partially overlaps with H3K4me3/H3K9ac regions and sites occupied by initiating and elongating RNA Pol II.
Figure 6. Histograms displaying the frequency of H3K4me3, H3K9ac, Pol II-S2P and Pol II-S5P peaks in the vicinity of mutp53 binding regions (mutp53-BR). The distance is displayed relative to the mutp53-BR center positions. The profiles were determined for control siRNA (blue) and p53 siRNA (red) treated samples and superimposed (combined color: black).
Focusing on the GAS1 and HTR2A genes, we detected a high incidence of H3K4me3 in the GAS1 () and HTR2A () genes with a peak around the TSS, marking both promoters as active. For the GAS1 gene, there is also a strong signal in the 3′-UTR, while the translated region of the gene is nearly devoid of H3K4me3. In the HTR2A gene, the H3K4me3 signal is restricted to an area stretching from the promoter to exon 3. A slight extension of the H3K4me3 signal upon mutp53 depletion can be observed in the HTR2A gene and in the 3′-UTR of the GAS1 gene, where it is accompanied by increased signal strength. Distribution of H3K9ac in the GAS1 gene reflects H3K4me3 distribution. For the HTR2A gene, a tendency for higher H3K9ac signals in mutp53-depleted cells can be observed. Taken together, the profiles for active histone marks H3K4me3 and H3K9ac correlate with an elevated transcription of the GAS1 and HTR2A genes upon mutp53 depletion, as they show a broadening and/or increase in signal intensity when transcription increases. The ChIP-chip profiles for Pol II S2-P and S5-P show a much more pronounced effect upon mutp53 depletion. Both signals for Pol II S2-P and S5-P are significantly increased (2–4 fold) in the promoter regions of GAS1 and HTR2A genes and also in the GAS1 3′-UTR, indicating a strong increase in initiated and also elongating Pol II complexes on the GAS1 and HTR2A genes. In total, 48 mutp53-BR (10.7% of 449 mutp53-BR associated with TSS) show either the appearance of Pol II S2-P-covered regions around the mutp53-BR, or expansion of the existing Pol II S2-P regions upon mutp53 depletion, indicating a repressive function of mutp53 at the elongation step in these genes.
Figure 7. (A, B) ChIP-chip profiles of H3K4me3, H3K9ac, Pol II S2-P and Pol II S5-P occupancy in the GAS1 (A) and the HTR2A (B) genes in scr- and sip53-transfected U251 cells. Mean log2-values of the signal ratio “ChIP-sample/input” for every probe on the tiling array from two biological replicates are plotted against the probe position. The transcription start site (TSS) is marked by an arrow and exons are highlighted. (C, D) A qPCR-based analysis of a representative experiment for the promoter regions of GAS1 (C) and HTR2A (D). Data for scr- and p53-siRNA-transfected cells are presented as percent recovery of input DNA by the specific antibodies. A control without addition of antibodies is shown. The results of the three replicate experiments are summarized as fold change of mutp53-depleted cells vs. control cells.
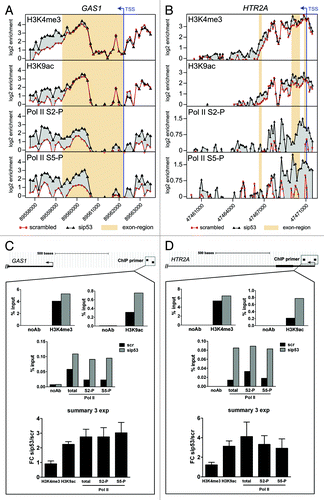
To validate the ChIP-chip results, we used qPCR, employing primers that map to the enriched areas observed in the promoter regions of the GAS1 and HTR2A genes. The same samples that had been used for the ChIP-chip experiments, and an additional replicate sample, were subjected directly to qPCR analysis without WGA pre-amplification. Furthermore, samples from ChIP experiments using an antibody against Pol II were included to assess total Pol II occupancy. By qPCR we could confirm the ChIP-chip data for both the GAS1 () and HTR2A () genes and extend their significance. While there was no differences between mutp53 depleted and control cells in the level of H3K4me3 at both, GAS1 and HTR2A genes, in the case of H3K9ac, owing to the higher sensitivity of qPCR compared with tiling array hybridization, a 2–3-fold increase at the GAS1 and HTR2A promoters can be observed upon mutp53 depletion, which correlates with increased transcription. Accordingly, Pol II occupancy is also increased 2–4-fold and is accompanied by a ~3-fold increase in Pol II S5-P/S2-P that indicates enhanced initiation and elongation. In another control experiment, we validated mutp53 binding within the GAS1 and HTR2A genes and confirmed the ChIP-chip data (Fig. S8).
In summary, the analysis of the transcriptional status of mutp53-regulated genes demonstrates that mutp53 generally modulates transcription from active promoters. An increase in the activating histone mark H3K9ac, connected to transcription initiation as well as increased recruitment and processivity of Pol II, can be detected. These results indicate that mutp53 has the ability to participate in several regulatory steps of active transcription by either interfering with early or superior events or by modulating each single step.
Discussion
Oncogenic functions of mutant p53 proteins and their underlying molecular basis have been the subject of extensive research over the past decade, and transcriptional regulation of target genes has been established as an important aspect of mutp53 activity.Citation5,Citation6 The current focus is on the search for co-operative interactions with sequence-specific TFs that recruit mutp53 to their target genes. In this scenario, DNA binding by mutp53 could potentially influence the dynamics of the transcription machinery on a target promoter, but is not necessarily essential for mutp53 function. Accordingly, the function of mutp53 would be restricted to the modulation (enhancement or repression) of transcription initiated and exerted by sequence-specific TFs that are able to interact with mutp53. As a consequence, mutp53 should regulate a set of target genes that is shared between different cellular contexts and is determined by these interaction partners of mutp53. However, evidence for the universality of mutp53 target genes across different experimental systems so far is missing,Citation5 and we here show that some genes consistently regulated in U251 cells are not regulated by the same mutp53 (R273H mutation) in other human tumor cell lines. Therefore, we believe that the range of mutp53 transcriptional modulation must be expanded. In this regard, direct binding of mutp53 to DNA might provide an additional means, as it could be critical for the selection of target genes.
Using a custom tiling array, we identified in U251 cells a significant number of reproducible mutp53 binding regions frequently located close to TSSs and overlapping with DNaseI-HS regions and CpG-islands. Notably, a number of promoters that were reported to be regulated by bound mutp53 in other studies also overlap with CpG islands, e.g., MYC,Citation41 ID2,Citation42 ID4,Citation16 TWIST1,Citation9 EGR1,Citation22 MAP2K3,Citation43 target genes of the transcription factors VDR (CYP24A1)Citation14 and NF-Y (CCNA1, CCNB2, CDK1, CDC25C),Citation13 genes containing sterol regulatory elements in their promoters (CYP51A1, FDPS, FDFT1, HMGCR, HMGCS1, MVK, SQLE)Citation17 and the TDP2 gene.Citation18 G/C-rich DNA stretches are of particular interest regarding gene regulation. They are not only subjected to epigenetic control via cytosine methylation, but under favorable conditions (e.g., superhelical tension and stabilization by bound proteins), are also prone to form left-handed (Z), triplex (H), cruciform and G-quadruplex structures, which have been implied in transcriptional regulation.Citation44 Although the existence and accessibility of these predicted non-B DNA structures in the context of chromatin are not yet sufficiently explored, G-quadruplex-forming sequences are overrepresented in gene-promoter regions,Citation45 especially of proto-oncogenes including the MYC gene.Citation46 Consequently, our findings that G4 motifs are enriched in mutp53R273H-binding sites, combined with significant binding of three p53 mutants to G-quadruplex structures in vitro (a property shared by wtp53) and promotion of G4-folding by mutp53, provide a basis for the regulation of a large set of cancer-relevant genes. At this point it is tempting to speculate that a decline in the histone modifications and Pol II S2-P/S5-P profiles within some mutp53 binding sites () might point toward binding of mutp53 to accessible DNA stretches that can adopt non-B DNA conformation. Our EMSA and CD spectroscopy data support direct binding of mutp53 and stabilization of the proposed G-quadruplex structures. However, further studies are needed to elucidate the mode of mutp53 binding to structured G/C-rich DNA of active genes, particularly in a chromatin setting and for different classes of mutants (i.e., contact mutations vs. conformation and protein structure destabilizing mutations).
In previous studies, we have established the important role of DNA structure for wtp53 and mutp53 DNA binding and proposed that retention of this feature in mutp53 serves as a means to modulate transcription.Citation21,Citation47 In this scenario, DNA structure-specific binding preserved from wtp53, combined with stabilization of the mutp53 protein and high-expression level, are drivers for different outcomes despite a common binding feature, ultimately resulting in mutp53 GOF. Whether this mode of action based on DNA structure recognition, especially G-quadruplexes, is a common feature of different p53 mutants, remains to be determined. It is possible that the capacity for structure-dependent interactions with DNA will vary for different p53 mutants depending on the degree of protein structure alterations. As a result, the mechanism of mutp53 GOF might be different depending on the mutation, and different mutp53 species may need cooperation with other regulators to achieve tumor-promoting effects. It would be interesting to determine in future studies whether there is a correlation between the degree of protein structure disruption and a decreasing capacity to bind structured DNA coupled with an increasing need for protein-protein interactions for the GOF of different p53 mutants.
Addressing the mechanistic basis of transcriptional regulation by mutp53R273H, we observed a significant transcriptional response upon mutp53 depletion that is characterized by considerable variability regarding the pattern of regulated genes. Only a fraction of the genes with mutp53 binding sites close to their TSS were regulated upon transient mutp53 depletion, indicating that mutp53-binding is a prerequisite rather than the cause for subsequent modulation of transcription. Furthermore, the effects seem to be cell type-specific, as several validated target genes were not regulated upon mutp53 depletion in two other cell lines carrying the same R273H mutation. Consequently, mutp53R273H does not act as a classical deterministic transcription factor, but rather functions as a co-factor that has the potential to regulate a large number of genes on the basis of numerous binding sites. However, the decision, whether transcription from a certain promoter is modulated, seems to depend on cell context and given stimuli. This notion is well in line with several recent publications on transcriptional regulation by mutp53. First, different p53 hotspot mutants seem to create comparable transcription signatures in the same cellular system, as has been shown for mutp53 ectopically expressed in p53-null H1299 cells,Citation48-Citation50 prostate cancer cellsCitation51 as well as in breast cancer,Citation52-Citation54 which strongly argues for cell type specificity. Second, several effects of mutp53 on transcription of target genes of the transcription factors NF-Y, VDR and NF-kB, seem to require specific stimuli, like DNA damage, vitamin D or TNF-α treatment, respectively.Citation13,Citation14,Citation55
The fact that 83% of mutp53-bound promoters in U251 cells carry H3K4me3 indicates an active chromatin status of genes potentially regulated by mutp53. A detailed analysis of the mutp53-regulated genes GAS1 and HTR2A, as well as further genes containing reproducible mutp53 binding sites around their TSS, revealed alterations in active histone marks as well as RNA Pol II occupancy and processivity upon mutp53 depletion. These results indicate that mutp53 almost exclusively modulates ongoing transcription, illustrating that it acts on a higher level of regulation by either affecting early steps of transcription or by impinging on multiple steps of transcriptional regulation. The molecular basis for this effect could be modulation of the activity of promoter-bound factors, as well as recruitment or exclusion of additional factors, or a combination of all three scenarios, resulting in alteration of the transcriptional program.
It is widely accepted that recruitment of mutp53 to promoters of regulated genes through protein-protein interactions is an important mode of mutp53 function. However, analyzing two TFs, SP1 and ETS1, known to bind G/C-rich consensus motifs, we did not find significant overlap of the respective binding sites and detected only weak interactions with mutp53R273H. Therefore, our results rather argue for at least SP1- and ETS1-independent binding of mutp53 to G/C-rich DNA in U251 cells. Low overlap of SP1 binding sites with CpG islands and G-quadruplex motifs in our study compared with an in silico analysis of a ChIP-chip data setCitation33 could result from our p53-specific array design, but could also be attributed to the special biological setting in U251 cells. Mutp53 is expressed at very high levels and might replace SP1 during competition for G-quadruplex structures. The study by Raiber et al.Citation33 is based on ChIP-chip data derived from colon cancer and leukemia patients,Citation56 but there is no information regarding p53 mutation status of these patients. It would be interesting to correlate the occurrence of p53 missense mutations in these tumor patients with the possible impact of mutp53 on SP1 binding to G-quadruplex structures. Considering the fact that SP1 is a ubiquitous TF, a competition with or even partly exclusion of SP1 from regulatory sequences could have a profound effect on a cell’s transcriptome and might provide a basis for large-scale modulation of transcription by mutp53. Differences in the prevalent transcriptome resulting from different biological background (i.e., glioblastoma, colon cancer and leukemia) would very likely create context-dependent effects. Importantly, however, autonomous DNA binding of mutp53 and the co-recruitment model are not mutually exclusive, and direct binding of mutp53 to structured DNA, added to the well-established modulation of transcription via interaction with other TFs, would provide a basis for the large biological spectrum of cancer settings where mutp53 GOF is observed. Interestingly, the SP1 ChIP-chip studyCitation56 demonstrates that TF binding correlates with resistance to de novo methylation in cancer cells. It is tempting to speculate that in our experimental system, mutp53R273H might exert a similar effect, which is supported by the observations that mutp53 binding strongly correlates with active chromatin marks (H3K9ac, H3K4me3) and that the majority of genes is downregulated upon mutp53 depletion.
Considering the large number of potential binding sites (e.g., G4 motifs within CpG-islands), mutp53 has the potential to contribute to the regulation of cell type/stimulus-specific transcription on a global scale as an epigenetic factor, supporting changes of the transcriptional program. Indeed, mutp53 contributes to induction of EMT over several passages by alleviation of the epigenetic repression of TWIST1, a master regulator of EMT.Citation9 Furthermore, mutp53 strongly enhances reprogramming efficiency and can replace one of the classical reprogramming factors, but the resulting cells are highly tumorigenic.Citation57 The “epigenetic alteration of transcription” by mutp53 would very likely be intrinsically stochastic due to slight differences in the transcriptional status of single cells. In fact, the observed variability in the gene expression pattern observed upon mutp53 depletion supports such a stochastic model for gene regulation by mutp53. Taking into account that DNA binding by mutp53 is non-selective and dynamic, the process of gradual mutp53 depletion by siRNA will lead to a gradual loss of mutp53 from its binding sites, accompanied by chromatin rearrangements on a global scale. Such rearrangements are likely to occur in a stochastic rather than a deterministic manner and will change the binding patterns of the residual mutp53, with concomitant stochastic alterations in the gene expression pattern, which, in the end, manifests in the observed heterogeneity in the transcriptional response upon mutp53 depletion.
The mutp53-supported transcriptional plasticity would give rise to multiple phenotypes from the same genotype and confer a considerable selective advantage to a tumor cell population by greatly enhancing adaptability. Indirect evidence for this model comes from our gene expression analysis of cells with transient depletion of mutp53. When comparing expression levels in mutp53-depleted cells and in control transfected cells to an internal reference value, the deviation in mutp53-depleted samples is smaller than in control samples (Fig. S9). This observation indicates that under our experimental conditions (lipophilic transfection of siRNA), depletion of mutp53 globally reduces the range of transcriptional variability. Based on this, we suggest a model that links mutp53 function to the maintenance of a heterogeneous transcriptional output. As a transcriptional co-factor, mutp53 contributes to multiple phenotypic states that are constrained by the differentiation program. In the case of U251 cells, the differentiation program is epigenetically inherited from the glioma-initiating cells, presumably from aberrant neural stem/progenitor cells.Citation58 By non-selective, preferential binding to G/C-rich sequences around TSS of active genes, mutp53 can help to keep them in an open, transcriptionally competent state, which may vary from a poised state to a steadily active state, thereby creating the tissue-specific phenotypic profile. Should the expression and activity of mutp53 be downregulated at the transcriptional and/or post-transcriptional level or impeded by interacting proteins, this would allow the drift of the tumor cell population along the differentiation program.
In summary, performing an integrative analysis on a model cell line, we conclude that an important aspect of mutp53 GOF is based on its direct binding to regulatory regions of active genes, where mutp53 contributes to the plasticity of the transcriptional output as well as to transcriptional competence of genes, and simultaneously maintains the phenotypic variability of tumor cells.
Material and Methods
Cell culture
The human glioblastoma cell line U251 was a kind gift from Dr. Kazuo Washiyama (Brain Research Institute, Niigata University). Mutation (R273H) in the TP53 gene has been confirmed by sequencing of PCR-amplified p53 cDNA. The cells were maintained in Dulbecco's modified Eagle medium (DMEM) (Invitrogen) supplemented with 10% fetal calf serum (FCS; PAA Laboratories GmbH) in a humid atmosphere containing 5% CO2. Generation of the mutp53-depleted U251 clone sh10 has been described elsewhere.Citation21 To analyze RNA stability, transcription was blocked by addition of actinomycin D (Sigma-Aldrich) to a final concentration of 1 µg/ml to the culture medium and samples for RNA preparation were collected over 6 h. Procedures for siRNA transfection, neurosphere culture and agar cloning are described in Supplementary Methods.
ChIP-chip on a custom tiling array
Cells were cultured in 15-cm diameter tissue culture dishes. At 80% confluence, cells were washed with warm PBS (pH 7.4) and were either treated directly with DMEM supplemented with 1% formaldehyde (Sigma-Aldrich) for 10 min at room temperature or subjected to a nuclear extraction procedure (see Supplementary Methods) to remove the cytosolic fraction before crosslinking. Procedures for chromatin immunoprecipitation (ChIP) are described in Supplementary Methods. Input and ChIP samples were amplified using the GenomePlex Whole Genome Amplification Kit (Sigma). Ten nanograms of input DNA and 10 µl of the ChIP samples were used for WGA performed according to the manufacturer’s instructions. For some ChIP samples, two rounds of amplification were necessary. After amplification, samples were purified by phenol-chloroform-isoamylalcohol (25:24:1; Biomol GmbH), extraction with subsequent precipitation with ethanol in the presence of NaCl (200 mM final concentration) and 20 μg glycogen (Roche), washed with 70% ethanol, air-dried and dissolved in water.
The Nimblegen custom 135K tiling array was designed to cover 1894 genomic intervals (Table S1) that comprise 902 coding and non-coding genes/regions and 1,040 non-coding regions in a length of 1 kb (for use in other p53-related projects). Eight hundred and twelve tiling intervals cover single genes; 36 tiling intervals cover two genes, and six tiling intervals cover three genes. Gene coordinates (GRCh37: Genome Reference Consortium Human genome built 37), including flanking regions in a length of 1,500 bp, were retrieved from Biomart (www.ensembl.org/biomart/martview). The gene list for the array (Table S1) was compiled from the pilot gene expression experiments performed to identify genes that are regulated dependent on mutp53 and wtp53 expression in U251 (endogenous mutp53-R273H expression) and H1299 (ectopic mutp53-R273H, mutp53-G245S and wtp53 expression) cell lines. Fabrication of 50-mer oligonucleotide arrays, DNA labeling, hybridization and image data processing was completed by Source BioScience imaGenes GmbH. Procedures for bioinformatic analysis of ChIP-chip data are described in Supplementary Methods.
ChIP-qPCR
PCR-based analysis of ChIP DNA was performed on an ABI 7900HT Fast thermal cycler (Applied Biosystems) in a standard program (10 min 95°C; 15 sec 95°C, 1 min 60°C; 40 cycles) using the Power SYBR Green PCR Master Mix (Applied Biosystems). Amplicons to measure mutp53 binding, histone modifications, Pol II, Pol II S5-P- and Pol II S2-P occupancy were selected in the respective peak regions determined by prior ChIP-chip experiments, and primers (Table S5) were designed using the Primer3 web tool. Input samples were diluted 1:10, IP samples 1:2 and run in duplicates. Quantification was performed with the help of a standard curve and results were calculated as percent recovery of input DNA. For mutp53 binding site validation, samples were run in triplicates and values from three mutp53-ChIPs were calculated as fold change over background by normalization to the β-globin gene (HBB).
Gene expression microarray analysis
Microarray hybridization was performed on Agilent-014850 Whole Human Genome Microarray 4x44K G4112F chips. Total RNA was treated with RNase-free DNase I (Qiagen) and purified. The quality and integrity of the total RNA was evaluated with the 2100 Bioanalyzer (Agilent Technologies). Further sample handling, labeling, hybridization on one channel arrays and data extraction was done by Source BioScience imaGenes GmbH. Raw gene expression signals were processed using the ComBat script for R statistical platform (ver. 2.11.00)Citation59 to remove batch effects observed in the data. For further analysis, the calculated expression values were restricted to those genes with a valid sequence mapping status defined by a manually revised platform annotation. Redundant expression values of genes with multiple probes on the array were collapsed by mean value calculation, and a threshold expression value of 1 was set preceding log2 transformation.
To detect genes differentially expressed in response to p53-siRNA treatment, samples were compared with their matched scr-siRNA-treated control samples. Genes were assumed potentially regulated if the expression value of either of the two samples compared exceeded a value of log2(100) and the absolute signal-log-ratio (SLR) was above 0.8 (Table S3). Statistical calculation and plotting of the Venn diagrams was done using R statistical platform (www.R-project.org/).
To visualize the global changes induced by mutp53 depletion, the scr- and p53-siRNA-treated samples were compared with an internal reference. This artificial reference sample was generated by calculating a robust Tukey’s biweight value for every gene over all six siRNA treated samples plus the non-transfected U251 sample. Genes were assumed potentially regulated if the expression value of either of the two samples compared exceeded a value of log2(100) and the SLR was above 1.
Additional methods
Procedures for quantitative real-time PCR, immunofluorescence microscopy, co-immunoprecipitation, immunoblotting, DNA binding assays and CD spectroscopy are described in Supplementary Methods.
Accession Numbers
The raw ChIP-chip and gene expression data used in our work have been made available in the GEO repository with accession number GSE35500.
| Keywords: | ||
| mutant p53 | = | transcription, chromatin, ChIP-chip, G-quadruplex |
Additional material
Download Zip (5.9 MB)Acknowledgments
We thank Marion Kühl for invaluable assistance in many experiments. This work was supported by funding from EC FP6 (to W.D.) and the German-Israeli Foundation (G.I.F. No. I-927–187.13/2006 to W.D.). The senior professorship of W.D. is supported by the Jung Foundation for Science and Research, Hamburg. The work by M.B. and I.K. was supported by the Czech Science Foundation (P301/10/2370 to M.B.), and by the project “CEITEC – Central European Institute of Technology” (CZ.1.05/1.1.00/02.0068) from European Regional Development Fund.
Related Research Data
References
- Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer 2009; 9:749 - 58; http://dx.doi.org/10.1038/nrc2723; PMID: 19776744
- Beckerman R, Prives C. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol 2010; 2:a000935; http://dx.doi.org/10.1101/cshperspect.a000935; PMID: 20679336
- Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2010; 2:a001008; http://dx.doi.org/10.1101/cshperspect.a001008; PMID: 20182602
- Kim E, Deppert W. Interactions of mutant p53 with DNA: guilt by association. Oncogene 2007; 26:2185 - 90; http://dx.doi.org/10.1038/sj.onc.1210312; PMID: 17401427
- Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer 2009; 9:701 - 13; PMID: 19693097
- Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol 2010; 2:a001107; http://dx.doi.org/10.1101/cshperspect.a001107; PMID: 20182618
- Blandino G, Levine AJ, Oren M. Mutant p53 gain of function: differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 1999; 18:477 - 85; http://dx.doi.org/10.1038/sj.onc.1202314; PMID: 9927204
- Bossi G, Lapi E, Strano S, Rinaldo C, Blandino G, Sacchi A. Mutant p53 gain of function: reduction of tumor malignancy of human cancer cell lines through abrogation of mutant p53 expression. Oncogene 2006; 25:304 - 9; PMID: 16170357
- Kogan-Sakin I, Tabach Y, Buganim Y, Molchadsky A, Solomon H, Madar S, et al. Mutant p53(R175H) upregulates Twist1 expression and promotes epithelial-mesenchymal transition in immortalized prostate cells. Cell Death Differ 2011; 18:271 - 81; http://dx.doi.org/10.1038/cdd.2010.94; PMID: 20689556
- Muller PA, Vousden KH, Norman JC. p53 and its mutants in tumor cell migration and invasion. J Cell Biol 2011; 192:209 - 18; http://dx.doi.org/10.1083/jcb.201009059; PMID: 21263025
- Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality?. Nat Med 2009; 15:1010 - 2; http://dx.doi.org/10.1038/nm0909-1010; PMID: 19734877
- Ram EV, Meshorer E. Transcriptional competence in pluripotency. Genes Dev 2009; 23:2793 - 8; http://dx.doi.org/10.1101/gad.1881609; PMID: 20008929
- Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A, et al. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 2006; 10:191 - 202; http://dx.doi.org/10.1016/j.ccr.2006.08.013; PMID: 16959611
- Stambolsky P, Tabach Y, Fontemaggi G, Weisz L, Maor-Aloni R, Siegfried Z, et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell 2010; 17:273 - 85; http://dx.doi.org/10.1016/j.ccr.2009.11.025; PMID: 20227041
- Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 2009; 137:87 - 98; http://dx.doi.org/10.1016/j.cell.2009.01.039; PMID: 19345189
- Fontemaggi G, Dell’Orso S, Trisciuoglio D, Shay T, Melucci E, Fazi F, et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat Struct Mol Biol 2009; 16:1086 - 93; http://dx.doi.org/10.1038/nsmb.1669; PMID: 19783986
- Freed-Pastor WA, Mizuno H, Zhao X, Langerød A, Moon S-H, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012; 148:244 - 58; http://dx.doi.org/10.1016/j.cell.2011.12.017; PMID: 22265415
- Do PM, Varanasi L, Fan S, Li C, Kubacka I, Newman V, et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev 2012; 26:830 - 45; http://dx.doi.org/10.1101/gad.181685.111; PMID: 22508727
- Göhler T, Jäger S, Warnecke G, Yasuda H, Kim E, Deppert W. Mutant p53 proteins bind DNA in a DNA structure-selective mode. Nucleic Acids Res 2005; 33:1087 - 100; http://dx.doi.org/10.1093/nar/gki252; PMID: 15722483
- Walter K, Warnecke G, Bowater R, Deppert W, Kim E. tumor suppressor p53 binds with high affinity to CTG.CAG trinucleotide repeats and induces topological alterations in mismatched duplexes. J Biol Chem 2005; 280:42497 - 507; http://dx.doi.org/10.1074/jbc.M507038200; PMID: 16230356
- Brázdová M, Quante T, Tögel L, Walter K, Loscher C, Tichý V, et al. Modulation of gene expression in U251 glioblastoma cells by binding of mutant p53 R273H to intronic and intergenic sequences. Nucleic Acids Res 2009; 37:1486 - 500; http://dx.doi.org/10.1093/nar/gkn1085; PMID: 19139068
- Weisz L, Zalcenstein A, Stambolsky P, Cohen Y, Goldfinger N, Oren M, et al. Transactivation of the EGR1 gene contributes to mutant p53 gain of function. Cancer Res 2004; 64:8318 - 27; http://dx.doi.org/10.1158/0008-5472.CAN-04-1145; PMID: 15548700
- Cesaroni M, Cittaro D, Brozzi A, Pelicci PG, Luzi L. CARPET: a web-based package for the analysis of ChIP-chip and expression tiling data. Bioinformatics 2008; 24:2918 - 20; http://dx.doi.org/10.1093/bioinformatics/btn542; PMID: 18945685
- Bock C, Halachev K, Büch J, Lengauer T. EpiGRAPH: user-friendly software for statistical analysis and prediction of (epi)genomic data. Genome Biol 2009; 10:R14; http://dx.doi.org/10.1186/gb-2009-10-2-r14; PMID: 19208250
- Cer RZ, Bruce KH, Mudunuri US, Yi M, Volfovsky N, Luke BT, et al. Non-B DB: a database of predicted non-B DNA-forming motifs in mammalian genomes. Nucleic Acids Res 2011; 39:Database issue D383 - 91; http://dx.doi.org/10.1093/nar/gkq1170; PMID: 21097885
- Siebenlist U, Hennighausen L, Battey J, Leder P. Chromatin structure and protein binding in the putative regulatory region of the c-myc gene in Burkitt lymphoma. Cell 1984; 37:381 - 91; http://dx.doi.org/10.1016/0092-8674(84)90368-4; PMID: 6327064
- Palumbo SL, Ebbinghaus SW, Hurley LH. Formation of a unique end-to-end stacked pair of G-quadruplexes in the hTERT core promoter with implications for inhibition of telomerase by G-quadruplex-interactive ligands. J Am Chem Soc 2009; 131:10878 - 91; http://dx.doi.org/10.1021/ja902281d; PMID: 19601575
- Friedlander P, Legros Y, Soussi T, Prives C. Regulation of mutant p53 temperature-sensitive DNA binding. J Biol Chem 1996; 271:25468 - 78; http://dx.doi.org/10.1074/jbc.271.41.25468; PMID: 8810317
- Kypr J, Kejnovská I, Renciuk D, Vorlícková M. Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res 2009; 37:1713 - 25; http://dx.doi.org/10.1093/nar/gkp026; PMID: 19190094
- Chicas A, Molina P, Bargonetti J. Mutant p53 forms a complex with Sp1 on HIV-LTR DNA. Biochem Biophys Res Commun 2000; 279:383 - 90; http://dx.doi.org/10.1006/bbrc.2000.3965; PMID: 11118296
- Sampath J, Sun D, Kidd VJ, Grenet J, Gandhi A, Shapiro LH, et al. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J Biol Chem 2001; 276:39359 - 67; http://dx.doi.org/10.1074/jbc.M103429200; PMID: 11483599
- Bailey TL, Bodén M, Whitington T, Machanick P. The value of position-specific priors in motif discovery using MEME. BMC Bioinformatics 2010; 11:179; http://dx.doi.org/10.1186/1471-2105-11-179; PMID: 20380693
- Raiber EA, Kranaster R, Lam E, Nikan M, Balasubramanian S. A non-canonical DNA structure is a binding motif for the transcription factor SP1 in vitro. Nucleic Acids Res 2012; 40:1499 - 508; http://dx.doi.org/10.1093/nar/gkr882; PMID: 22021377
- Myers RM, ENCODE Project Consortium. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 2011; 9:e1001046; http://dx.doi.org/10.1371/journal.pbio.1001046; PMID: 21526222
- Rosenbloom KR, Dreszer TR, Long JC, Malladi VS, Sloan CA, Raney BJ, et al. ENCODE whole-genome data in the UCSC Genome Browser: update 2012. Nucleic Acids Res 2012; 40:Database issue D912 - 7; http://dx.doi.org/10.1093/nar/gkr1012; PMID: 22075998
- Ciccarelli C, Philipson L, Sorrentino V. Regulation of expression of growth arrest-specific genes in mouse fibroblasts. Mol Cell Biol 1990; 10:1525 - 9; PMID: 1690845
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell 2007; 129:823 - 37; http://dx.doi.org/10.1016/j.cell.2007.05.009; PMID: 17512414
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007; 130:77 - 88; http://dx.doi.org/10.1016/j.cell.2007.05.042; PMID: 17632057
- Buratowski S. Progression through the RNA polymerase II CTD cycle. Mol Cell 2009; 36:541 - 6; http://dx.doi.org/10.1016/j.molcel.2009.10.019; PMID: 19941815
- Sims RJ 3rd, Belotserkovskaya R, Reinberg D. Elongation by RNA polymerase II: the short and long of it. Genes Dev 2004; 18:2437 - 68; http://dx.doi.org/10.1101/gad.1235904; PMID: 15489290
- Frazier MW, He X, Wang J, Gu Z, Cleveland JL, Zambetti GP. Activation of c-myc gene expression by tumor-derived p53 mutants requires a discrete C-terminal domain. Mol Cell Biol 1998; 18:3735 - 43; PMID: 9632756
- Yan W, Liu G, Scoumanne A, Chen X. Suppression of inhibitor of differentiation 2, a target of mutant p53, is required for gain-of-function mutations. Cancer Res 2008; 68:6789 - 96; http://dx.doi.org/10.1158/0008-5472.CAN-08-0810; PMID: 18701504
- Gurtner A, Starace G, Norelli G, Piaggio G, Sacchi A, Bossi G. Mutant p53-induced up-regulation of mitogen-activated protein kinase kinase 3 contributes to gain of function. J Biol Chem 2010; 285:14160 - 9; http://dx.doi.org/10.1074/jbc.M109.094813; PMID: 20223820
- Ohyama T. DNA conformation and transcription. Landes Bioscience, New York, NY, 2005.
- Balasubramanian S, Hurley LH, Neidle S. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy?. Nat Rev Drug Discov 2011; 10:261 - 75; http://dx.doi.org/10.1038/nrd3428; PMID: 21455236
- Eddy J, Maizels N. Gene function correlates with potential for G4 DNA formation in the human genome. Nucleic Acids Res 2006; 34:3887 - 96; http://dx.doi.org/10.1093/nar/gkl529; PMID: 16914419
- Kim E, Deppert W. The versatile interactions of p53 with DNA: when flexibility serves specificity. Cell Death Differ 2006; 13:885 - 9; http://dx.doi.org/10.1038/sj.cdd.4401909; PMID: 16543936
- Scian MJ, Stagliano KE, Anderson MA, Hassan S, Bowman M, Miles MF, et al. Tumor-derived p53 mutants induce NF-kappaB2 gene expression. Mol Cell Biol 2005; 25:10097 - 110; http://dx.doi.org/10.1128/MCB.25.22.10097-10110.2005; PMID: 16260623
- Scian MJ, Stagliano KE, Deb D, Ellis MA, Carchman EH, Das A, et al. Tumor-derived p53 mutants induce oncogenesis by transactivating growth-promoting genes. Oncogene 2004; 23:4430 - 43; http://dx.doi.org/10.1038/sj.onc.1207553; PMID: 15077194
- Scian MJ, Stagliano KE, Ellis MA, Hassan S, Bowman M, Miles MF, et al. Modulation of gene expression by tumor-derived p53 mutants. Cancer Res 2004; 64:7447 - 54; http://dx.doi.org/10.1158/0008-5472.CAN-04-1568; PMID: 15492269
- Tepper CG, Gregg JP, Shi XB, Vinall RL, Baron CA, Ryan PE, et al. Profiling of gene expression changes caused by p53 gain-of-function mutant alleles in prostate cancer cells. Prostate 2005; 65:375 - 89; http://dx.doi.org/10.1002/pros.20308; PMID: 16037992
- Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA 2005; 102:13550 - 5; http://dx.doi.org/10.1073/pnas.0506230102; PMID: 16141321
- Troester MA, Herschkowitz JI, Oh DS, He X, Hoadley KA, Barbier CS, et al. Gene expression patterns associated with p53 status in breast cancer. BMC Cancer 2006; 6:276; http://dx.doi.org/10.1186/1471-2407-6-276; PMID: 17150101
- Langerød A, Zhao H, Borgan O, Nesland JM, Bukholm IR, Ikdahl T, et al. TP53 mutation status and gene expression profiles are powerful prognostic markers of breast cancer. Breast Cancer Res 2007; 9:R30; http://dx.doi.org/10.1186/bcr1675; PMID: 17504517
- Weisz L, Damalas A, Liontos M, Karakaidos P, Fontemaggi G, Maor-Aloni R, et al. Mutant p53 enhances nuclear factor kappaB activation by tumor necrosis factor alpha in cancer cells. Cancer Res 2007; 67:2396 - 401; http://dx.doi.org/10.1158/0008-5472.CAN-06-2425; PMID: 17363555
- Gebhard C, Benner C, Ehrich M, Schwarzfischer L, Schilling E, Klug M, et al. General transcription factor binding at CpG islands in normal cells correlates with resistance to de novo DNA methylation in cancer cells. Cancer Res 2010; 70:1398 - 407; http://dx.doi.org/10.1158/0008-5472.CAN-09-3406; PMID: 20145141
- Sarig R, Rivlin N, Brosh R, Bornstein C, Kamer I, Ezra O, et al. Mutant p53 facilitates somatic cell reprogramming and augments the malignant potential of reprogrammed cells. J Exp Med 2010; 207:2127 - 40; http://dx.doi.org/10.1084/jem.20100797; PMID: 20696700
- Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, et al. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell 2009; 15:45 - 56; http://dx.doi.org/10.1016/j.ccr.2008.12.006; PMID: 19111880
- Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007; 8:118 - 27; http://dx.doi.org/10.1093/biostatistics/kxj037; PMID: 16632515