Abstract
DNA interstrand cross-links (ICLs) present a major challenge to cells, preventing separation of the two strands of duplex DNA and blocking major chromosome transactions, including transcription and replication. Due to the complexity of removing this form of DNA damage, no single DNA repair pathway has been shown to be capable of eradicating ICLs. In eukaryotes, ICL repair is a complex process, principally because several repair pathways compete for ICL repair intermediates in a strictly cell cycle-dependent manner. Yeast cells require a combination of nucleotide excision repair, homologous recombination repair and postreplication repair/translesion DNA synthesis to remove ICLs. There are also a number of additional ICL repair factors originally identified in the budding yeast Saccharomyces cerevisiae, called Pso1 though 10, of which Pso2 has an apparently dedicated role in ICL repair. Mammalian cells respond to ICLs by a complex network guided by factors mutated in the inherited cancer-prone disorder Fanconi anemia (FA). Although enormous progress has been made over recent years in identifying and characterizing FA factors as well as in elucidating certain aspects of the biology of FA, the mechanistic details of the ICL repair defects in FA patients remain unknown. Dissection of the FA DNA damage response pathway has, in part, been limited by the absence of FA-like pathways in highly tractable model organisms, such as yeast. Although S. cerevisiae possesses putative homologs of the FA factors FANCM, FANCJ and FANCP (Mph1, Chl1 and Slx4, respectively) as well as of the FANCM-associated proteins MHF1 and MHF2 (Mhf1 and Mhf2), the corresponding mutants display no significant increase in sensitivity to ICLs. Nevertheless, we and others have recently shown that these FA homologs, along with several other factors, control an ICL repair pathway, which has an overlapping or redundant role with a Pso2-controlled pathway. This pathway acts in S-phase and serves to prevent ICL-stalled replication forks from collapsing into DNA double-strand breaks.
DNA is constantly challenged by agents that can inflict structural damage either on one or both strands of the DNA duplex. Lesions affecting one strand present a limited challenge, as the complementary undamaged DNA strand provides the repair machinery with a template to accurately restore the original sequence in the damaged strand. By contrast, lesions affecting both DNA strands are far more deleterious, as they remove the template for cut-and-patch repair reactions. This class of DNA lesions includes DNA interstrand cross-links (ICLs), complex DNA lesions that are extremely toxic owing to the covalent linkage between the two DNA strands. Such linkage prevents DNA strand separation during all fundamental metabolic processes ongoing on cellular DNA.Citation1
As ICLs can arise endogenously, cells have adapted to this threat throughout evolution by the co-ordination of several repair activities in a complex network, termed ICL repair. The first models of ICL repair emerged from studies in Escherichia coli, where the sequential action of nucleotide excision repair (NER) and homologous recombination repair (HRR) appears to be sufficient to repair ICLs in an error-free manner.Citation2 When HRR cannot act during this process, as a consequence of genetic inactivation of the pathway or unavailability of a homologous DNA template, an alternative pathway, dependent on DNA polymerase II (Polβ)-mediated translesion DNA synthesis (TLS), is utilized.Citation3
Although the basic mechanisms and biochemical activities involved in ICL repair appear to be conserved throughout evolution, the overall process is more complex in eukaryotic cells. First, there is considerable redundancy in certain ICL repair activities in eukaryotic cells, and these need to be strictly coordinated. Second, cell cycle phase directs the use of different ICL repair mechanisms and this is dependent on substrate availability. Finally, genome organization per se contributes to an increased sophistication of the ICL repair process in eukaryotic cells. In Saccharomyces cerevisiae, important roles for NER, HRR and postreplication repair (PRR)/TLS in ICL repair have all been identified. Additionally, a number of factors that affect ICL sensitivity, but not sensitivity to other types of DNA damage, have been identified in genetic screens.Citation4,Citation5 Of these, Pso2 (previously referred to as Snm1, but subsequently renamed due to a naming conflict)Citation6 is of greatest interest, as pso2 mutant cells are specifically and highly sensitive to 8-methoxypsoralen photoadditionCitation7 and nitrogen mustard,Citation8,Citation9 two classic ICL-inducing treatments, but not to any other DNA damage types tested to date. Pso2 is thought to act downstream of ICL incision, which is primarily controlled by the NER apparatus in yeast.Citation10-Citation12 The Pso2 protein is the founder member of family of eukaryotic proteins with a conserved domain structure containing two highly conserved motifs: the metallo-β-lactamase (MBL) structural domain (found in hydrolytic enzymes with wide-ranging functions from antibiotic detoxification to nucleic acid metabolism) and the β-CASP [a conserved motif limited to CPSF, an mRNA processing factor; Artemis, a nuclease acting in V(D)J recombination and non-homologous end-joining—NHEJ; SNM1; Pso2] domain.Citation13-Citation16 This domain architecture is typical for β-CASP family members, representing a subfamily of canonical MBLs, and suggested that the Pso2 protein may have nuclease activity. Indeed, biochemical data has shown that Pso2 possesses both 5' to 3' exonuclease activityCitation17,Citation18 and a structure-specific endonuclease activity that is able to open DNA hairpins.Citation18 Both MBL and β-CASP domains are essential for the activity of Pso2, and catalytic site inactivating mutations in these domains (D252A and H611A substitutions, respectively) abolish nuclease activity. In addition, cells carrying these mutations display null-mutant phenotype regarding repair of ICLsCitation19 and of transposon-induced DNA hairpins.Citation18 Hairpin opening activity may suggest a novel role for Pso2 outside ICL repair, although the relevant physiological processes that generate DNA hairpin substrates for Pso2 remain to be elucidated.Citation4,Citation18,Citation20
There are three Pso2 orthologs in vertebrates, SNM1A, SNM1B/Apollo and SNM1C/Artemis.Citation21 Like Pso2, SNM1A and SNM1B display 5' to 3' exonuclease activity, whereas SNM1C possesses endonuclease activity, cleaving 5' and 3' overhangs and hairpins.Citation22 SNM1C probably does not play a major role in ICL repair and rather acts in pathway required for the processing of a subset of DNA double-strand breaks (DSBs) induced by ionizing radiation prior to rejoining by NHEJ.Citation23 However, both SNM1A and SNM1B depletion increase sensitivity to ICL-inducing agents such as mitomycin C,Citation21,Citation24 while the latter seemingly sensitizes the cells to a broader spectrum of DNA damage.Citation25 Notably, co-disruption of SNM1A and SNM1B in chicken DT40 cells leads to an additional increase in ICL sensitivity compared with either single disruptant, suggesting that SNM1A and SNM1B have different or redundant functions in ICL repair.Citation24 SNM1B is believed to promote DSB formation in response to ICLs, possibly through the collapse of replication forks following ICL treatment,Citation26 while SNM1A suppresses ICL-induced DSBs during replication.Citation12,Citation25 Studies aimed at defining the genetic relationship between SNM1-dependent and -independent ICL repair pathways strengthened the notion that SNM1A and SNM1B have diverse roles in ICL repair. SNM1B has been shown to act epistatically with the central Fanconi anemia (FA) factor FANCD2,Citation27 indicating that this Pso2 ortholog functions within the FA pathway during ICL repair, and we have reported collaboration between SNM1A and XPF-ERCC1 in initiating ICL repair in replicating cells.Citation12 The major functional homolog of yeast Pso2 appears to be SNM1A, as expression of SNM1A in pso2 disruptant cells can partially complement ICL sensitivity and elevated ICL-associated DSBs.Citation28
In vertebrates, a major DNA damage response pathway triggered by ICL exposure involves proteins mutated in FA. Mutations in FA pathway lead to a rare autosomal recessive disorder characterized by defects in all stages of ICL repair.Citation29,Citation30 These repair defects are associated with congenital malformations, progressive bone marrow failure, profound genomic instability and a highly elevated risk of hematological malignancies and solid tumors.Citation31 The disease is genetically heterogeneous, and patients with mutations in 15 genes have been reported to show traits associated with FA (FANCA, B, C, D1/BRCA2, D2, E, F, G, H, I, J/BACH1/BRIP1, L, M, N/PALB2, P/SLX4/BTBD12 and O/RAD51C). Mutations in these genes have been found in over 95% of all known FA patients, while cultured cells with defects in a number of other genes, including FAAP24, FAAP100, FAN1, MHF1, MHF2, exhibit FA-like defects in ICL repair. It has been proposedCitation32 that the FA pathway is regulated by three factors, FANCD2-FANCI, SLX4 and RAD18, which act both in parallel and in concert. FANCD2-FANCI and SLX4 likely play a key role in controlling the incision step of ICL repair, with the latter an attractive candidate for a regulator of the recruitment of dedicated nucleases. The role for FANCD2-FANCI in this process still remains largely unknown, although further examination of the relevance of the physical association of FANCD2 with FAN1 (Fanconi anemia-associated nuclease 1) may provide clues. RAD18 controls the FA pathway by regulating FANCD2 monoubiquitylation and chromatin loading of FANCD2-FANCI. Additionally, it contributes to targeting SNM1A to stalled replication forks via monoubiquitylating proliferating cell nuclear antigen (PCNA) and mediating its direct interaction with this nuclease at ICL-stalled replication forks, suggesting it might be an important node for co-ordinating ICL repair factors acting in parallel or sequentially. It is worth noting that Rad18-dependent FA pathway activation appears not to be ICL-specific, but also in response to other DNA damage types.Citation33
Among the known FA factors, FANCA, FANCB, FANCC, FANCE, FANCF, FANG, FANCL, FANCM, two FA-associated proteins (FAAP24 and FAAP100) and FANCM-associated histone-fold proteins, MHF1 and MHF2, constitute what has become known as the FA “core complex.” This core complex is activated upon ICL treatment during S-phase and acts as a ubiquitin E3 ligase, monoubiquitylating FANCD2 and FANCI,Citation30,Citation34 which ultimately leads to their retention on chromatin.Citation35 ICL damage recognition in this cell cycle phase requires FANCM,Citation36 a protein capable of binding to structures generated when replication forks encounter an ICL. FANCM belongs to the ERCC1/XPF(ERCC4) family of structure-specific DNA binding proteinsCitation37 and contains two conserved domains: an apparently inactive ERCC4 nuclease domain that resides at the C terminus and is responsible for binding to branched DNA structure in vitro,Citation38 and an internal domain that is required for interaction with the FA core complex.Citation39 FANCM forms a stable heterodimer with another protein containing an ERCC4 domain, FAAP24.Citation38 This heterodimer mediates recruitment of the FA core complex to chromatin. Due to its interaction with MHF1 and MHF2, FANCM itself appears to be constitutively associated with chromatin. FANCM functions early in the FA pathway, following stalling of a replication fork at an ICL. It is thought that ICL-induced fork collapse can be prevented by FANCM-mediated fork regression or stabilization, and indeed, in vitro data indicates that FANCM can promote reversal of replication forks via concerted displacement and annealing of the nascent DNA strands.Citation40 Multiple nucleases are also recruited at an early step during S-phase-specific ICL repair. The FAN1 nuclease is recruited to stalled replication forks by monoubiquitylated FANCD2 and possesses 5' flap and 5' to 3' exonuclease activities. Weaker activity, at replication fork structures and the cleavage of DNA opposite a nicked strand, has also been demonstrated, as recently recapitulated in references Citation32 and Citation41. SLX4 is recruited to stalled replication forks, directly owing to its tandem ubiquitin-binding zinc finger domains,Citation42,Citation43 recruiting MUS81-EME1, XPF-ERCC1 and SLX1 nucleases as part of the SLX4 complex.Citation44 MUS81-EME1 and XPF-ERCC1 are members of the XPF/MUS81 family of structure-specific endonucleases, while SLX1 is a structurally distinct endonuclease possessing an N-terminal UvrC-intron-endonuclease and a C-terminal plant homeodomain-type zinc finger domains. Biochemically, MUS81-EME1 cleaves 3' flaps, replication fork structures, Holliday junction (HJ) resolution intermediates and splayed arms, and XPF1-ERCC1 acts on splayed-arms, bubbles, stem-loop structures and 3' flaps. SLX1 in complex with SLX4 exhibits hydrolytic activity toward 5' flaps, stem loops, replication fork structures and HJs, as summarized in references Citation32 and Citation41. The coordinated action of FAN1, MUS81-EME1, XPF-ERCC1 and SLX1 nucleases is thought to result in ICL unhooking, although which nucleases create the critical incisions remains unconfirmed. Subsequently, TLS polymerases ensure lesion bypass past the unhooked cross-link. HRR is required to complete repair and DNA replication can subsequently be re-established. In late S-phase, there is a greater probability that two replication forks could converge on a single ICL. Studies using a Xenopus cell-free extract system, in conjunction with plasmid substrates bearing site-specific ICLs, suggest that following unhooking incisions, TLS polymerases extend the converging leading strand past the exonucleolytically processed ICL lesion.
The FA pathway had initially been thought to be restricted to vertebrates. However, the identification of some individual FA counterparts in other metazoans such as Caenorhabditis elegansCitation45-Citation47 and Drosophila melanogaster,Citation48-Citation50 as well as in both buddingCitation50 and fissionCitation51 yeast, challenges this concept. Nevertheless, the whole pathway in its entirety does not seem to exist outside vertebrates. In particular, the FA core complex factors are not clearly identifiable or annotated outside this kingdom. It would appear that lower eukaryotes do, however, possess a “stripped-down” pathway, which lacks the majority of the FA core complex apart from two components, FANCM and FANCL. Three other members of the FA pathway are also well-conserved, FAND1, FANCD2 and FANCJ. The question then becomes how does this substantially stripped-down version of the FA pathway, lacking the core complex, operate? Solving this question would clearly accelerate progress in our understanding of the mechanistic basis of the ICL repair defect in FA.
Saccharomyces cerevisiae is a highly attractive model system owing to its genetic tractability, which has revealed many important biological insights, including within the field of eukaryotic DNA repair. NER, HRR and PRR/TLS have all been suggested to act in the yeast ICL repair, with a significant contribution from a pathway controlled by Pso2, following the initial ICL incisions produced, in all likelihood, by NER factors.Citation1 Budding yeast cells also possess putative homologs of FANCM,Citation34,Citation52-Citation55 FANCJ and FANCPCitation54,Citation55 (Mph1, Chl1 and Slx4, respectively) as well as of the FANCM-associated histone-fold proteins MHF1 and MHF2 (Mhf1 and Mhf2).Citation54,Citation56 Until recently, the existence of the FA-like ICL repair in this organisms has been discounted, since the disruption of these FA-like factors does not sensitize yeast cells to ICL-inducing agents. However, work from our own laboratories and that of K.J. Myung report that these factors constitute an S-phase-specific branch of the ICL repair pathway, obscured by the pathway or pathways controlled by Pso2 and Srs2. Furthermore, we have shown that other factors including Mgm101, a mitochondrial genome maintenance factor that also now appears to play a role in nuclear repair, Mhf1-Mhf2, a complex having heterotetrameric architecture similar to that of the histones (H3-H4)2 heterotetramer, the MutSα (Msh2-Msh6) mismatch repair complex, Exo1, an exonuclease with multiple roles in DNA repair, Smc5-Smc6, a complex playing a role in chromosome organization and dynamics, and PCNA (Pol30), involved in many metabolic processes ongoing on DNA, are also required for this pathway.Citation54,Citation55 It seems that this pathway primarily prevents ICL-stalled replication forks from collapsing into unrepairable DSBs. Rad5, an E3 ubiquitin ligase associated with the error-free branch of PRR, directs this FA-like pathway, possibly to restart replication by a fork reversal mechanism. Importantly, this pathway acts independently of the Pso2-controlled pathway,Citation54,Citation55,Citation57 suggesting that there might be a functional overlap between Pso2 and Exo1 where both are able to exonucleolytically degrade the tethered oligonucleotide associated with an unhooked ICL. Furthermore, the pathway is independent of Rad6-Rad18 error-prone branch of PRR.Citation54
Based on data available and on our own and the Myung group’s new findings,Citation54,Citation55 we propose a modified model for ICL repair in replicating yeast cells (). NER may initiate ICL processing in S-phase yeast cells prior to the arrival of the fork, since it is well established that ICLs are efficiently incised in G1 phase yeast cells. Therefore, the pathway controlled by NER and Pso2 might not be replication-coupled in the classical sense, or might operate both away from and at stalled replication forks to repair ICLs in S-phase (). In pso2 cells, where the FA-like pathway now becomes necessary, the evidence suggests that replication stalls at the ICL and, in the absence of efficient nucleolytic processing by Pso2, produces a signal which Rad5 responds to. As a consequence, Rad5 polyubiquitinates PCNA and causes recruitment of Mph1 to site of the ICL. Mph1 stabilizes or regresses the stalled fork (regression is shown), and Mgm101, Smc5-Smc6 and Mhf1-Mhf2, likely representing Mph1 accessory factors, may help to protect and stabilize the structure of the generated ICL repair intermediate. Msh2-Msh6 (MutSα) also participates in this pathway, potentially acting to sense the aberrant DNA structure at the fork. Subsequently, Exo1 is recruited, possibly by its association with MutSα, and digests the tethered oligonucleotide to produce a substrate for downstream processing events and gap-filling. Gap-filling is achieved by TLS, and DNA replication is subsequently restored by HRR (). In cells disabled for both Pso2-controlled and FA-like pathways, the replication fork collapses into an unrepairable DSB due to an inability to further process the cross-linked structure remaining after initial incision generated by NER, ultimately leading to cell death ().
Figure 1. Proposed model for ICL repair in replicating Saccharomyces cerevisiae cells. (A) A situation in wild-type cells, where Pso2-controlled and FA-like pathways have overlapping or redundant roles in ICL repair, with a major contribution of the former. (B) In pso2 cells, FA-like pathway constitutes branch of the ICL repair pathway, which prevents replication forks from collapsing into DSBs. In addition, this pathway protects and stabilizes the generated ICL repair intermediate structure. (C) In the absence of both pathways, replication forks collapse into unrepairable DSBs upon ICL treatment leading to cell death. For more details, see text.
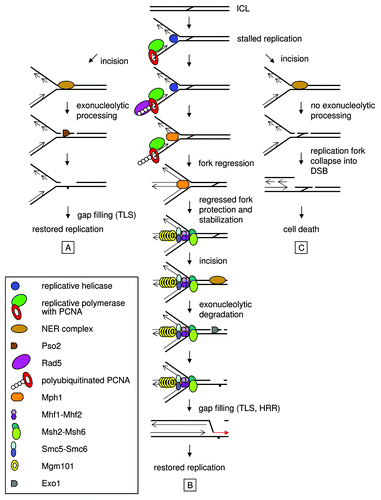
Mechanistically, this pathway is reminiscent of FA pathway in mammals, indicating that functional conservation of the FA pathway probably exceeds the kingdom of vertebrates despite an apparent lack of FA core complex factors. In both yeast and mammalian pathways, Mph1/FANCM-mediated fork regression or stabilization induced by DNA replication stalled at the ICL is an apparently sine qua non step, suggesting it might be essential for replication-coupled ICL repair. Such steps have been suggested to protect ICL repair intermediates from inappropriate repair and/or processing.Citation54
Acknowledgments
Work in the laboratory of M.C. is supported by the VEGA Grant Agency of the Slovak Republic (grant no. 2/0165/09) and by the project TRANSMED that is part of the Research and Development Operational Programme funded by the European Regional Development Fund. Work performed in the laboratory of P.J.M. was supported by Cancer Research UK.
Related Research Data
References
- Lehoczký P, McHugh PJ, Chovanec M. DNA interstrand cross-link repair in Saccharomyces cerevisiae.. FEMS Microbiol Rev 2007; 31:109 - 33; http://dx.doi.org/10.1111/j.1574-6976.2006.00046.x; PMID: 17096663
- Cole RS. Repair of DNA containing interstrand crosslinks in Escherichia coli: sequential excision and recombination. Proc Natl Acad Sci USA 1973; 70:1064 - 8; http://dx.doi.org/10.1073/pnas.70.4.1064; PMID: 4577788
- Dronkert MLG, Kanaar R. Repair of DNA interstrand cross-links. Mutat Res 2001; 486:217 - 47; http://dx.doi.org/10.1016/S0921-8777(01)00092-1; PMID: 11516927
- Brendel M, Bonatto D, Strauss M, Revers LF, Pungartnik C, Saffi J, et al. Role of PSO genes in repair of DNA damage of Saccharomyces cerevisiae.. Mutat Res 2003; 544:179 - 93; http://dx.doi.org/10.1016/j.mrrev.2003.06.018; PMID: 14644320
- Henriques JAP, Brozmanová J, Brendel M. Role of PSO genes in the repair of photoinduced interstrand cross-links and photooxidative damage in the DNA of the yeast Saccharomyces cerevisiae.. J Photochem Photobiol B 1997; 39:185 - 96; http://dx.doi.org/10.1016/S1011-1344(97)00020-1; PMID: 9253198
- Cassier-Chauvat C, Moustacchi E. Allelism between pso1-1 and rev3-1 mutants and between pso2-1 and snm1 mutants in Saccharomyces cerevisiae.. Curr Genet 1988; 13:37 - 40; http://dx.doi.org/10.1007/BF00365754; PMID: 3282695
- Henriques JA, Moustacchi E. Isolation and characterization of pso mutants sensitive to photo-addition of psoralen derivatives in Saccharomyces cerevisiae.. Genetics 1980; 95:273 - 88; PMID: 7009316
- Ruhland A, Kircher M, Wilborn F, Brendel M. A yeast mutant specifically sensitive to bifunctional alkylation. Mutat Res 1981; 91:457 - 62; http://dx.doi.org/10.1016/0165-7992(81)90052-X; PMID: 7027037
- Ruhland A, Haase E, Siede W, Brendel M. Isolation of yeast mutants sensitive to the bifunctional alkylating agent nitrogen mustard. Mol Gen Genet 1981; 181:346 - 51; http://dx.doi.org/10.1007/BF00425609; PMID: 7017347
- McHugh PJ, Sones WR, Hartley JA. Repair of intermediate structures produced at DNA interstrand cross-links in Saccharomyces cerevisiae.. Mol Cell Biol 2000; 20:3425 - 33; http://dx.doi.org/10.1128/MCB.20.10.3425-3433.2000; PMID: 10779332
- De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol 2000; 20:7980 - 90; http://dx.doi.org/10.1128/MCB.20.21.7980-7990.2000; PMID: 11027268
- Wang AT, Sengerová B, Cattell E, Inagawa T, Hartley JM, Kiakos K, et al. Human SNM1A and XPF-ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev 2011; 25:1859 - 70; http://dx.doi.org/10.1101/gad.15699211; PMID: 21896658
- Callebaut I, Moshous D, Mornon JP, de Villartay JP. Metallo-β-lactamase fold within nucleic acids processing enzymes: the β-CASP family. Nucleic Acids Res 2002; 30:3592 - 601; http://dx.doi.org/10.1093/nar/gkf470; PMID: 12177301
- Aravind L, Walker DR, Koonin EV. Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res 1999; 27:1223 - 42; http://dx.doi.org/10.1093/nar/27.5.1223; PMID: 9973609
- Dominski Z. Nucleases of the metallo-beta-lactamase family and their role in DNA and RNA metabolism. Crit Rev Biochem Mol Biol 2007; 42:67 - 93; http://dx.doi.org/10.1080/10409230701279118; PMID: 17453916
- Cattell E, Sengerová B, McHugh PJ. The SNM1/Pso2 family of ICL repair nucleases: from yeast to man. Environ Mol Mutagen 2010; 51:635 - 45; PMID: 20175117
- Li X, Hejna J, Moses RE. The yeast Snm1 protein is a DNA 5′-exonuclease. DNA Repair (Amst) 2005; 4:163 - 70; http://dx.doi.org/10.1016/j.dnarep.2004.08.012; PMID: 15590324
- Tiefenbach T, Junop M. Pso2 (SNM1) is a DNA structure-specific endonuclease. Nucleic Acids Res 2012; 40:2131 - 9; http://dx.doi.org/10.1093/nar/gkr1059; PMID: 22102580
- Li X, Moses RE. The β-lactamase motif in Snm1 is required for repair of DNA double-strand breaks caused by interstrand crosslinks in S. cerevisiae.. DNA Repair (Amst) 2003; 2:121 - 9; http://dx.doi.org/10.1016/S1568-7864(02)00192-1; PMID: 12509272
- Bonatto D, Revers LF, Brendel M, Henriques JA. The eukaryotic Pso2/Snm1/Artemis proteins and their function as genomic and cellular caretakers. Braz J Med Biol Res 2005; 38:321 - 34; http://dx.doi.org/10.1590/S0100-879X2005000300002; PMID: 15761611
- Dronkert MLG, de Wit J, Boeve M, Vasconcelos ML, van Steeg H, Tan TLR, et al. Disruption of mouse SNM1 causes increased sensitivity to the DNA interstrand cross-linking agent mitomycin C. Mol Cell Biol 2000; 20:4553 - 61; http://dx.doi.org/10.1128/MCB.20.13.4553-4561.2000; PMID: 10848582
- Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 2002; 108:781 - 94; http://dx.doi.org/10.1016/S0092-8674(02)00671-2; PMID: 11955432
- Riballo E, Kühne M, Rief N, Doherty A, Smith GC, Recio MJ, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to γ-H2AX foci. Mol Cell 2004; 16:715 - 24; http://dx.doi.org/10.1016/j.molcel.2004.10.029; PMID: 15574327
- Ishiai M, Kimura M, Namikoshi K, Yamazoe M, Yamamoto K, Arakawa H, et al. DNA cross-link repair protein SNM1A interacts with PIAS1 in nuclear focus formation. Mol Cell Biol 2004; 24:10733 - 41; http://dx.doi.org/10.1128/MCB.24.24.10733-10741.2004; PMID: 15572677
- Demuth I, Digweed M, Concannon P. Human SNM1B is required for normal cellular response to both DNA interstrand crosslink-inducing agents and ionizing radiation. Oncogene 2004; 23:8611 - 8; http://dx.doi.org/10.1038/sj.onc.1207895; PMID: 15467758
- Bae JB, Mukhopadhyay SS, Liu L, Zhang N, Tan J, Akhter S, et al. Snm1B/Apollo mediates replication fork collapse and S Phase checkpoint activation in response to DNA interstrand cross-links. Oncogene 2008; 27:5045 - 56; http://dx.doi.org/10.1038/onc.2008.139; PMID: 18469862
- Mason JM, Sekiguchi JM. Snm1B/Apollo functions in the Fanconi anemia pathway in response to DNA interstrand crosslinks. Hum Mol Genet 2011; 20:2549 - 59; http://dx.doi.org/10.1093/hmg/ddr153; PMID: 21478198
- Hazrati A, Ramis-Castelltort M, Sarkar S, Barber LJ, Schofield CJ, Hartley JA, et al. Human SNM1A suppresses the DNA repair defects of yeast pso2 mutants. DNA Repair (Amst) 2008; 7:230 - 8; http://dx.doi.org/10.1016/j.dnarep.2007.09.013; PMID: 18006388
- Su X, Huang J. The Fanconi anemia pathway and DNA interstrand cross-link repair. Protein Cell 2011; 2:704 - 11; http://dx.doi.org/10.1007/s13238-011-1098-y; PMID: 21948210
- Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer 2011; 11:467 - 80; http://dx.doi.org/10.1038/nrc3088; PMID: 21701511
- Joenje H, Patel KJ. The emerging genetic and molecular basis of Fanconi anaemia. Nat Rev Genet 2001; 2:446 - 57; http://dx.doi.org/10.1038/35076590; PMID: 11389461
- Sengerová B, Wang AT, McHugh PJ. Orchestrating the nucleases involved in DNA interstrand cross-link (ICL) repair. Cell Cycle 2011; 10:3999 - 4008; http://dx.doi.org/10.4161/cc.10.23.18385; PMID: 22101340
- Palle K, Vaziri C. Rad18 E3 ubiquitin ligase activity mediates Fanconi anemia pathway activation and cell survival following DNA Topoisomerase 1 inhibition. Cell Cycle 2011; 10:1625 - 38; http://dx.doi.org/10.4161/cc.10.10.15617; PMID: 21478670
- Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet 2007; 8:735 - 48; http://dx.doi.org/10.1038/nrg2159; PMID: 17768402
- Matsushita N, Kitao H, Ishiai M, Nagashima N, Hirano S, Okawa K, et al. A FancD2-monoubiquitin fusion reveals hidden functions of Fanconi anemia core complex in DNA repair. Mol Cell 2005; 19:841 - 7; http://dx.doi.org/10.1016/j.molcel.2005.08.018; PMID: 16168378
- Niedernhofer LJ. The Fanconi anemia signalosome anchor. Mol Cell 2007; 25:487 - 90; http://dx.doi.org/10.1016/j.molcel.2007.02.002; PMID: 17317622
- Ciccia A, McDonald N, West SC. Structural and functional relationships of the XPF/MUS81 family of proteins. Annu Rev Biochem 2008; 77:259 - 87; http://dx.doi.org/10.1146/annurev.biochem.77.070306.102408; PMID: 18518821
- Ciccia A, Ling C, Coulthard R, Yan Z, Xue Y, Meetei AR, et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol Cell 2007; 25:331 - 43; http://dx.doi.org/10.1016/j.molcel.2007.01.003; PMID: 17289582
- Deans AJ, West SC. FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol Cell 2009; 36:943 - 53; http://dx.doi.org/10.1016/j.molcel.2009.12.006; PMID: 20064461
- Gari K, Décaillet C, Delannoy M, Wu L, Constantinou A. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc Natl Acad Sci USA 2008; 105:16107 - 12; http://dx.doi.org/10.1073/pnas.0804777105; PMID: 18843105
- Cybulski KE, Howlett NG. FANCP/SLX4: a Swiss army knife of DNA interstrand crosslink repair. Cell Cycle 2011; 10:1757 - 63; http://dx.doi.org/10.4161/cc.10.11.15818; PMID: 21527828
- Kim Y, Lach FP, Desetty R, Hanenberg H, Auerbach AD, Smogorzewska A. Mutations of the SLX4 gene in Fanconi anemia. Nat Genet 2011; 43:142 - 6; http://dx.doi.org/10.1038/ng.750; PMID: 21240275
- Yamamoto KN, Kobayashi S, Tsuda M, Kurumizaka H, Takata M, Kono K, et al. Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc Natl Acad Sci U.SA 2011; 108:6492 - 6; http://dx.doi.org/10.1073/pnas.1018487108; PMID: 21464321
- Muñoz IM, Hain K, Déclais AC, Gardiner M, Toh GW, Sanchez-Pulido L, et al. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol Cell 2009; 35:116 - 27; http://dx.doi.org/10.1016/j.molcel.2009.06.020; PMID: 19595721
- Collis SJ, Barber LJ, Ward JD, Martin JS, Boulton SJ. C. elegans FANCD2 responds to replication stress and functions in interstrand cross-link repair. DNA Repair (Amst) 2006; 5:1398 - 406; http://dx.doi.org/10.1016/j.dnarep.2006.06.010; PMID: 16914393
- Petalcorin MI, Sandall J, Wigley DB, Boulton SJ. CeBRC-2 stimulates D-loop formation by RAD-51 and promotes DNA single-strand annealing. J Mol Biol 2006; 361:231 - 42; http://dx.doi.org/10.1016/j.jmb.2006.06.020; PMID: 16843491
- Youds JL, Barber LJ, Ward JD, Collis SJ, O’Neil NJ, Boulton SJ, et al. DOG-1 is the Caenorhabditis elegans BRIP1/FANCJ homologue and functions in interstrand cross-link repair. Mol Cell Biol 2008; 28:1470 - 9; http://dx.doi.org/10.1128/MCB.01641-07; PMID: 18086896
- Marek LR, Bale AE. Drosophila homologs of FANCD2 and FANCL function in DNA repair. DNA Repair (Amst) 2006; 5:1317 - 26; http://dx.doi.org/10.1016/j.dnarep.2006.05.044; PMID: 16860002
- Lo T, Pellegrini L, Venkitaraman AR, Blundell TL. Sequence fingerprints in BRCA2 and RAD51: implications for DNA repair and cancer. DNA Repair (Amst) 2003; 2:1015 - 28; http://dx.doi.org/10.1016/S1568-7864(03)00097-1; PMID: 12967658
- Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat Genet 2005; 37:958 - 63; http://dx.doi.org/10.1038/ng1626; PMID: 16116422
- Sun W, Nandi S, Osman F, Ahn JS, Jakovleska J, Lorenz A, et al. The FANCM ortholog Fml1 promotes recombination at stalled replication forks and limits crossing over during DNA double-strand break repair. Mol Cell 2008; 32:118 - 28; http://dx.doi.org/10.1016/j.molcel.2008.08.024; PMID: 18851838
- Patel KJ, Joenje H. Fanconi anemia and DNA replication repair. DNA Repair (Amst) 2007; 6:885 - 90; http://dx.doi.org/10.1016/j.dnarep.2007.02.002; PMID: 17481966
- McVey M. Strategies for DNA interstrand crosslink repair: insights from worms, flies, frogs, and slime molds. Environ Mol Mutagen 2010; 51:646 - 58; PMID: 20143343
- Daee DL, Ferrari E, Longerich S, Zheng XF, Xue X, Branzei D, et al. Rad5-dependent DNA repair functions of the Saccharomyces cerevisiae FANCM homolog Mph1. J Biol Chem 2012; 287:26563 - 75; http://dx.doi.org/10.1074/jbc.M112.369918; PMID: 22696213
- Ward T, Dudášová Z, Sarkar S, Bhide M, Vlasáková D, Chovanec M, et al. Components of a Fanconi-like pathway control Pso2-independent DNA interstrand crosslink repair in yeast. PLoS Genet 2012; 8:e1002884; http://dx.doi.org/10.1371/journal.pgen.1002884; PMID: 22912599
- Yang H, Zhang T, Tao Y, Wu L, Li HT, Zhou JQ, et al. Saccharomyces cerevisiae MHF complex structurally resembles the histones (H3-H4)₂ heterotetramer and functions as a heterotetramer. Structure 2012; 20:364 - 70; http://dx.doi.org/10.1016/j.str.2011.12.012; PMID: 22325783
- Barber LJ, Ward TA, Hartley JA, McHugh PJ. DNA interstrand cross-link repair in the Saccharomyces cerevisiae cell cycle: overlapping roles for PSO2 (SNM1) with MutS factors and EXO1 during S phase. Mol Cell Biol 2005; 25:2297 - 309; http://dx.doi.org/10.1128/MCB.25.6.2297-2309.2005; PMID: 15743825