Abstract
TRAIL, a putative anticancer cytokine, induces extrinsic cell death by activating the caspase cascade directly (Type I cells) via the death-inducing signaling complex (DISC) or indirectly (Type II cells) by caspase-8 cleavage of Bid and activation of the mitochondrial cell death pathway. Cancer cells are characterized by their dependence on aerobic glycolysis, which, although inefficient in terms of ATP production, facilitates tumor metabolism. Our studies show that TRAIL-induced cell death is significantly affected by the metabolic status of the cell. Inhibiting glycolysis with 2-deoxyglucose potentiates TRAIL-induced cell death, whereas glucose deprivation can paradoxically inhibit apoptosis. These conflicting responses to glycolysis inhibition are modulated by the balance between the Akt and AMPK pathways and their subsequent downstream regulation of mTORC1. This results in marked changes in protein translation, in which the equilibrium between anti- and pro-apoptotic Bcl-2 family member proteins is decided by their individual degradation rates. This regulates the mitochondrial cell death pathway and alters its sensitivity not only to TRAIL, but to ABT-737, a Bcl-2 inhibitor. Taken together, our studies show that the sensitivity of cancer cells to apoptosis can be modulated by targeting their unique metabolism in order to enhance sensitivity to apoptotic agents.
The Role of Metabolism in Tumor Cell Death
Typically, anticancer agents kill by inducing apoptotic cell death, either through the extrinsic (death receptor) or the intrinsic (mitochondrial) pathway. The extrinsic pathway is activated when TNF ligand family members, such as tumor necrosis factor (TNFα), CD95L/FasL and TNF-related apoptosis-inducing ligand (TRAIL) bind to their respective death receptors.Citation1 Ligation initiates recruitment of the Fas-associated death domain (FADD) adaptor molecule to the cytoplasmic death domain of the trimerized/clustered receptor, which, in turn, recruits procaspase-8 to form the death-inducing signaling complex (DISC, ). In both CD95L and TRAIL-induced cell death, the DISC kills by directly activating effector caspases-3/7 (Type I cells) and/or by cleaving Bid to generate tBid, which activates Bak/Bax at the mitochondrial membrane to induce cytochrome c release, apoptosome formation and cell death (Type II cells). Thus, TRAIL according to cell type can employ two separate but interlinked pathways for executing cell death. Originally, TRAIL was thought to be specific for all cancer cells, but most primary neoplasms, notably leukemias and several epithelial cell tumors are resistant, potentially limiting the therapeutic potential of this cytokine.Citation2 TRAIL resistance can be due to either defects in DISC assembly and/or the downstream proteins involved in the mitochondrial cell death pathway. Thus, the mitochondria and attendant pro- and anti-apoptotic proteins can play a critical role in both the intrinsic and extrinsic cell death pathways. Mitochondria, by generating ATP, which is used by anabolic processes such as protein translation, can potentially have other profound effects on apoptotic cell death pathways. In this respect, cancer cells, as originally described by Otto Warburg, predominantly use aerobic glycolysis rather than mitochondrial oxidative phosphorylation to fuel and support their rapid growth and proliferation.Citation3 This is in spite of the fact that glycolysis generates only two ATP molecules per glucose precursor, which is a very poor return in comparison to the 36 ATP molecules that can be produced during mitochondrial oxidative phosphorylation. Some cancer cells undergo metabolic switching that allow very high levels of glucose uptake (upregulated glucose transporters) and utilization, which not only provides energy but, crucially, via the pentose phosphate pathway, produces ribose sugars, nucleotides, glycerol, citrate (lipid synthesis) and non-essential amino acids.Citation4 This dependence on the “Warburg effect” has attracted interest as a specific cancer hallmark and a therapeutic target.Citation5 Already, clinical practice makes use of the Warburg effect, as tumors can be imaged using fluorodeoxygluocose in FDG-PET scanning. But potentially, the Warburg effect can also be exploited to kill cancer cells using their weakness for glycolytic metabolism.Citation5 In our recent studies and this report, we explore metabolic mechanisms and strategies for enhancing glucose-dependent cell death in cancer cells.
Figure 1. 2DG potentiates cell death. Cells treated with 5 mM 2DG are more sensitive to both TRAIL and ABT-737-induced cell death. This scheme shows that in the presence of 2DG, the direct activation of effector caspases by the DISC is synergised by 2DG leading to more rapid and extensive processing of caspases-8 and -3 (A). In addition, 2DG strongly inhibits glycolysis (ECAR) and reduces mitochondrial respiration (OCR) and oxidative phosphorylation (B). ATP levels are reduced (C), leading to increased AMP levels and activation of AMPK, which inhibits mTORC1, thereby depressing protein translation. In this model, Akt is not hyperactive, and hence, GSK3β would be dephosphorylated, thus promoting GSK3β activity and phosphorylation of Mcl-1, leading to its degradation via the proteasome. 2DG inhibited protein translation as judged by polysome profiling (D), and the net result is to change the balance of pro- and anti-apoptotic Bcl-2 proteins, in favor of pore (MOMP) formation, cytochrome c release and apoptosome formation.
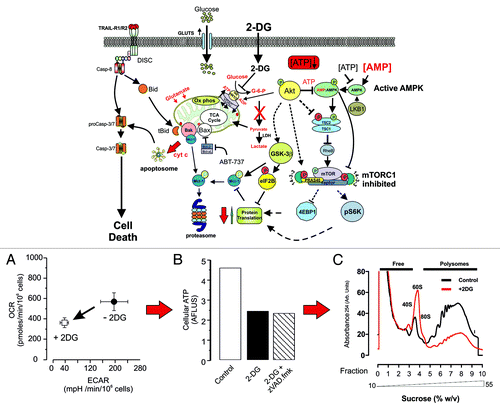
2-Deoxyglucose Potentiates Cell Death
Apoptosis induced by many chemotherapeutic agents (and TRAIL/CD95L in Type II cells) requires activation of the mitochondrial cell death pathway, which is believed to require high levels of ATP.Citation6 As tumors invariably depend on the “Warburg effect” for survival and growth, we investigated whether or not metabolism can alter the cell death response of leukemic cells to TRAIL. We therefore determined whether the ATP levels and cellular response to TRAIL can be modulated by altering the balance between mitochondrial oxidative phosphorylation and aerobic glycolysis. Cancer metabolism has been targeted by using either glucose deprivation or 2-deoxyglucose (2DG, a competitive inhibitor of hexokinase) to abrogate glycolysis in the belief that either anti-glycolytic treatment would produce similar effects. However, when we compared the effects of 2DG or glucose deprivation on cellular metabolism and TRAIL-induced cell death in a mantle lymphoma cell (Z138) line,Citation6 we found, rather surprisingly, that in the presence of pyruvate and glutamine, 2DG treatment and glucose deprivation have quite different and opposing effects. First, metabolic characterization using extra-cellular flux (XF) analysis showed that Z138 cells are competent for both oxidative phosphorylation and glycolysis as measured by the oxygen consumption (OCR) and extra-cellular acidification rates (ECAR, measures lactate release), respectively. Treating cells for 20 h with 2DG abolished ECAR and reduced OCR by 30–35%, indicating a strong but not absolute dependence on glycolysis (). However, cellular ATP levels decreased by 50% and were accompanied by key changes in the cellular cell death machinery and increased sensitivity to TRAIL-induced cell death ( and ). First, TRAIL DISC formation was normal, but there was a more rapid cleavage/activation of caspase-8, caspase-3 and the canonical death caspase-3/-7 substrate, PARP.Citation6 After 2DG treatment, cFLIPL levels were maintained, but the anti-apoptotic short form cFLIPS was markedly downregulated.Citation6 This change in the ratio of long and short isoforms of cFLIP would enhance DISC-mediated caspase-8 activation and subsequently caspase-3 processing. However, 2DG treatment induced significant changes in the pro- and anti-Bcl-2 apoptotic proteins, which, in general, decreased after 2DG treatment. Furthermore, individual proteins showed variable/opposing changes; notably, Bak levels, for example, remained relatively high, whereas its antagonist Mcl-1 was significantly downregulated. Activation of the TRAIL DISC in Type II cells results in Bid cleavage, releasing tBid, which activates Bak by displacing Mcl-1Citation7 to induce cytochrome c release, apoptosome formation and caspase-3/7 activation (). This change in the Bak/Mcl-1 ratio would sensitize cells to tBid-mediated mitochondrial cell death. Furthermore, Z138 cells pre-treated with 2DG were also approximately 6–8-fold more sensitive to ABT-737, a Bad mimetic and specific inducer of the intrinsic pathway (). Other recent studies have shown that 2DG suppresses glucose metabolism and potentiates ABT-263/737 killing in a mouse xenograft model of hormone-independent, chemo-resistant human prostate cancer.Citation8 Similarly, Bcl-2 antagonises 2DG-induced toxicity in mouse B cell lymphoma cell lines and is, in turn, neutralized by ABT-737.Citation9 Thus, targeting glycolysis in cancer cells may offer a promising strategy for specifically enhancing the therapeutic efficacy of anti-Bcl-2 compounds such as ABT-737. This implies a link between Bcl-2 family proteins and aerobic glycolysis; the key question is, what is the link?
Table 1. Effects of anti-glycolytic treatments on TRAIL and ABT-737 induced cell death and loss of mitochondrial membrane potential
Increasingly, evidence points toward energy-linked changes in the Akt, mTOR and AMPK signaling pathways, which control metabolism, cell growth and protein synthesis. In our studies with 2DG, we found that global protein translation, as judged by sucrose density polysome analysis, was markedly downregulated (). As degradation rates of key proteins are different, a slowdown in protein synthesis leads to changes in the relative levels of anti- and pro-apoptotic proteins. Thus, there was a marked loss of Mcl-1 (T½ ~1 h) in contrast to other Bcl-2 family members, such as Bcl-2 (T½ ~10 h), which have much longer half-lives.Citation10 AMPK is believed to be more sensitive to ATP/AMP levels than mTORC1, and 2DG (25 mM) has been shown to activate AMPK in HEK293 cells, leading to dephosphorylation/deactivation of the downstream targets S6K1 and 4EBP1 and inhibition of protein translation.Citation11 Additionally, decreased Akt activation leads to reduced phosphorylation and, hence, activation of GSK-3β, which is active in its dephosphorylated state. This kinase phosphorylates a large number of downstream targets,Citation12 including Mcl-1, targeting it for proteasomal degradation,Citation13 and also eIF2B, which in its phosphorylated state inhibits eIF2 and disrupts formation of the eIF2.GTP.Met-tRNAi ternary complex, which is required for the initiation of protein translation.Citation14 These effects on protein translation in part support the model shown in , where 2DG predominantly activates the AMPK pathway with little or reduced effect on the parallel Akt pathway and inhibits downstream mTORC1 signaling.
Glucose Deprivation Inhibits Cell Death
Intriguingly, using a chronic glucose deprivation model, a different scenario comes into play in that glucose removal and depression of glycolysis results in a corresponding increase in mitochondrial oxidative phosphorylation and maintenance of cellular ATP levelsCitation6 (). Under these conditions, TRAIL DISC formation/activation was reduced, coupled with a decreased sensitivity to TRAIL, ABT-737 () and radiation-induced cell death. Cells maintained without glucose were characterized by well-coupled, functional mitochondria, with ultra-structurally, extensively developed cristae. This suggests that the absence of glucose induces re-structuring of the mitochondria to allow the cells to utilize pyruvate and glutamine to maintain ATP levels and cell viability. Significantly, mitochondria from glucose-deprived cells showed increased levels of cytochrome c, perhaps reflecting the increased demand for oxidative phosphorylation and/or changes in mitochondrial or associated proteins, which control the release of cytochrome c. Glucose deprivation induces a rapid and transient phosphorylation of p53,Citation15 and activation of p53 affects both glycolysis and oxidative phosphorylation. Thus, while hexokinase 2 is upregulated to stimulate glycolysis, the enzyme TP53-induced glycolysis and apoptosis regulator (TIGAR) is also upregulated, which decreases the levels of the glycolytic activator fructose-2,6-biphosphate and diverts glycolytic intermediates into the pentose phosphate pathway (for review, see refs. Citation16 and Citation17). In addition, p53 also regulates the SCO2 gene (synthesis of cytochrome c oxidase 2), which encodes an assembly factor for the 13-subunit cytochrome c oxidase (COX IV complex).Citation18 Upregulation of COX IV would lead to increased binding of cytochrome c, and this may explain the increased levels of cytochrome c found in mitochondria isolated from glucose-deprived cells. Possibly, the increased binding of cytochrome c to COX IV may inhibit the release of cytochrome c after an apoptotic stimulus. However, our results clearly show that in glucose-deprived cells, the levels of the anti-apoptotic proteins Bcl-2, Bcl-XL, and Mcl-1 are either maintained or were higher than in glucose cultured cells. This would antagonize the pro-apoptotic effects of Bax/Bak (and ABT-737), thereby inhibiting cytochrome c release. In this context, Obatoclax, a pan Bcl-2 antagonist enhances TRAIL-induced cell death in Ramos cells by a mechanism involving Bak/Bim release from Bcl-2/Bcl-XL complexes, Mcl-1 inhibition and upregulation of TRAIL-R2, resulting from Obatoclax-induced inhibition of the TRAIL-R2 repressor Yin Yang 1.Citation19 Maintenance of Mcl-1 levels would also antagonize the effects of Bim, Noxa and Puma. Bim has been suggested in other studies to be a key effector in 2DG and low glucose-induced toxicity by upregulating the BH3-only proteins Bim and Bmf.Citation9,Citation20 However, Z138 cells reportedly do not express Bim,Citation21 and we could not detect Bim by immunoblotting in whole Z138 cell lysates, suggesting that in some cell types, other Bim-independent mechanisms may come into play.
Figure 2. Glucose withdrawal inhibits cell death. In this model, glycolysis is inhibited by conditioning Z138 cells for 2 wk on glucose-free (plus pyruvate and glutamine) media. Under these conditions, the cells adapt and continue to grow, but are now resistant to TRAIL and ABT-737 induced cell death. In glucose-free media, the cells have slightly reduced expression of TRAIL-R1/R2 and less DISC formation and activity in response to TRAIL stimulation (A). The cells also upregulate oxidative phosphorylation to compensate for the lack of glycolysis (B), maintaining ATP levels (C). Under these conditions, the Akt/mTORC1 pathway is activated, and key substrates such as GSK3β and S6K1 are phosphorylated (D). This results in a shift in the balance between the pro- and anti-apoptotic Bcl-2 proteins, inhibiting MOMP and the action of ABT-737.
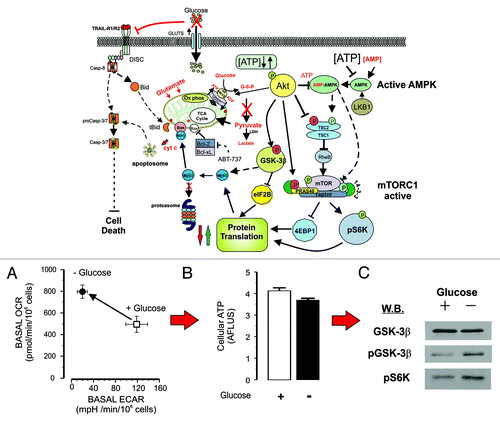
In more recent experiments we have investigated the effects of glucose removal on the AMPK, Akt and mTOR pathways in Z138 cells by blotting for phosphorylated proteins and kinases involved in these pathways. Our results suggest that under chronic, glucose-free conditions, the Akt pathway is hyper-activated as pAkt levels are increased (data not shown), leading to downstream phosphorylation of GSK-3β and S6K1 (). In normal cells, glucose deprivation downregulates S6K1 phosphorylation and activation, thus inactivation of GSK-3β would inhibit Mcl-1 phosphorylation and ubiquitin-dependent degradation, leading to stabilization of the protein, as observed in our earlier studies. The activation of S6K1 (via mTOR) and inhibition of 4EBP1 would maintain protein translation. Activated Akt also phosphorylates key proteins in the mTOR pathway, specifically the TSC1/2 complex, which is a negative regulator of mTORC1 (for review, see ref. Citation22). TSC2 is phosphorylated by Akt (Ser939/989 and Thr1462) promoting 14-3-3 binding and inhibition of TSC2, which, in turn, inhibits the GTPase activity of Rheb. GTP-Rheb activates mTORC1 and stimulates protein synthesis, the energy (ATP) demands of which would be met by the increased oxidative phosphorylation. Akt also directly phosphorylates PRAS40, which binds to 14-3-3 and activates mTORC1, while, conversely, AMPK directly phosphorylates Raptor, which also binds to 14-3-3 and inhibits mTORC1.Citation23 The maintenance of ATP levels in glucose-free cells suggests that AMP levels would be low and the AMPK pathway would not be significantly activated. Taken together, these data suggest that in glucose-free cells, the Akt pathway predominates, whereas with 2DG, the AMPK pathway controls the downstream mTORC1 complex and its effector proteins.
Targeting Aerobic Glycolysis: A Universal Potentiator of Tumor Cell Death?
It is clear from our studies that 2DG and glucose deprivation both block aerobic glycolysis, but can potentially have different outcomes on the sensitivity of tumor cells to apoptosis. Thus, metabolic switching to oxidative phosphorylation can protect against cell death by promoting the maintenance of the anti-apoptotic Bcl-2 family members, such as Bcl-2, Bcl-XL and Mcl-1. In this respect, our studies with TRAIL and ABT-737 indicate that the Akt pathway is probably the most important driver in this resistance mechanism. Akt has been implicated in a variety of proposed survival mechanisms, and clearly its effects via regulation of protein translation support the idea that it maintains the correct balance between anti- and pro-apoptotic proteins to control cytochrome c release. In this context, Mcl-1 may be the key player, although in some studies, glucose deprivation has also been shown to induce Mcl-1 degradation and upregulation of Bim, which promotes apoptosis.Citation20 However, in these studies, the cells were maintained for 36 h in glucose-free media, whereas we have looked at the effects of chronic glucose deprivation in the presence of alternative mitochondrial substrates. Interestingly, when Z138 cells are first cultured in glucose-free media, they are slightly more sensitive to TRAIL, exhibiting an increased necrotic cell death, but after 20 h of glucose deprivation, they become resistant to TRAIL and ABT-737.Citation6 This suggests that the cellular response to inducers of apoptosis varies between acute and chronic glucose deprivation.
Following on from this, the use of 2DG to synergize chemotherapeutic agents is an attractive proposition, as, in theory, you could specifically target cancer cells (which in comparison to normal cells have elevated glucose uptake and utilization) with chemotherapeutic drugs in combination with a compound with known pharmacology and toxicology. However, 2DG has also been reportedCitation24 to protect against cell death in some cell types via Akt phosphorylation and activation, independent of LKB1/AMPK activation and inhibition of glycolysis. Thus, an emerging picture is that dependent on cell type and signaling repertoire, 2DG can enhance or de-sensitize a particular cancer cell to other agents. Interestingly, 2DG has been used as a single agent to induce apoptosis in alveolar, but not embryonal rhabdomyosarcoma cell lines by downregulating Mcl-1 and upregulating Noxa.Citation25 Thus, some cells appear to be very sensitive to 2DG treatment, while others are not, depending on the activation balance between the Akt and AMPK pathways. A further complication arises from the fact that it is still not clear how 2DG synergizes cell death. Recent studies have reported that 2DG treatment can synergize ABT-737/263 cell death without depleting ATP or inhibiting protein translation and Mcl-1 levels.Citation8 In this study, enhanced apoptosis was induced in cells treated with 2DG for up to 6 h in the presence of glucose before treatment with ABT-263. This study reported that a combination of 2DG and ABT-737/263 disrupted the interactions between Mcl-1 and Bak/Bax, implying a hitherto unknown effect of 2DG. However, in our recent studies, 2DG inhibited glycolysis within 2 h leading to a rapid depletion of cellular ATP levels, which was not concurrent with the increased sensitivity to TRAIL (G. Miles, M.M. and K.C., unpublished results). Thus, while in some cell types, 2DG may synergize with agents like ABT-737 to activate the mitochondrial cell death pathway, in other cell types, its effects on Akt activation may promote cell survival.
Nevertheless, the observation that low glucose levels can increase resistance to apoptotic cell death has implications for the potential effects of nutritional and metabolic status on cancer initiation and progression. The finding that switching from aerobic glycolysis to oxidative phosphorylation can alter the response to some apoptosis-inducing agents is a surprising finding. However, other studies suggest that this may not be an uncommon event. For example, MCF-7 cells in single cultures are metabolically glycolytic and sensitive to Tamoxifen-induced cell death, but in co-culture with immortalized human fibroblasts switch to oxidative phosphorylation and become resistant to the drug.Citation26 Furthermore, in this study, Dasatinib, a tyrosine kinase inhibitor, reversed the metabolically switched co-cultured MCF-7 cells back to aerobic glycolysis, and the cells regained their sensitivity to Tamoxifen. In other studies, cell death induced in the HEK293-T cell line by serum withdrawal is prevented by glucose and amino acid deprivation or 2DG treatment.Citation27 Nutrient restriction was associated with reduced activity of the mTOR/S6 kinase cascade, which could be reversed by pharmacological or genetic inhibition. Typically, low ATP levels caused by glucose deprivation and anti-glycolytic inhibition (e.g., 2DG) or oxidative phosphorylation inhibition by metformin lead to AMPK activation and inhibition of mTORC1 (see for review, ref. Citation28). Thus, hyperglycaemia and obesity, which are associated with enhanced risk of cancer, may be due to suppression of the LKB1/AMPK pathway. Conversely, activation of AMPK and inhibition of mTORC1 might play a role in the anticancer effects associated with exercise and low-calorie diets. However, our results suggest that in some cell types, glucose deprivation in the presence of pyruvate and glutamine is compensated by an increase in oxidative phosphorylation. This results in normal ATP levels and resistance to apoptotic stimuli. In conclusion, the use of 2DG or other anti-glycolytic strategies may have the potential to synergize with anti-chemotherapy therapies. However, this is likely to be tumor cell-specific, and we need to understand the complex interplay between metabolism and translation mechanisms in greater detail in order to predict or select which cancer is likely to respond to this type of combination therapy.
Acknowledgments
The authors appreciate many helpful discussions with Dr. Ewan Smith on developing the schemes shown in and . This work was supported by the UK Medical Research Council.
Related Research Data
References
- Dickens LS, Powley IR, Hughes MA, MacFarlane M. The ‘complexities’ of life and death: death receptor signalling platforms. Exp Cell Res 2012; 318:1269 - 77; http://dx.doi.org/10.1016/j.yexcr.2012.04.005; PMID: 22542855
- MacFarlane M, Harper N, Snowden RT, Dyer MJ, Barnett GA, Pringle JH, et al. Mechanisms of resistance to TRAIL-induced apoptosis in primary B cell chronic lymphocytic leukaemia. Oncogene 2002; 21:6809 - 18; http://dx.doi.org/10.1038/sj.onc.1205853; PMID: 12360407
- Warburg O. On respiratory impairment in cancer cells. Science 1956; 124:269 - 70; PMID: 13351639
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 2008; 7:11 - 20; http://dx.doi.org/10.1016/j.cmet.2007.10.002; PMID: 18177721
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646 - 74; http://dx.doi.org/10.1016/j.cell.2011.02.013; PMID: 21376230
- Robinson GL, Dinsdale D, Macfarlane M, Cain K. Switching from aerobic glycolysis to oxidative phosphorylation modulates the sensitivity of mantle cell lymphoma cells to TRAIL. Oncogene 2012; http://dx.doi.org/10.1038/onc.2012.13; PMID: 22310286
- Gillissen B, Wendt J, Richter A, Richter A, Müer A, Overkamp T, et al. Endogenous Bak inhibitors Mcl-1 and Bcl-xL: differential impact on TRAIL resistance in Bax-deficient carcinoma. J Cell Biol 2010; 188:851 - 62; http://dx.doi.org/10.1083/jcb.200912070; PMID: 20308427
- Yamaguchi R, Janssen E, Perkins G, Ellisman M, Kitada S, Reed JC. Efficient elimination of cancer cells by deoxyglucose-ABT-263/737 combination therapy. PLoS One 2011; 6:e24102; http://dx.doi.org/10.1371/journal.pone.0024102; PMID: 21949692
- Zagorodna O, Martin SM, Rutkowski DT, Kuwana T, Spitz DR, Knudson CM. 2-deoxyglucose-induced toxicity is regulated by Bcl-2 family members and is enhanced by antagonizing Bcl-2 in lymphoma cell lines. Oncogene 2012; 31:2738 - 49; http://dx.doi.org/10.1038/onc.2011.454; PMID: 21986940
- Merino R, Ding L, Veis DJ, Korsmeyer SJ, Nuñez G. Developmental regulation of the Bcl-2 protein and susceptibility to cell death in B lymphocytes. EMBO J 1994; 13:683 - 91; PMID: 8313913
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003; 115:577 - 90; http://dx.doi.org/10.1016/S0092-8674(03)00929-2; PMID: 14651849
- Sutherland C.. What Are the bona fide GSK3 Substrates? Int J Alzheimers Dis. 2011; 2011:505607; PMID: 21629754
- Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 2006; 21:749 - 60; http://dx.doi.org/10.1016/j.molcel.2006.02.009; PMID: 16543145
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci 2003; 116:1175 - 86; http://dx.doi.org/10.1242/jcs.00384; PMID: 12615961
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA 2005; 102:8204 - 9; http://dx.doi.org/10.1073/pnas.0502857102; PMID: 15928081
- Maddocks ODK, Vousden KH. Metabolic regulation by p53. J Mol Med (Berl) 2011; 89:237 - 45; http://dx.doi.org/10.1007/s00109-011-0735-5; PMID: 21340684
- Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P, Mahdi AA. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget 2011; 2:948 - 57; PMID: 22248668
- Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, et al. p53 regulates mitochondrial respiration. Science 2006; 312:1650 - 3; http://dx.doi.org/10.1126/science.1126863; PMID: 16728594
- Martínez-Paniagua MA, Baritaki S, Huerta-Yepez S, Ortiz-Navarrete VF, González-Bonilla C, Bonavida B, et al. Mcl-1 and YY1 inhibition and induction of DR5 by the BH3-mimetic Obatoclax (GX15-070) contribute in the sensitization of B-NHL cells to TRAIL apoptosis. Cell Cycle 2011; 10:2792 - 805; http://dx.doi.org/10.4161/cc.10.16.16952; PMID: 21822052
- Coloff JL, Macintyre AN, Nichols AG, Liu T, Gallo CA, Plas DR, et al. Akt-dependent glucose metabolism promotes Mcl-1 synthesis to maintain cell survival and resistance to Bcl-2 inhibition. Cancer Res 2011; 71:5204 - 13; http://dx.doi.org/10.1158/0008-5472.CAN-10-4531; PMID: 21670080
- Tagawa H, Karnan S, Suzuki R, Matsuo K, Zhang X, Ota A, et al. Genome-wide array-based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM. Oncogene 2005; 24:1348 - 58; http://dx.doi.org/10.1038/sj.onc.1208300; PMID: 15608680
- Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J 2008; 412:179 - 90; http://dx.doi.org/10.1042/BJ20080281; PMID: 18466115
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008; 30:214 - 26; http://dx.doi.org/10.1016/j.molcel.2008.03.003; PMID: 18439900
- Zhong D, Liu X, Schafer-Hales K, Marcus AI, Khuri FR, Sun SY, et al. 2-Deoxyglucose induces Akt phosphorylation via a mechanism independent of LKB1/AMP-activated protein kinase signaling activation or glycolysis inhibition. Mol Cancer Ther 2008; 7:809 - 17; http://dx.doi.org/10.1158/1535-7163.MCT-07-0559; PMID: 18413794
- Ramírez-Peinado S, Alcázar-Limones F, Lagares-Tena L, El Mjiyad N, Caro-Maldonado A, Tirado OM, et al. 2-deoxyglucose induces Noxa-dependent apoptosis in alveolar rhabdomyosarcoma. Cancer Res 2011; 71:6796 - 806; http://dx.doi.org/10.1158/0008-5472.CAN-11-0759; PMID: 21911456
- Martinez-Outschoorn UE, Lin Z, Ko YH, Goldberg AF, Flomenberg N, Wang C, et al. Understanding the metabolic basis of drug resistance: therapeutic induction of the Warburg effect kills cancer cells. Cell Cycle 2011; 10:2521 - 8; http://dx.doi.org/10.4161/cc.10.15.16584; PMID: 21768775
- Panieri E, Toietta G, Mele M, Labate V, Ranieri SC, Fusco S, et al. Nutrient withdrawal rescues growth factor-deprived cells from mTOR-dependent damage. Aging (Albany NY) 2010; 2:487 - 503; PMID: 20739737
- Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer 2009; 9:563 - 75; http://dx.doi.org/10.1038/nrc2676; PMID: 19629071