Abstract
Comment on: Panneerselvam J, et al. Cell Cycle 2012; 11:2947-55.
Fanconi anemia (FA) is a rare inherited disease. Besides anemia and other symptoms, the patients also show a high cancer penetrance, due to a defect in one of the genes that repair DNA crosslink, and, thus, constitute a potent tumor-suppressive pathway. These genes include BRCA2, also called FA-BRCA pathway, of which the FANCL is a key member, and the catalytic subunit of a protein complex of E3 ubiquitin ligase. Deficiency in crosslink repairs also makes cancer cells in FA patients supersensitive to crosslink-causing agents, such as the chemotherapeutic drug cisplatin.
A paper in the August 1, 2012 issueCitation1 from Fei’s lab, together with a previous one,Citation2 shows that a shorter alternative splice variant of FANCL, dubbed FAVL, is highly expressed in sporadic bladder cancer as an oncoprotein capable of promoting cancer formation and rendering cells resistant to cisplatin. Restated, while the long form of FANCL, considered as the wild-type (wt), is a tumor suppressor, the short one (FAVL) resembles an oncogene, somewhat opposite to the case of Bcl-x wherein the long splice form (Bcl-xL) is oncogenic, but the short splice form (Bcl-xS) is tumor-suppressive. Hence, in bladder cancer, the FA-BRCA-suppressive pathway can be flipped to an oncogenic one simply by alternative splicing of a key component. This exciting finding raises an intriguing question as to whether a similar flip, which fortunately may be reversible and manageable, occurs also in other types of sporadic cancer. Relevant studies in sporadic malignancies have hitherto been focused on the BRCA1 and BRCA2 in some reproductive cancers. Fei’s study incites us to interrogate FANCL and other FA members in other types of sporadic malignancy, not only for mechanistic insights, but also for therapeutic purposes, because tumors with a normal or abnormal FA-BRCA-suppressive pathway may respond differently to crosslink-causing drugs, as seen in bladder cancer cells.
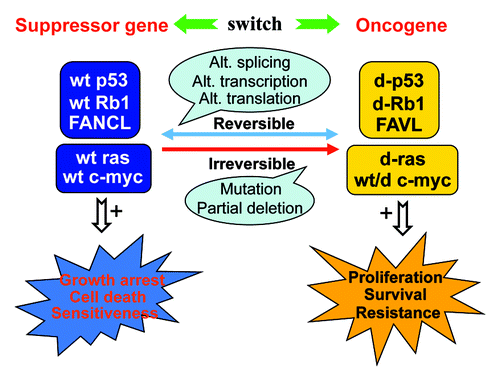
Besides Bcl-x and FANCL, many other genes can also be switched between an oncogenic and a suppressive status to meet cells’ needs in different situations. The switches can occur via many reversible mechanisms including alternative splicing, alternative transcription or translation initiation, etc., or via irreversible mechanisms like mutation or partial deletion (). Some genes, like c-myc,Citation3 may not be switched often, because their wt is versatile, e.g., can cause proliferation or cell death, although some of their variants or mutants pro-and-con proliferation or apoptosis.
Figure 1. The wild-type (wt) of tumor suppressor genes exemplified by p53, Rb1 and FANCL can be switched to a different (d) form with oncogenic features, whereas the wt of canonical oncogenes, exemplified by k-ras and c-myc, are versatile or can be switched to a form with suppressive traits. The switch may occur via reversible mechanisms such as alternative (Alt.) transcription, splicing or translation, or via irreversible mechanisms such as mutation or partial deletion. Tumor-suppressive functions are typically manifested as enhanced (+) growth arrest, cell death or sensitivity to cancer treatments, whereas oncogenic traits include increased (+) proliferation, survival or resistance to therapies.
Nobody was born as a good or bad person; environment makes what we are, basically. Similarly, none of our genes initially favors or disfavors cancer. In other words, there probably is no such thing called tumor suppressor gene or oncogene, although such dichotomy of cancer-related genes has helped in delineating our research. For instance, although p53 represents tumor suppressor genes and k-ras represents oncogenes, they are actually much alike: overexpression of their wt form often causes cell death,Citation4 but overexpression of their mutants promotes cancer formation. Ras-induced cell death is a paradigm of oncogene-induced senescence.Citation5 p53 was initially classified as an oncogene for its high level in cancer, but later reclassified as a suppressor, because the alleles in cancer were found to be mutants. Probably the reclassification is unnecessary, since some of its 30,000 mutantsCitation6 and some of its normal variants derived from alternative splicing or transcription initiationCitation7 are oncogenic. Another suppressor gene, Rb1, which sometimes also helps cell proliferation or survival, is overexpressed or amplified in some cancers and induces some tumors in animals,Citation8 although whether some of its 932 mutantsCitation9 are oncogenic is understudied. Why do cancer cells not simply delete the p53 or Rb1 (which occurs only at low frequency), but instead undergo complicated mutations? Probably, switching these genes to a less suppressive or more oncogenic form helps their survival, since genes are actually more multi-faceted dice than two-sided coins, and cells holding more of those switches live better. This “switch” hypothesis, with the FANCL-FAVL flip as a paradigm, inspires us to conceptualize, and thus act, differently. For instance, those of us who consider p53 also an oncogene may use knockout models more cautiously because of a greater concern of going too far from the reality where most cancers do not lack the whole gene, but instead, highly express (mutated) p53.
References
- Panneerselvam J, et al. Cell Cycle 2012; 11:2947 - 55; http://dx.doi.org/10.4161/cc.21400; PMID: 22828653
- Zhang J, et al. J Clin Invest 2010; 120:1524 - 34; http://dx.doi.org/10.1172/JCI40908; PMID: 20407210
- Wang C, et al. Cancer Biol Ther 2011; 11:615 - 26; http://dx.doi.org/10.4161/cbt.11.7.14688; PMID: 21278493
- Singh A, et al. FASEB J 2005; 19:161 - 9; http://dx.doi.org/10.1096/fj.04-2584hyp; PMID: 15677339
- McDuff FK, et al. Cell Signal 2011; 23:6 - 13; http://dx.doi.org/10.1016/j.cellsig.2010.07.004; PMID: 20633638
- Soussi T, et al. Hum Mutat 2010; 31:1020 - 5; http://dx.doi.org/10.1002/humu.21313; PMID: 20572016
- Marcel V, et al. Cell Death Differ 2011; 18:1815 - 24; http://dx.doi.org/10.1038/cdd.2011.120; PMID: 21941372
- Viatour P, et al. Dis Model Mech 2011; 4:581 - 5; http://dx.doi.org/10.1242/dmm.008060; PMID: 21878458
- Valverde JR, et al. BMC Genet 2005; 6:53 - 61; http://dx.doi.org/10.1186/1471-2156-6-53; PMID: 16269091