Abstract
Comment on: Beier UH, et al. Sci Signal 2012; 5:ra45.
Keywords: :
Renal transplantation is curative for end-stage renal disease. To ensure patient and graft survival, the recipient’s immune system must be suppressed in a carefully controlled manner, avoiding allograft rejection while maintaining protective immunity against infection and cancer. Current immunosuppressive regimens, involving induction with T cell-depleting antibodies and maintenance with calcineurin inhibitors, are effective at reducing acute cellular rejection. However, such reduction of acute rejection has not led to improved long-term allograft survival,Citation1 and there are persistent risks of malignancies and infections arising from maintenance immunosuppression. Long-term allograft loss is largely attributable to chronic allograft nephropathy, a multifactorial disease process involving immune- and non-immune factors, leading to a gradual decline in renal function. T-regulatory (Treg) cells, characterized by expression of the transcription factor Foxp3, are a subset of T cells capable of attenuating immune responses in an antigen-specific manner, and can help prevent long-term allograft loss.Citation2 Unfortunately, the induction agent Thymoglobulin targets both effector T cells and Tregs, and Basiliximab (CD25 monoclonal antibody) depletes Tregs as a result of their constitutive CD25 expression. Likewise, maintenance agents such as calcineurin inhibitors and the newly introduced Belatacept (CTLA4-Ig) impair Treg function.Citation3
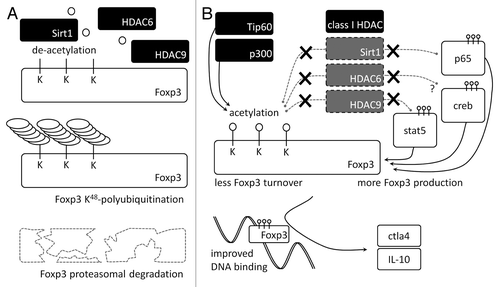
We have shown that Treg-suppressive function can be selectively enhanced by targeting of the histone/protein deacetylases (HDAC)-9, HDAC6 and Sirtuin-1 (Sirt1).Citation4-Citation6 Indeed, all three HDAC enzymes can deacetylate Foxp3, and combined genetic or pharmacologic targeting of these HDACs can be additive in improving Treg function.Citation7 Foxp3 acetylation is essential at regulating the amount of available protein, as Foxp3 is subject to rapid turnover via ubiquitination at unacetylated lysine residues ().Citation8 In addition, we identified individual transcription factors subject to deacetylation by these HDACs, and which are more transcriptionally active when acetylated (). Sirt1 can deacetylate lysine 310 of the p65 subunit of nuclear factor κB, also known as RelA.Citation5 Deletion of HDAC9 leaves signal transducer and activator of transcription 5 (Stat5) more acetylated, and acetylated Stat5 is stabilized in its transcriptionally active phosphorylated dimer.Citation7 Furthermore, we have evidence that HDAC6 can deacetylate cyclic AMP-responsive element-binding protein (CREB). HDAC6 is normally located in the cytosol, but can translocate into the nucleus upon T cell activation.Citation7 Taken together, both increased Foxp3 gene transcription and translation, as well as delayed proteasomal turnover, increase Foxp3 expression in Treg cells. In addition, acetylation of certain lysine residues can promote the DNA binding and transcriptional activity of Foxp3 ().Citation9 At present, many details are lacking as to which specific HDACs and histone acetyltransferases (HATs) control the acetylation of individual lysine residues of Foxp3. Recently, Kwon et al. reported K31, K262 and K267 act as Sirt1-dependent acetylation sites.Citation10 We hypothesize that HDAC6 might deacetylate different lysine residues on Foxp3, and are currently investigating this question.
Figure 1. HDACs control Foxp3+Treg function. (A) HDAC6, HDAC9 and Sirt1 deacetylate Foxp3 lysine residues, enabling ubiquitination and proteasomal degradation. (B) Pharmacologic targeting of HDAC isoforms facilitating Foxp3 deacetylation favors Foxp3 acetylation by histone acetyltransferases, preserving Foxp3 protein. Furthermore, acetylation of certain lysine residues improves DNA binding and transcriptional activity of Foxp3. In addition, Foxp3 translation is increased due to removal of inhibitory effects on transcription factors promoting Foxp3 gene expression. Taken together, these effects can improve Treg function and number. Toxic effects on other HDACs are minimized due to isoform-selective HDAC inhibitors. Abbreviations: Tip60, 60 kDa Tat-interactive protein; p300, histone acetyltransferase p300; Sirt1, Sirtuin-1; HDAC, histone/protein deacetylase; Foxp3, forkhead box P3; K, lysine; ctla4, Cytotoxic T-lymphocyte protein 4; IL, interleukin; stat5, signal transducer and activator of transcription 5; creb, Cyclic AMP-responsive element-binding protein; p65, transcription factor p65.
Remarkably, we found that combined inhibition and/or deletion of HDAC6 and Sirt1, and to a lesser extent HDAC6/HDAC9 and HDAC9/Sirt1, were additive in improving Treg function.Citation7 Combining isoform-specific inhibitors of the biologically relevant HDAC offers advantages beyond maximizing therapeutic efficacy. Non-selective HDAC inhibitors have been studied in cancer therapy, and their use is limited by their toxicities. Avoiding class I HDAC inhibition altogether by using selective HDAC inhibitors may bypass similar limitations for HDAC inhibition aimed at strengthening Treg-suppressive function. Of note, Sirt1 and HDAC6 can already be targeted with isoform-selective inhibitors, while no HDAC9-specific pharmacologic inhibitors are yet available.
In conclusion, we have demonstrated that HDAC6, HDAC9 and Sirt1 negatively regulate Foxp3+ Treg, and that combined isoform-specific targeting of these HDAC has additive therapeutic effects. This may be an interesting therapeutic option for enhancing Treg function in transplant recipients.
Related Research Data
References
- Meier-Kriesche HU, et al. Am J Transplant 2004; 4:378 - 83; http://dx.doi.org/10.1111/j.1600-6143.2004.00332.x; PMID: 14961990
- Tsaur I, et al. Kidney Int 2011; 79:1005 - 12; http://dx.doi.org/10.1038/ki.2010.533; PMID: 21270769
- Riella LV, et al. Am J Transplant 2012; 12:846 - 55; http://dx.doi.org/10.1111/j.1600-6143.2011.03929.x; PMID: 22300534
- de Zoeten EF, et al. Mol Cell Biol 2011; 31:2066 - 78; http://dx.doi.org/10.1128/MCB.05155-11; PMID: 21444725
- Beier UH, et al. Mol Cell Biol 2011; 31:1022 - 9; http://dx.doi.org/10.1128/MCB.01206-10; PMID: 21199917
- Tao R, et al. Nat Med 2007; 13:1299 - 307; http://dx.doi.org/10.1038/nm1652; PMID: 17922010
- Beier UH, et al. Sci Signal 2012; 5:ra45; http://dx.doi.org/10.1126/scisignal.2002873; PMID: 22715468
- van Loosdregt J, et al. Blood 2010; 115:965 - 74; http://dx.doi.org/10.1182/blood-2009-02-207118; PMID: 19996091
- Liu Y, et al. PLoS One 2012; 7:e29035; http://dx.doi.org/10.1371/journal.pone.0029035; PMID: 22247766
- Kwon HS, et al. J Immunol 2012; 188:2712 - 21; http://dx.doi.org/10.4049/jimmunol.1100903; PMID: 22312127