Abstract
Comment on: Qian Y, et al. Proc Natl Acad Sci USA 2012; 109:11300-5.
The p53 tumor suppressor is an essential barrier against cancer, which is evidenced by loss of p53 activity in the majority of human cancers and unexceptionally early onset of tumors in p53-knockout mice and human Li-Fraumeni syndrome patients. p53 is a sequence-specific transcription factor that regulates gene expression via binding to the consensus p53-responsive element (p53-RE). p53 acts as a central signaling coordinator that receives upstream signals, including DNA damage, aberrant oncogenic activity, hypoxia and ribosomal stress, and consequently activates its targets leading to downstream cellular outcomes, including cell cycle arrest, DNA repair, apoptosis and cellular senescence. However, it is not yet clear how p53 dictates a cell to survival vs. death in response to a specific stress.
It has been shown that the ability of p53 to regulate target genes can be modulated by posttranslational modifications. For example, acetylation of p53 on lysine 120 by Tip60 is crucial for p53-mediated apoptosis but not cell cycle arrest.Citation1 Consistently, p90, a cofactor of Tip60, is required for Tip60-dependent p53 acetylation and, thus, specifically promotes p53-mediated apoptosis.Citation2 Interestingly, evidence showed that p53 activity is also regulated by binding partners that do not affect p53 modifications. For example, ASPP proteins interact with p53 and enhance the DNA binding and transactivation function of p53 on the promoters of pro-apoptotic genes, such as Bax and PIG-3.Citation3 Similarly, p53β preferentially enhances p53 transcriptional activity on the Bax but not p21 promoter.Citation4 In addition, hCAS/CSE1L, a factor identified in the p53 transcription complex, specifically associates with the PIG-3 promoter to induce p53-mediated apoptosis.Citation5 By contrast, BRCA1Citation6 and HZFCitation7 direct p53-dependent transactivation of pro-arrest target genes over pro-apoptotic target genes.
Recently, we found that DEC1, a target of the p53 family and a basic helix-loop-helix (bHLH) transcription factor, modulates p53-depedent cell survival vs. cell death in response to genotoxic stress.Citation8 In addition, DEC1 is capable of inducing cell cycle arrest in a p53-independent manner.Citation9 As a transcription factor, DEC1 is able to repress gene expression through class B E-box elements, but activate gene expression through Sp1 sites.10 However, DEC1 does not regulate p53 expression. Instead, DEC1 specifically attenuates p53-dependent transactivation of MIC-1, but not other p53 target genes, including two cell cycle regulators (p21 and GADD45α), four pro-apoptotic genes (Puma, Bax, PolH and FDXR) and Mdm2. Consistent with this, knockdown of DEC1 enhances p53-dependent induction of MIC-1 and apoptosis in response to DNA damage. Moreover, upon knockdown of MIC-1, the effect of DEC1 knockdown on p53-dependent cell death is abrogated. These findings suggest that DEC1 forms a feedback loop with p53 to control the response of DNA damage-induced cell survival vs. cell death via MIC-1.
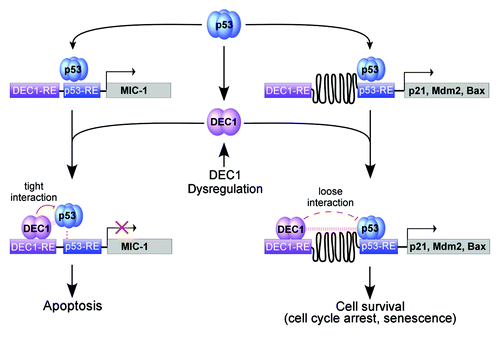
To characterize the mechanism by which DEC1 differentially affects the ability of p53 to regulate its target genes, we found that DEC1 associates with the p53 tetramerization domain through its bHLH motif. Importantly, we noticed that the distance between the DEC1-RE and p53-RE on the MIC-1 promoter is only 17 nt, but more than 280 nt on the p21 and Mdm2 promoters. ChIP and ChIP-reChIP assays showed that DEC1 binds to the MIC-1, p21 and Mdm2 promoters but co-localizes with p53 only on the MIC-1 promoter. In addition, DEC1 preferentially suppresses p53 binding to the MIC-1 promoter upon DNA damage. Thus, we hypothesize that both DEC1 and p53 have to bind to the same promoter in close proximity to allow DEC1-p53 interaction, which is prerequisite for DEC1 to decrease p53 DNA-binding affinity and consequently reduce MIC-1 expression (). This unique regulatory loop between DEC1 and p53 provides a clue for investigating other p53 target genes potentially regulated by DEC1.
Figure 1. A model for DEC1 to differentially modulate p53-dependent gene expression. Upon binding to the p53-responsive element (p53-RE), p53 induces an arrary of pro-survival and pro-apoptotic genes, including p21, Mdm2, Bax, MIC-1 and DEC1. Since the p53-RE is adjacent to the DEC1-RE on the MIC-1 promoter, p53 and DEC1 interact tightly on the MIC-1 promoter, which then weakens the ability of p53 to bind to the MIC-1 promoter and consequently p53-induction of MIC-1. By contrast, due to a large space between the DEC1-RE and the p53-RE on the promoters of other p53 target genes, including p21, Mdm2 and Bax, the interaction between DEC1 and p53 on the target gene promoters is too weak to inhibit p53 DNA-binding activity. Therefore, DEC1 does not inhibit the ability of p53 to induce p21, Mdm2 and Bax. Together, we hypothesize that DEC1 forms a feedback loop with p53 to control the response of DNA damage-induced cell survival vs. cell death via MIC-1 and that dysregulation of DEC1 alters the sensitivity of tumors to cancer therapies via the p53-DEC1-MIC-1 loop.
p53 is frequently mutated in tumors, and most tumor-derived p53 mutations are located in the DNA-binding domain but not the tetramerization domain. Importantly, tumor-derived p53 mutants have gain-of-function activities contributing to tumorigenesis and chemotherapeutical resistance. Since p53 tetramerization domain is found to interact with DEC1, it is likely that DEC1 and mutant p53 physically interact on a promoter that carries DEC1-RE and mutant p53-RE. Thus, it would be of great interest to examine whether DEC1 modulates the function of mutant p53 in tumor progression and the resistance of tumors to therapy. In addition, as a member of the p53 family, p63 and p73 share a high-sequence identity with p53 in the tetramerization domain along with the transactivation and DNA-binding domains. Interestingly, p63 and p73 not only possess p53-like functions in tumor suppression, but also play a key role in development. Thus, it would be interesting to examine whether DEC1 physically interacts with p63 and p73 on a promoter that carries DEC1-RE and p53-RE, and whether DEC1 modulates the function of p63 and p73 in tumor suppression and development.
Acknowledgments
This work is supported in part by NIH grant CA076069.
References
- Tang Y, et al. Mol Cell 2006; 24:827 - 39; http://dx.doi.org/10.1016/j.molcel.2006.11.021; PMID: 17189186
- Dai C, et al. Proc Natl Acad Sci USA 2011; 108:18937 - 42; http://dx.doi.org/10.1073/pnas.1110988108; PMID: 22084066
- Samuels-Lev Y, et al. Mol Cell 2001; 8:781 - 94; http://dx.doi.org/10.1016/S1097-2765(01)00367-7; PMID: 11684014
- Bourdon JC, et al. Genes Dev 2005; 19:2122 - 37; http://dx.doi.org/10.1101/gad.1339905; PMID: 16131611
- Tanaka T, et al. Cell 2007; 130:638 - 50; http://dx.doi.org/10.1016/j.cell.2007.08.001; PMID: 17719542
- MacLachlan TK, et al. Mol Cell Biol 2002; 22:4280 - 92; http://dx.doi.org/10.1128/MCB.22.12.4280-4292.2002; PMID: 12024039
- Das S, et al. Cell 2007; 130:624 - 37; http://dx.doi.org/10.1016/j.cell.2007.06.013; PMID: 17719541
- Qian Y, et al. Proc Natl Acad Sci USA 2012; 109:11300 - 5; http://dx.doi.org/10.1073/pnas.1203185109; PMID: 22723347
- Qian Y, et al. J Biol Chem 2008; 283:2896 - 905; http://dx.doi.org/10.1074/jbc.M708624200; PMID: 18025081
- Qian Y, et al. J Biol Chem 2011; 286:12033 - 41; http://dx.doi.org/10.1074/jbc.M110.207241; PMID: 21317427