Pore-forming toxins (PFT) form the largest family of secreted toxins from pathogenic bacteria.Citation1,Citation2 Staphylococcus aureus produces different hemolysins, and α-hemolysin is one of the well-studied PFTs for which the X-ray structure of the pore is available.Citation1 These toxins form heptameric pores of 1–2 nm in size in cell membranes, leading to various outcomes, including apoptotic cell death, in the host cells.Citation2 As epithelial cells and keratinocytes are the major sites of pathogen entry, we have set out to investigate the molecular machinery driving cell death mediated by α-hemolysin in these cell types. To our surprise, we have detected an initiator caspase role for caspase-2 during PFT-mediated apoptosis, and further investigations revealed that caspase-2 is activated in a PIDDosome-independent manner under these conditions.Citation3
Caspases are effector proteases of the apoptotic cascade and broadly divided into two major classes depending on the structure and chronology of their activation as initiator and effector caspases.Citation4 Despite being the first mammalian caspases to be cloned and highly conserved, the physiological role of this caspase remains unclear.Citation5,Citation6 Caspase-2-deficient mice develop normally, with mild defects, and emerging evidence suggests a role for this caspase in regulating DNA damage response, cell cycle regulation and tumor suppression.Citation6 Like other initiator caspases, caspase-2 is activated by dimerization mediated through the CARD domains, and initial studies revealed that caspase-2 is recruited to a high molecular weight complex distinct from the apoptosome in cell lysates upon transfer from 4°C to 37°C.Citation7 During genotoxic stress, caspase-2 has been shown to be recruited to a multimeric protein complex, PIDDosome, constituting a Death Domain (DD)-containing PIDD (P53-induced protein with death domain) and a DD and CARD domain-containing adaptor protein RAIDD (RIP-associated ICH-1/CED-3 homologous protein with death domain).Citation8 However, recent studies reveal that caspase-2 can be activated in the absence of PIDDosome, which warrants for the presence of additional caspase-2 activation platforms.Citation9 However, whether such caspase-2 complexes are formed in apoptotic cells and the unique apoptotic stimuli where caspase-2 is activated as an initiator caspase remain unclear.
By exploiting an “in situ caspase trapping” approach employing biotin-VAD, we identified that caspase-2, but not caspase-8 or caspase-9, is precipitated as a proximal caspase during PFT-mediated apoptosis in HeLa cells.Citation3 Consistently, loss of caspase-2 prevented PFT-mediated apoptosis in HeLa cells and mouse embryonic fibroblasts. Interestingly, loss of either PIDD or RAIDD has failed to render protection to PFT-mediated apoptosis, and caspase-2 is activated in RAIDD-deficient cells. Caspase-2 is activated in caspase-3/7 DKO MEFs, suggesting that the activation and processing of caspase-2 under these settings is independent of effector caspases.
Intriguingly, PFT-mediated cell death is partially inhibited by loss of Bax and Bak, the major regulators of the mitochondrial outer membrane permeabilization or by the loss of effector caspases 3 and 7 (). It is currently unclear how caspase-2 accomplishes Bax/Bak activation, and further, it is tempting to propose that caspase-2 might forgo the loss of these two effector caspases and cleave the relevant substrates directly to accomplish cell death (). Recent studies employing N-terminal COFRADIC (combined fractional diagonal chromatography) to study the degradome of caspases revealed that the protease specificities of caspase-2 largely overlap with the effector caspases.Citation10 By applying Bimolecular Fluorescence Complementation (BiFc), we have checked for the intracellular localization of dimerized caspase-2-CARD domains in toxin-treated cells. Consistent with Bouchier-Hayes et al., we have detected a cytosolic staining of caspase-2-CARD-BiFc signals in HeLa cells after treatment with α-toxin.Citation3,Citation11 The BiFc signals for caspase-2-CARD domain exhibited a punctuate staining under these settings.Citation3
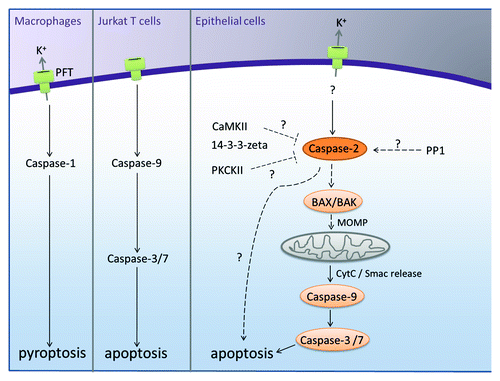
Figure 1. Pore-forming toxins induce various forms of cell death depending on the cell type and the extent of toxin insult. In epithelial cells, caspase-2 is activated as an initiator caspase in a PIDDosome-independent manner to regulate PFT-mediated cell death. The PFT-mediated cell death is partially dependent on Bax and Bak as well as on effector caspases, revealing that caspase-2 might also exhibit features of effector caspases. Activation of caspase-2 has been shown to be regulated by PP1, 14-3-3 zeta and PKCK2 under various settings, and it would be interesting to characterize if caspases-2 phosphorylation regulates its activation in response to PFTs.
By employing high-resolution gel filtration chromatography, we detected that caspase-2 is consistently detected in a high molecular weight fraction larger than a single ribosome (approx. 3 MDa) in the toxin-treated cells. This complex is devoid of RAIDD or PIDD, and we detect either the full-length or the processed fragments of caspase-2 in this high molecular weight complex in the apoptotic cells. Detection of full-length caspase-2 to this HMW complex suggests that caspase-2 is possibly recruited to this complex for its activation during PFT-mediated apoptosis in these cell types.
Previous studies revealed that PFTs induce K+ efflux in the cells, leading to various outcomes ranging from inflammasome activation to pyroptosis to autophagy depending on the cell type and the extent of the toxin insult (). As expected, inhibition of PFT-mediated K+ efflux prevented the recruitment and activation of caspase-2 to the HMW complex, thus preventing cell death. Further, physiological concentrations of K+ and Na+ ions have been shown to prevent the assemblage of apoptosome as well as the activation of caspase-2 in cell-free systems.Citation7,Citation12 Whether reduction in the physiological concentrations of K+ ions upon toxin treatment spontaneously led to the direct dimerization of caspase-2 needs further studies. Further, it is currently unclear if there is a concurrent increase in the intracellular calcium ion concentrations upon toxin treatment, and if there is any role for Ca2+ influx in mediating caspase-2 activation in these cell types.
Interestingly, caspase-2 is regulated by phosphorylation under various settings, and it would be interesting to test if phosphorylation/dephosphorylation events regulate the activation of caspase-2 upon PFT treatment.Citation5 In conclusion, our studies reveal unique apoptotic stimuli where caspases-2 is activated as an initiator caspase to mediate apoptotic cell death in non-lymphoid cells. These observations possibly open up new avenues to decipher potential PIDDosome-independent caspase-2 activation platforms.
References
- Bischofberger M, et al. Curr Opin Cell Biol 2009; 21:589 - 95; http://dx.doi.org/10.1016/j.ceb.2009.04.003; PMID: 19442503
- Gonzalez MR, et al. Cell Mol Life Sci 2008; 65:493 - 507; http://dx.doi.org/10.1007/s00018-007-7434-y; PMID: 17989920
- Imre G, et al. EMBO J 2012; 31:2615 - 28; http://dx.doi.org/10.1038/emboj.2012.93; PMID: 22531785
- Thornberry NA, et al. Science 1998; 281:1312 - 6; http://dx.doi.org/10.1126/science.281.5381.1312; PMID: 9721091
- Krumschnabel G, et al. Oncogene 2009; 28:3093 - 6; http://dx.doi.org/10.1038/onc.2009.173; PMID: 19581929
- Kumar S. Nat Rev Cancer 2009; 9:897 - 903; http://dx.doi.org/10.1038/nrc2745; PMID: 19890334
- Read SH, et al. J Cell Biol 2002; 159:739 - 45; http://dx.doi.org/10.1083/jcb.200209004; PMID: 12460989
- Tinel A, et al. Science 2004; 304:843 - 6; http://dx.doi.org/10.1126/science.1095432; PMID: 15073321
- Manzl C, et al. J Cell Biol 2009; 185:291 - 303; http://dx.doi.org/10.1083/jcb.200811105; PMID: 19364921
- Wejda M, et al. J Biol Chem 2012; http://dx.doi.org/10.1074/jbc.M112.384552; PMID: 22825847
- Bouchier-Hayes L, et al. Mol Cell 2009; 35:830 - 40; http://dx.doi.org/10.1016/j.molcel.2009.07.023; PMID: 19782032
- Cain K, et al. J Biol Chem 2001; 276:41985 - 90; http://dx.doi.org/10.1074/jbc.M107419200; PMID: 11553634