Abstract
FBXW7, a component of E3 ubiquitin ligase, plays an important role in mitotic checkpoint, but its role remains unclear. Aurora B is a mitotic checkpoint kinase that plays a pivotal role in mitosis by ensuring correct chromosome segregation and normal progression through mitosis. Whether Aurora B and FBXW7 are coordinately regulated during mitosis is not known. Here, we show that FBXW7 is a negative regulator for Aurora B. Ectopic expression of FBXW7 can suppress the expression of Aurora B. Accordingly, FBXW7 deficiency leads to Aurora B elevation. Mechanistic studies show that all FBXW7 isoforms are negative regulators of Aurora B expression through ubiquitination-mediated protein degradation. Aurora B interacts with R465 and R505 residues of WD 40 domain of FBXW7. Significantly, inverse correlation between FBXW7 and Aurora B elevation is translated into the deregulation of mitosis. FBWX7 expression mitigates Aurora B-mediated cell growth and mitotic deregulation. In addition, FBXW7 reduces the percentage of multinucleated cells caused by Aurora B overexpression. These data suggest that FBXW7 is an important negative regulator of Aurora B, and that the loss or mutation of FBXW7 as seen in many types of cancer could lead to an abnormal elevation of Aurora B and result in deregulated mitosis, which accelerates cancer cell growth.
Keywords: :
Introduction
Aurora kinases play important roles in mitosis.Citation1-Citation7 There are three Aurora kinase family members (A, B and C) that are expressed at maximum levels during mitosis. Aurora A and Aurora B differ in subcellular localization, and each performs distinct tasks during mitosis.Citation1 Aurora A is located at the centrosomes during prophase and later relocates to the spindle poles during prometaphase and metaphase. Aurora B is part of the chromosomal passenger protein complex (CPC) and is located on the chromosome arms during prophase and at the centromeres during prometaphase and metaphase.Citation1,Citation8 During cytokinesis Aurora B subsequently localizes to the midbody. Aurora C is also a chromosomal passenger protein and, as such, behaves like Aurora B.Citation9 The protein levels of each Aurora kinase are strictly controlled during each stage of mitosis, primarily through ubiquitination-mediated degradation. Although Aurora B degradation has been studied, a detailed mechanistic regulatory pathway for Aurora B stability remains unclear.
The ubiquitin-mediated protein degradation pathway is important for controlling the abundance of proteins and plays an essential role in maintaining normal cellular function, including cell cycle. Dysregulation of ubiquitin-mediated proteolysis results in the development of a variety of human cancers.Citation10 The SCF (SKP1-CUL1-F-box protein) ubiquitin ligases are best characterized as mammalian cullin RING ubiquitin ligases; the F-box protein provides the substrate targeting specificity of the complex. Sixty-nine F-box proteins have been identified in humans. FBXW7 (F-box and WD repeat domain-containing 7) is one of the F-box proteins that function as substrate-recognition subunits of SCF complexes. Although SCFFBXW7 is known to regulate the degradation of several important substrates, such as Notch, c-Myc, cyclin E and c-Jun,Citation11,Citation12 many of its substrates have yet to be identified. Importantly, FBXW7 is a tumor suppressor.Citation13-Citation15 It is found mutated in numerous cancersCitation13,Citation16-Citation18 and is mapped to 4q32, which is a site of heterozygosity loss in a number of cancers.Citation19 Furthermore, it is one of the p53-dependent genes involved in tumorigenesis.Citation16 The FBXW7 gene encodes three protein isoforms, the FBXW7α, -β and -γ form, which are translated from mRNAs transcribed from distinct 5′ exons with individual and unique promoters and joined with 10 shared exons.Citation17,Citation20 Loss of FBXW7 activity leads to chromosomal instability.Citation18 FBXW7 deregulation has been implicated as a cause of mitotic defects including the formation of micronuclei and abnormal chromosome numbers,Citation18 but the mechanism that relates FBXW7 to these defects is not completely understood. In this study, we found that FBXW7 and Aurora B interact and were able to further indicate that FBXW7 negatively regulates Aurora B via the ubiquitination pathway. Significantly, we demonstrate the role of the FBXW7 in controlling Aurora B during mitosis. Our studies indicate that the FBXW7-Aurora B axis has the potential to serve as a therapeutic intervention target in cancer treatment.
Results
FBXW7 negatively regulates Aurora B protein stability
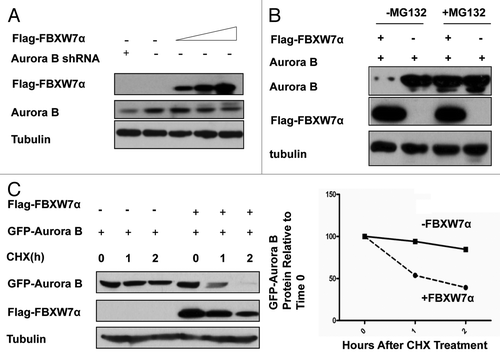
We previously showed that Aurora B is a kinase that can negatively regulate p53 stability.Citation21 Because the stability of both p53 and Aurora B is regulated through ubiquitination, ubiquitin-mediated stabilization could play an important role for coordinating this inverse relationship. We examined whether FBXW7, a p53 target gene product and an E3 ubiquitin ligase component, has a biological impact on Aurora B. The Aurora B levels decreased in a dose-dependent manner when cells were transfected with FBXW7 (). FBXW7-mediated Aurora B downregulation was suppressed by MG132, a proteasome inhibitor, suggesting the involvement of the 26S proteasome (). To investigate if FBXW7 downregulates Aurora B at the post-transcriptional level, we examined the turnover rate of Aurora B in the presence of the de novo protein synthesis inhibitor, cycloheximide (CHX) (). Indeed, overexpression of FBXW7 increases the turnover rate of Aurora B when compared with non-overexpression control group (). This result suggests that FBXW7 is a negative regulator of Aurora B.
Figure 1. FBXW7 regulates Aurora B stability. (A) FBXW7 negatively regulates the steady-state expression of Aurora B. 293T cells were co-transfected with the indicated plasmids and increasing amounts of FBXW7. Equal amounts of cell lysates were immunoblotted with the indicated antibodies. (B) FBXW7-mediated degradation of Aurora B is proteasome-dependent. 293T cells were co-transfected with the indicated plasmids. Cells were treated with MG132 for 6 h before harvesting. Equal amounts of cell lysates were immunoblotted with the indicated antibodies. (C) Aurora B turnover rate is increased in FBXW7 expressing cells. 293T cells transfected with the indicated plasmids were treated with cycloheximide (CHX) (100 µg/ml) for the indicated times. Cell lysates were immunoblotted with the indicated antibodies. Integrated OD values of GFP-Aurora B bands at each time point were measured using a densitometer. Levels of GFP-Aurora B at time 0 were set at 100%. Remaining GFP-Aurora B is indicated graphically (right).
FBXW7 regulates Aurora B protein stability through ubiquitination
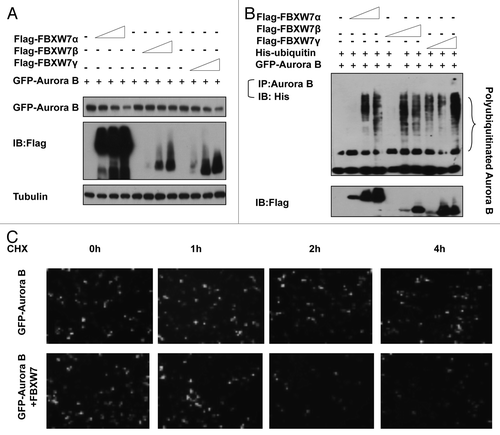
FBXW7 family has three isoforms: α (nulear), β (cytoplasmic) and γ (nucleaolar), each with distinct subcellular localization.Citation22 To see if all three of the isoforms have a similar impact on Aurora B, we examined the steady-state expression of Aurora B in the presence of increasing amounts of each FBXW7 isoform. Indeed, overexpression of FBXW7 α and γ isoforms can reduce the steady-state expression of Aurora B in a dose-dependent manner (). FBXW7β is less efficient in this assay. Nevertheless, we found that each FBXW7 isoform increased the ubiquitination level of Aurora B in a dose-dependent manner (). Also, we examined the turnover of GFP-Aurora B in the presence of cycloheximide (CHX) when cotransfected with increasing amounts of FBXW7 isoforms. As expected, the green signals of GFP-Aurora B decreased faster when GFP-Aurora B was cotransfected with FBXW7 isoforms (), reconfirming that FBXW7 can regulate Aurora B turnover. Representative pictures of FBXW7α transfectants are shown.
Figure 2. FBXW7 isoforms increase Aurora B turnover via increasing Aurora B ubiquitination. (A) Different FBXW7 isoforms regulate the steady-state expression of Aurora B in a dose-dependent manner. Lysates of 293T cells transfected with increasing amounts of the indicated FBXW7 isoforms and GFP-Aurora B were immunoblotted with the indicated antibodies. (B) FBXW7 downregulates Aurora B by increasing Aurora B ubiquitination. 293T cells were transfected with the indicated plasmids. The proteasome inhibitor MG132 was added 6 h prior to cell harvesting. The amount of ubiquitinated Aurora B was analyzed by immunoprecipitated (IP) with anti-Aurora B followed by immunoblotting (IB) with anti-His. (C) FBXW7α dereases the turnover of Aurora B. Indicated 293T cells were transfected with GFP-Aurora B with or without FBXW7 and then treated with cycloheximide (CHX) (100 µg/ml) for the indicated times. Signals of GFP-Aurora B from cells were observed under microscope.
FBXW7 is required for Aurora B degradation
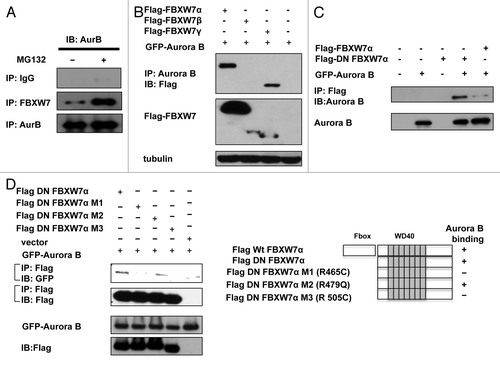
We have shown that FBWX7 overexpression leads to Aurora B degradation. To further study the role of FBXW7 in the regulation of Aurora B under physiological conditions, we employed HCT 116 FBXW7−/− cellsCitation18 for the studies. We analyzed wt and FBXW7−/− HCT116 cell lysates and found that HCT 116 FBXW7−/− cells had elevated endogenous Aurora B when compared with wt HCT116 cells (). Again, regulation of Aurora B stability was found to be proteasome-dependent, as MG132 treatment leads to stabilization of endogenous Aurora B (). We also examined the turnover of endogenous Aurora B in the presence of cycloheximide (CHX) in HCT 116 FBXW7−/− cells, and found that FBXW7 deficiency decelerated the turnover of endogenous Aurora B (). These results suggest that FBXW7 is a novel E3 ligase component involved in regulation of Aurora B stability. Although FBXW7 is required for the degradation of Aurora B, an interaction between FBXW7 and Aurora B has not been characterized. We demonstrated the endogenous interaction between FBXW7α and Aurora B (), and we also analyzed cell lysates cotransfected with FBXW7 isoforms and Aurora B. Indeed, FBXW7 isoforms were able to associate with Aurora B as assayed by co-ip (). We then mapped the Aurora B binding region on FBXW7. The results showed that Aurora B binds to the C terminus of FBXW7 (DN FBXW7), which contains WD40 repeatsCitation23(). We also mapped the Aurora B binding site on FBXW7. A co-ip assay suggests that FBXW7 R465 and R505 sites are responsible for binding Aurora B, as the mutation of these two sites (R465 C and R505C) resulted in loss of binding (). R479Q mutant is not critical. These results demonstrate that the WD40 repeats of CSN6 bind to Aurora B.
Figure 3. FBXW7 deficiency leads to Aurora B stabilization. (A) FBXW7 deficiency increases the steady-state expression of Aurora B. Indicated wt and FBXW7−/− HCT116 cells were immunoblotted with anti-Aurora B or actin antibodies. (B) Aurora B stability regulation is proteasome-dependent. Indicated wt and FBXW7−/− HCT116 cells were treated with MG132 6 h before harvesting. Equal amounts of cell lysates were immunoblotted with anti-Aurora B or Actin antibodies. (C) Aurora B turnover rate is reduced in FBXW7 deficient cells. Indicated wt and FBXW7−/− HCT116 cells were treated with cycloheximide (CHX) (100 µg/ml) for the indicated times. Cell lysates were immunoblotted with the indicated antibodies. Integrated OD values of bands at each time point were measured using a densitometer. Levels of Aurora B at time 0 were set at 100%. Remaining Aurora B is indicated graphically (right).
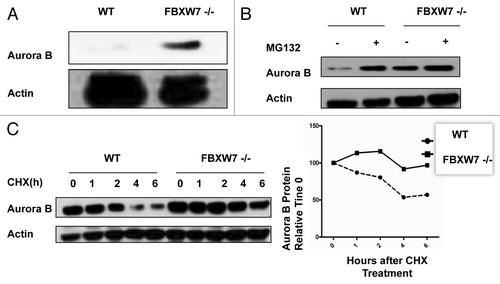
Figure 4. FBXW7 interacts with Aurora B through WD 40 repeat. (A) Endogenous assciation between FBXW7 and Aurora B. 293T cells were immunoprecipitated with indicated antibodies or IgG and immunoblotted with anti-Aurora B in the presence or absence of MG132. (B) Interaction of FBXW7 isoforms with Aurora B. Equal amounts of 293T cell lysates transfected with indicated FBXW7 isoforms were immunoprecipitated with anti-Aurora B and immunoblotted with anti-Flag. (C) Mapping of Aurora B binding domains on FBXW7. Indicated Flag-FBXW7α or C terminus FBXW7 (DN-FBXW7α) was cotransfected with Aurora B into 293T cells. Cell lysates were immunoprecipitated with anti-Flag and immunoblotted with anti-Aurora B. (D) Mapping of Aurora B binding sites on FBXW7α. Indicated Flag-DN FBXW7α or DN FBXW7α mutants were cotransfected with GFP-Aurora B into 293T cells. Cell lysates were immunoprecipitated with anti-Flag and immunoblotted with the indicated antibodies. Schematic representation of the mutants is shown.
FBXW7-AuroraB axis regulates cell growth and mitosis
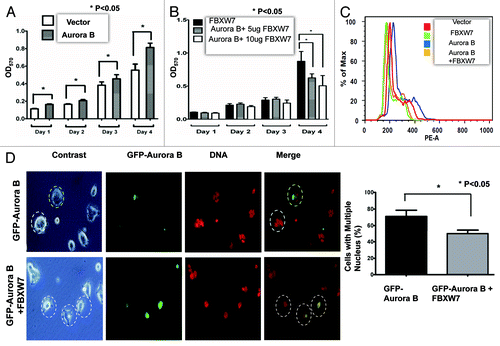
Since FBXW7 downregulates and thus antagonizes the activity of Aurora B, we reasoned that FBXW7 would have a role in cell proliferation. We were able to show that Aurora B overexpression does indeed facilitate cell growth (). Since FBXW7 can mediate Aurora B degradation, we sought to examine the impact of FBXW7 expression on growth in terms of cell proliferation and DNA histogram analysis of the cell cycle in Aurora B-overexpressing cells. We found that Aurora B-overexpressing 293T cells transfected with FBXW7 showed inhibited cell proliferation in a dose-dependent manner (). Because Aurora B is involved in the mitotic spindle checkpoint, either overexpression or downregulation of Aurora B will cause mitotic catastrophe. DNA histogram analysis by FACS showed that Aurora B overexpression led to increased DNA content well above normal levels, while Aurora B overexpression in the presence of FBXW7 prevented such a dysregulation from occurring (). Also, we showed that FBXW7 antagonized Aurora B-induced multinucleated cell formation (). 293T cells were transfected with GFP-Aurora B in the absence or presence of FBXW7 (plasmid ratio of GFP-Aurora B: FBXW7 = 1:3), and nuclear DNA was observed under fluorescence microscope. GFP-Aurora B expression alone led to mitotic defects and subsequently caused the formation of multinucleated cells as demonstrated by fluorescence microscopy (). Significantly, GFP-Aurora B cotransfected with FBXW7 had fewer multinucleated cells than GFP-Aurora B alone (). Thus, FBXW7 is able to reduce the percentage of multinucleated cells caused by Aurora B expression.
Figure 5. FBXW7 mitigates Aurora B-mediated cell proliferation and reduces multi-nuclei formation. (A) Aurora B promotes cell proliferation. Cells transfected with Aurora B were estimated by MTT assay every day for a total of 4 d. Results are expressed as an OD570. (B) FBXW7 inhibits Aurora B-mediated cell proliferation. Cell cotransfected with Aurora B and FBXW7 were estimated by MTT assay every day for a total of 4 d. Results are expressed as an OD570. (C) FBXW7 inhibits Aurora B-mediated increased DNA content during cell cycle. Cells cotransfected with Aurora B and FBXW7 were analyzed by FCCS. DNA histogram was investigated by FLOWJO program. PE-A indicates the DNA content. % of max is the % of cell analyzed. (D) FBXW7 antagonizes Aurora B-induced multinucleate cell formation. 293T cells were transfected with GFP-Aurora B or GFP-Aurora B + FBXW7 (in a plasmid ratio of 1:3) for 48 h. Phase-contrast images and the corresponding fluorescent micrographs are shown. DNA was stained with DAPI and was pseudocolored red. GFP-Aurora B-expressing cells are circled in the color-merged micrographs. Note that GFP-Aurora B cells cotransfected with FBXW7 exhibited less multinucleation than GFP-Aurora B cells. Random areas of the green cells were counted, and % multinucleate cells were plotted. Error bars: 95% confidence.
Since FBWX7 overexpression mitigates Aurora B-mediated mitotic deregulation, we then conducted further studies on the role of FBXW7 in regulating Aurora B and mitosis under physiological condition by employing HCT116 FBXW7−/− cells. We synchronized wt and FBXW7−/− HCT116 cells to the G2→M phase by nocodazole treatment. Cells were then released from nocodazole block for certain time points and analyzed by FACS for DNA content (). Interestingly, HCT 116 FBXW7−/− cells accumulated a higher DNA content than wt HCT 116 cells ( and ), suggesting that FBXW7 deficiency leads to accumulation of Aurora B, which, in turn, causes mitotic defects. On the other hand, overexpression of FBWX7 in HCT116 also led to mitotic defects as demonstrated by a high percentage of cells with abnormally high DNA content ( and ), suggesting that FBXW7 overexpression leads to excessive downregulation of Aurora B and subsequently causes mitotic defects as well.
Figure 6. FBXW7 deficiency leads to polyploidy. Indicated wt and FBXW7−/− HCT116 cells were synchronized to the G2→M phase by nocodazole. Cell samples, at labeled time points after release from nocodazole block, were stained with propidium iodide (PI) and analyzed by FACS for DNA content. DNA content histograms are shown for the time points as labeled.
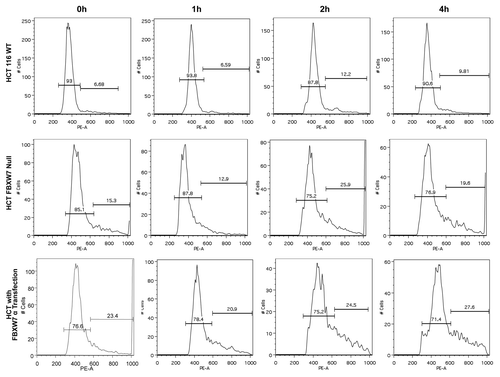
Table 1. Summary of percentage of polyploidy cells after release from nocodazole block
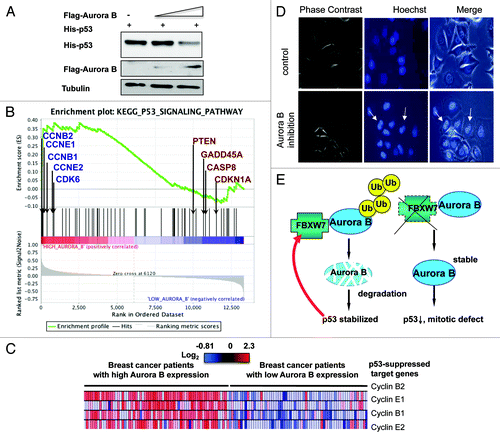
To understand the consequences of Aurora B accumulation due to FBXW7 deficiency, as in the case of HCT116 FBXW7−/− cells, we overexpressed Aurora B in cells and found that Aurora B caused the downregulation of p53 in a dose-dependent manner (). This data suggests that p53 downregulation may account for mitotic defects in FBXW7−/− HCT116 cells. Interestingly, our Gene Set Enrichment Analysis (Broad Institute) data showed a positive correlation between Aurora B expression and the levels of p53 suppressed genes such as cyclin E, CDK6 and cyclin B, involved in cell cycle (). The heat map from transcriptomics analysis shows that p53-suppressed target genes were upregulated in breast cancer patients highly expressing Aurora B (). To assess the impact of excessive downregulation of Aurora B as a result of overexpression of FBXW7, as in the case of FBXW7 expression in HCT116 cells, we treated cells with Aurora B inhibitor AZD1152, which inhibits Aurora B and causes Aurora B downregulation.Citation24 We found that Aurora B inhibitor, AZD1152, led to the formation of multinucleated cells (). Together, these data illustrate that the FBXW7-AurorB axis is critical to the control of cell growth and mitosis. Deregulation could be caused by FBXW7 deficiency or Aurora B overexpression. On the other hand, FBXW7 overexpression can also cause mitotic defects. Thus, a proper balance between Aurora B and FBXW7 is critical to sustaining mitotic progression.
Figure 7. FBXW7-Aurora B axis is involved in the regulation of p53 stability and mitotic progression. (A) Aurora B decreased the steady-state protein level of p53. H1299 were transfected with the indicated plasmids. Cell lysates were immunoblotted with the indicated antibodies. (B) Enrichment score graph and Ranked list metric graph showing the upregulation of p53-suppressed target genes in Aurora B-highly expressing patients (Gene Set Enrichment Analysis, Broad Institute, MIT). Cohort: GSE-20194 consisting of 255 untreated stage I–stage III breast cancer patients at MD Anderson Cancer Center. (C) Heat map from transcriptomics analysis showing that p53-suppressed target genes were upregulated in Aurora B-highly expressing breast cancer patients. Cohort: GSE-20194 consisting of 255 untreated stage I–stage III breast cancer patients at MD Anderson Cancer Center. (D) Aurora B inhibition leads to multi-nucleated cells. MCF7 cells were treated with AZD1152-HQPA for 24 h and stained with DAPI. (E) Model of the FBXW7-Aurora B axis in regulating p53 and mitosis.
Discussion
In mitosis, the chromosomal passenger complex, composed of Aurora B, Survivin, Borealin and inner centromere protein (INCENP), controls chromosome alignment, histone modification and cytokinesis.Citation25 Presence of this complex at the right place at the right time is the key to precise control of Aurora B kinase.Citation26-Citation28 Regulation of the protein amounts of Aurora B determines the timing of Aurora B activity. Here, we discover a critical role of FBXW7 in the control Aurora B homeostasis by regulating its ubiquitin-proteasomal degradation. Our results provide unique insights into the consequences of FBXW7 loss, specifically, its effects on Aurora B expression during carcinogenesis and cancer progression. This new layer of Aurora B post-translational regulation also offers additional insight into the delicate regulation of mitotic kinase Aurora B.
So far, there is evidence for the involvement of two ubiquitin E3 ligases involved in ubiquitin-mediated degradation of Aurora B: anaphase-promoting cyclosome complex (APC/c)Citation29,Citation30 and a Cullin 3 (Cul3)-based E3 ligase with the substrate-specific adaptors KLHL9 and KLHL13.Citation31 Aurora B interacts with APC/c through the Cdc27 subunit. Aurora B also binds to the APC/c activator proteins Cdh1 and Cdc20, the overexpression of which accelerates Aurora B degradation. Here we showed for the first time that FBXW7 is involved in Aurora B ubiquitination and degradation. It remains to be seen whether these E3 ligases function independently or collaboratively during the cell cycle.
Aurora B is overexpressed in many types of cancers, including multiple myeloma and colorectal, prostate and pancreatic cancers.Citation32 This overexpression has made Aurora B or its substrate an attractive target for therapeutic cancer drugs.Citation33,Citation34 However, although high levels of Aurora B are associated with advanced clinical stages and poor prognosis in several cancers, the mechanism of aberrant overexpression of Aurora B remains unclear. Our studies show that loss of FBXW7 leads to the accumulation of Aurora B, thus providing critical insight into Aurora B overexpression in cancer, as FBXW7 is a tumor suppressor gene and is frequently mutated in cancer.Citation18
In , we find that Aurora B overexpression results in polyploidy, and that FBXW7 overexpression can correct such a phenotype. This, however, occurred in a p53-inactive background (293T cells), as overexpression of FBXW7 in p53 wt background cells (HCT116) leads to polyploidy cells (), supposedly a manifestation of excessive Aurora B downregulation caused by FBXW7 overexpression. Additionally, our data showing that AZD1152, an Aurora B inhibitor and downregulator,Citation24 causes multinucleated cell formation () lends support to this argument.
The proteins known to interact with FBXW7, including but not limited to cyclin E, Myc, c-Jun and Aurora A all play important roles in tumorigenesis.Citation11,Citation15,Citation35 In our studies we find that Aurora B is a new FBXW7-associating protein (), which is regulated by FBXW7 during mitosis (). We determine that FBXW7 regulates Aurora B posttranscriptionally by enhancing Aurora B ubiquitination. Most of the known protein substrates of FBXW7 contain a phosphorylated phosphodegron with the consensus sequence LXTPX.Citation22,Citation36,Citation37 Arginine (R) residues in the FBXW7 eight-bladed WD40 propeller domain make up the bulk of the surface that interacts with the phosphodegron. This conclusion is based on a structural analysis of FBXW7 bound to the cyclin E phosphodegron.Citation18,Citation37 It is important to note that the arginines that make up this binding pocket are mutated in human cancers.Citation18 Several targets of FBXW7 have mutations within the phosphodegron that either abolish the capacity of these sequences to be phosphorylated or block the ability of the phosphorylated proteins to be recognized by FBXW7.Citation38 Interestingly, our binding assay indicated that the arginines of FBXW7 R465 and R505 within the binding pockets are necessary for binding to Aurora B, as R465 and R505 mutants are unable to associate with Aurora B (). It is important to point out that FBXW7 R465 mutations are frequently observed in cancersCitation18; therefore, the mutant would not be able to interact with Aurora B and mediate its degradation, which may lead to Aurora B accumulation and mitotic defects, thereby providing a growth advantage. Our data showed that FBXW7-deficient cells undergo extensive mitotic slippage after release from nocodazole treatment (). Additional data also showed that FBXW7 can correct phenotype of Aurora B overexpression-induced polyploidy (). When taken together, these data suggest that FBXW7 may be a master regulator of mitotic checkpoints.
We previously showed that p53 deficiency causes the downregulation of FBXW7α.Citation39Additionally, Aurora B can phosphorylate p53 and subsequently enhance MDM2-mediated p53 degradationCitation21,Citation40 or suppress p53 transcriptional activity.Citation41 Our study of FBXW7-mediated Aurora B degradation indicates that a feedback loop exists to maintain the equilibrium between Aurora B, p53 and FBXW7 (). This mutual regulation is very critical, as any signaling that tilts the balance, such as Aurora B overexpression, p53 mutation or FBXW7 mutations that prevent Aurora B binding for degradation, may lead to mitotic catastrophe, which, in turn, may result in cancer growth. Our mechanistic studies of FBXW7-mediated Aurora B downregulation explain how the loss/mutation of FBXW7 can lead to deregulation of Aurora B in our proposed model (). This study shows that the p53-FBXW7 axis negatively regulates Aurora B and keeps Aurora B protein levels in check to prevent mitotic deregulation (). Clearly, the FBXW7-Aurora B link will be an important molecular target for rational cancer therapy. Since FBXW7 expression can correct mitotic defects mediated by Aurora B activity (), targeting the FBXW7-Aurora B axis may be a useful therapeutic strategy for cancer intervention in Aurora B-overexpressing cancer.
Material and Methods
Cell lines and reagents
293T cells were obtained from the ATCC and were maintained in DME/F12 media (in-house supplier) supplemented with 5% or 10% (v/v) fetal bovine serum. HCT116 FBXW7 +/+ and HCT116 FBXW7 −/− (a kind gift from Dr. Bert Vogelstein) were cultured in McCoy’s 5A media (Hyclone) supplemented with 5% or 10% (v/v) fetal bovine serum, 2 mM L-glutamine (Cellgro) and 1% antibiotic-antimycotic solution (Invitrogen). All cells were incubated in a humidified incubator at 37°C with 5% CO2. MG132 is obtained from Sigma. MTT assay were performed as previously described.Citation42 AZD1152 (orally bioavailable pro-drug) and AZD1152-HQPA (active specific inhibitor of Aurora B kinase) were kindly provided by Dr. Kirsten Mundt (Astra Zeneca).Citation21 AZD1152-HQPA was dissolved in 100% DMSO at a 10-mM concentration as a stock solution prior to further dilution in appropriate aqueous solutions/media at final concentrations as indicated. Antibodies: Flag (M2, Sigma), tubulin (Sigma), Aurora B (Abcam) and actin (Sigma). Flag-Aurora B, GFP-Aurora B, Flag-FBXW7, DNFBWX7, DNFBWX7R mutants and His-p53 were previously described.Citation21,Citation23,Citation43-Citation45 Aurora B shRNA was previously described.Citation21
Immunofluorescence
Indicated cell lines after transfection were grown in either chamber slides, tissue culture dishes or on cover glasses to 50–75% confluence. For GFP-Aurora B-tranfected cells, the green fluorescence was observed directly under microscope. For Aurora B inhibition experiment, cells were treated with vehicle or 20 nM AZD1152-HQPA, for 48 h. Cells were stained with DAPI and mounted on microscope slides using Fluoromount G (Southern Biotech) or Prolong Gold (Invitrogen) that contained DAPI. Immunostained cells were visualized with an Olympus IX81 confocal microscope.
Fluorescence activated cell sorting
Cells to be analyzed were plated in 6-well tissue culture dishes and grown to log phase. Appropriate treatments by transfection of plasmids were performed for 24–48 h. Monolayers were rinsed twice with PBS, and the cells were scraped into microcentrifuge tubes. Cells were centrifuged at low speed and rinsed once with PBS. Pellets were resuspended in 0.5 ml hypotonic PI solution [0.85 mg/ml sodium citrate (Sigma), 0.1 mg/ml RNase A, 0.1% Triton X-100, 20 mg/ml PI (Roche)] and incubated in the dark for 30 min. Cell cycle/PI analysis was performed using a Becton Dickinson FACStar PLUS.
Immunoblotting and immunoprecipitation
Total-cell lysates were processed as previously described.Citation42 Cell lysates for western blot or immunoprecipitation were collected from tissue culture dishes after two rinses with cold PBS. Cells were centrifuged at low speed for 10 min and supernatants discarded. Pellets were then either frozen at -80°C for further processing later, or they were lysed with 100–300 μl 1x lysis buffer [0.5 L batch: 7.5g 1 M Tris (Fisher), 15 ml 5M NaCl (Fisher), 0.5 ml NP-40 (USB Corp.), 0.5 ml Triton X-100 (Sigma) and 1 ml 0.5M EDTA (Fisher)] for 20 min at 4°C. Lysis buffer also contained a cocktail of protease/phosphatase inhibitors: 5 mM NaV, 1 mM NaF, 1 μM DTT, 0.1 mg/ml Pepstatin A, 1 mM PMSF and 1000x Complete Cocktail Protease Inhibitor (Roche). Lysates were immunoblotted with indicated antibodies. For immunoprecipitations, cell lysates were prepared and standardized as before, and 1 mg protein was immunoprecipitated with appropriately diluted antibody in lysis buffer overnight. Antibody was pulled down with 50 μl of either Protein A or G beads (Santa Cruz Biotechnology) for 1 h. Beads were centrifuged at low speed for 10 min and supernatant discarded. Dried beads were mixed with 2x loading dye and boiled for 5 min. Lysate samples were loaded onto gels and SDS-PAGE was performed as before.
Ubiquitination assay
293T cells were transiently co-transfected with indicated plasmids and were treated with 5 µg/mL MG132 for 6 h before harvesting. Ubiquitinated Aurora B was immunoprecipitated with anti-Aurora B, followed by immunoblotting with anti-His.
Acknowledgments
This work was supported by grants in part from the National Institutes of Health (NIH) (R01CA089266), Directed Medical Research Programs (DOD SIDA BC062166 to S.J.Y. and M.H.L.) and Susan G. Komen Breast Cancer Foundation (KG081048). This research was supported in part by a cancer prevention fellowship for G.V.T (R25T CA57730). The University of Texas M. D. Anderson Cancer Center is supported by NIH core grant CA16672. We would like to thank Michael McGuire and Jessica Chromheecke for critical reading.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol 2003; 4:842 - 54; http://dx.doi.org/10.1038/nrm1245; PMID: 14625535
- Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol 2007; 8:798 - 812; http://dx.doi.org/10.1038/nrm2257; PMID: 17848966
- Lindqvist A, Rodríguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol 2009; 185:193 - 202; http://dx.doi.org/10.1083/jcb.200812045; PMID: 19364923
- Fu J, Bian M, Jiang Q, Zhang C. Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res 2007; 5:1 - 10; http://dx.doi.org/10.1158/1541-7786.MCR-06-0208; PMID: 17259342
- Ducat D, Zheng Y. Aurora kinases in spindle assembly and chromosome segregation. Exp Cell Res 2004; 301:60 - 7; http://dx.doi.org/10.1016/j.yexcr.2004.08.016; PMID: 15501446
- Katayama H, Brinkley WR, Sen S. The Aurora kinases: role in cell transformation and tumorigenesis. Cancer Metastasis Rev 2003; 22:451 - 64; http://dx.doi.org/10.1023/A:1023789416385; PMID: 12884918
- Lens SM, Voest EE, Medema RH. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev Cancer 2010; 10:825 - 41; http://dx.doi.org/10.1038/nrc2964; PMID: 21102634
- Sampath SC, Ohi R, Leismann O, Salic A, Pozniakovski A, Funabiki H. The chromosomal passenger complex is required for chromatin-induced microtubule stabilization and spindle assembly. Cell 2004; 118:187 - 202; http://dx.doi.org/10.1016/j.cell.2004.06.026; PMID: 15260989
- Slattery SD, Moore RV, Brinkley BR, Hall RM. Aurora-C and Aurora-B share phosphorylation and regulation of CENP-A and Borealin during mitosis. Cell Cycle 2008; 7:787 - 95; http://dx.doi.org/10.4161/cc.7.6.5563; PMID: 18239465
- Hoeller D, Hecker CM, Dikic I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat Rev Cancer 2006; 6:776 - 88; http://dx.doi.org/10.1038/nrc1994; PMID: 16990855
- Onoyama I, Nakayama KI. Fbxw7 in cell cycle exit and stem cell maintenance: insight from gene-targeted mice. Cell Cycle 2008; 7:3307 - 13; http://dx.doi.org/10.4161/cc.7.21.6931; PMID: 18948752
- Malyukova A, Dohda T, von der Lehr N, Akhoondi S, Corcoran M, Heyman M, et al. The tumor suppressor gene hCDC4 is frequently mutated in human T-cell acute lymphoblastic leukemia with functional consequences for Notch signaling. Cancer Res 2007; 67:5611 - 6; http://dx.doi.org/10.1158/0008-5472.CAN-06-4381; PMID: 17575125
- Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res 2007; 67:9006 - 12; http://dx.doi.org/10.1158/0008-5472.CAN-07-1320; PMID: 17909001
- Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 2008; 8:83 - 93; http://dx.doi.org/10.1038/nrc2290; PMID: 18094723
- Minella AC, Clurman BE. Mechanisms of tumor suppression by the SCF(Fbw7). Cell Cycle 2005; 4:1356 - 9; http://dx.doi.org/10.4161/cc.4.10.2058; PMID: 16131838
- Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, et al. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 2004; 432:775 - 9; http://dx.doi.org/10.1038/nature03155; PMID: 15592418
- Spruck CH, Strohmaier H, Sangfelt O, Müller HM, Hubalek M, Müller-Holzner E, et al. hCDC4 gene mutations in endometrial cancer. Cancer Res 2002; 62:4535 - 9; PMID: 12183400
- Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, et al. Inactivation of hCDC4 can cause chromosomal instability. Nature 2004; 428:77 - 81; http://dx.doi.org/10.1038/nature02313; PMID: 14999283
- Knuutila S, Aalto Y, Autio K, Björkqvist AM, El-Rifai W, Hemmer S, et al. DNA copy number losses in human neoplasms. Am J Pathol 1999; 155:683 - 94; http://dx.doi.org/10.1016/S0002-9440(10)65166-8; PMID: 10487825
- Grim JE, Gustafson MP, Hirata RK, Hagar AC, Swanger J, Welcker M, et al. Isoform- and cell cycle-dependent substrate degradation by the Fbw7 ubiquitin ligase. J Cell Biol 2008; 181:913 - 20; http://dx.doi.org/10.1083/jcb.200802076; PMID: 18559665
- Gully CP, Velazquez-Torres G, Shin JH, Fuentes-Mattei E, Wang E, Carlock C, et al. Aurora B kinase phosphorylates and instigates degradation of p53. Proc Natl Acad Sci USA 2012; 109:E1513 - 22; http://dx.doi.org/10.1073/pnas.1110287109; PMID: 22611192
- Ye X, Nalepa G, Welcker M, Kessler BM, Spooner E, Qin J, et al. Recognition of phosphodegron motifs in human cyclin E by the SCF(Fbw7) ubiquitin ligase. J Biol Chem 2004; 279:50110 - 9; http://dx.doi.org/10.1074/jbc.M409226200; PMID: 15364936
- Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, et al. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med 2007; 204:1825 - 35; http://dx.doi.org/10.1084/jem.20070872; PMID: 17646408
- Gully CP, Zhang F, Chen J, Yeung JA, Velazquez-Torres G, Wang E, et al. Antineoplastic effects of an Aurora B kinase inhibitor in breast cancer. Mol Cancer 2010; 9:42; http://dx.doi.org/10.1186/1476-4598-9-42; PMID: 20175926
- Knauer SK, Mann W, Stauber RH. Survivin’s dual role: an export’s view. Cell Cycle 2007; 6:518 - 21; http://dx.doi.org/10.4161/cc.6.5.3902; PMID: 17361097
- Lens SM, Rodriguez JA, Vader G, Span SW, Giaccone G, Medema RH. Uncoupling the central spindle-associated function of the chromosomal passenger complex from its role at centromeres. Mol Biol Cell 2006; 17:1897 - 909; http://dx.doi.org/10.1091/mbc.E05-08-0727; PMID: 16436504
- Jelluma N, Brenkman AB, van den Broek NJ, Cruijsen CW, van Osch MH, Lens SM, et al. Mps1 phosphorylates Borealin to control Aurora B activity and chromosome alignment. Cell 2008; 132:233 - 46; http://dx.doi.org/10.1016/j.cell.2007.11.046; PMID: 18243099
- Ricke RM, van Deursen JM. Aurora B hyperactivation by Bub1 overexpression promotes chromosome missegregation. Cell Cycle 2011; 10:3645 - 51; http://dx.doi.org/10.4161/cc.10.21.18156; PMID: 22033440
- Nguyen HG, Chinnappan D, Urano T, Ravid K. Mechanism of Aurora-B degradation and its dependency on intact KEN and A-boxes: identification of an aneuploidy-promoting property. Mol Cell Biol 2005; 25:4977 - 92; http://dx.doi.org/10.1128/MCB.25.12.4977-4992.2005; PMID: 15923616
- Stewart S, Fang G. Destruction box-dependent degradation of aurora B is mediated by the anaphase-promoting complex/cyclosome and Cdh1. Cancer Res 2005; 65:8730 - 5; http://dx.doi.org/10.1158/0008-5472.CAN-05-1500; PMID: 16204042
- Sumara I, Quadroni M, Frei C, Olma MH, Sumara G, Ricci R, et al. A Cul3-based E3 ligase removes Aurora B from mitotic chromosomes, regulating mitotic progression and completion of cytokinesis in human cells. Dev Cell 2007; 12:887 - 900; http://dx.doi.org/10.1016/j.devcel.2007.03.019; PMID: 17543862
- Yeung SC, Gully C, Lee MH. Aurora-B kinase inhibitors for cancer chemotherapy. Mini Rev Med Chem 2008; 8:1514 - 25; http://dx.doi.org/10.2174/138955708786786480; PMID: 19075809
- Keen N, Taylor S. Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer 2004; 4:927 - 36; http://dx.doi.org/10.1038/nrc1502; PMID: 15573114
- Sanhaji M, Friel CT, Wordeman L, Louwen F, Yuan JP. Mitotic centromere-associated kinesin (MCAK): a potential cancer drug target. Oncotarget 2011; 2:935 - 47; PMID: 22249213
- Wang Z, Inuzuka H, Zhong J, Wan L, Fukushima H, Sarkar FH, et al. Tumor suppressor functions of FBW7 in cancer development and progression. FEBS Lett 2012; 586:1409 - 18; http://dx.doi.org/10.1016/j.febslet.2012.03.017; PMID: 22673505
- Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J 2004; 23:2116 - 25; http://dx.doi.org/10.1038/sj.emboj.7600217; PMID: 15103331
- Orlicky S, Tang X, Willems A, Tyers M, Sicheri F. Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell 2003; 112:243 - 56; http://dx.doi.org/10.1016/S0092-8674(03)00034-5; PMID: 12553912
- Jin J, Ang XL, Shirogane T, Wade Harper J. Identification of substrates for F-box proteins. Methods Enzymol 2005; 399:287 - 309; http://dx.doi.org/10.1016/S0076-6879(05)99020-4; PMID: 16338364
- Wu CC, Yang TY, Yu CT, Phan L, Ivan C, Sood AK, et al. p53 negatively regulates Aurora A via both transcriptional and posttranslational regulation. Cell Cycle 2012; 11:3433 - 42; PMID: 22894933
- Chiang CM. p53-Aurora A mitotic feedback loop regulates cell cycle progression and genomic stability. Cell Cycle 2012; 11; In press PMID: 22982999
- Wu LM, Ma CA, Jain A. When Aurora B met p53: newly revealed regulatory phosphorylation in an old protein. Cell Cycle 2011; 10:171 - 2; http://dx.doi.org/10.4161/cc.10.2.14349; PMID: 21212738
- Laronga C, Yang HY, Neal C, Lee MH. Association of the cyclin-dependent kinases and 14-3-3 sigma negatively regulates cell cycle progression. J Biol Chem 2000; 275:23106 - 12; http://dx.doi.org/10.1074/jbc.M905616199; PMID: 10767298
- Yang HY, Wen YY, Chen CH, Lozano G, Lee MH. 14-3-3 sigma positively regulates p53 and suppresses tumor growth. Mol Cell Biol 2003; 23:7096 - 107; http://dx.doi.org/10.1128/MCB.23.20.7096-7107.2003; PMID: 14517281
- Yang HY, Wen YY, Lin YI, Pham L, Su CH, Yang H, et al. Roles for negative cell regulator 14-3-3sigma in control of MDM2 activities. Oncogene 2007; 26:7355 - 62; http://dx.doi.org/10.1038/sj.onc.1210540; PMID: 17546054
- Yang H, Zhao R, Lee MH. 14-3-3sigma, a p53 regulator, suppresses tumor growth of nasopharyngeal carcinoma. Mol Cancer Ther 2006; 5:253 - 60; http://dx.doi.org/10.1158/1535-7163.MCT-05-0395; PMID: 16505098