Abstract
Activation of adaptive mechanisms plays a crucial role in cancer progression and drug resistance by allowing cell survival under stressful conditions. Therefore, inhibition of the adaptive response is considered as a prospective therapeutic strategy. The PERK-eIF2α phosphorylation pathway is an important arm of the unfolded protein response (UPR), which is induced under conditions of endoplasmic reticulum (ER) stress. Our previous work showed that ER stress is induced in chronic myeloid leukemia (CML) cells. Herein, we demonstrate that the PERK-eIF2α phosphorylation pathway is upregulated in CML cell lines and CD34+ cells from CML patients and is associated with CML progression and imatinib resistance. We also show that induction of apoptosis by imatinib results in the downregulation of the PERK-eIF2α phosphorylation arm. Furthermore, we demonstrate that inactivation of the PERK-eIF2α phosphorylation arm decreases the clonogenic and proliferative capacities of CML cells and sensitizes them to death by imatinib. These findings provide evidence for a pro-survival role of PERK-eIF2α phosphorylation arm that contributes to CML progression and development of imatinib resistance. Thus, the PERK-eIF2α phosphorylation arm may represent a suitable target for therapeutic intervention for CML disease.
Introduction
The response of tumor cells to various stress stimuli is an important determinant of cancer development. Tumor cells respond to microenvironmental stressors by evolutionarily conserved systems that regulate the protective phenotype.Citation1 In contrast to the mild chronic stress, which allows for adaptation through permanent changes in cellular functioning and activation of pro-survival mechanisms, excessive stress activates cell death.Citation1 Thus, determining how the transition from adaptation to the cell death is regulated has a tremendous physiological importance for cancer biology and patient treatment.
One of the organelles that senses alterations in homeostasis is the endoplasmic reticulum (ER).Citation2 A number of stimuli can disrupt ER homeostasis and impose stress.Citation2 The ER has evolved a highly specific signaling network called the unfolded protein response (UPR) to adapt to stress or to activate cell death when protective mechanisms are not sufficient.Citation3,Citation4 UPR initially leads to the inhibition of global protein synthesis and consequently results in the transcriptional induction of genes encoding proteins that allow cells to adapt to ER stress. UPR pathways consist of three main signaling cascades initiated by ER-localized stress sensors: the inositol-requiring kinase 1 (IRE1), the PKR-like ER-resident kinase (PERK) and the activating transcription factor 6 (ATF6).Citation3,Citation4 The effects of UPR on translation inhibition are mediated by the phosphorylation of the α subunit of translation initiation factor eIF2 at serine 51 (S51) as a result of PERK activation.Citation5,Citation6 Phosphorylation of eIF2α at the S51 impairs global mRNA translation, but at the same time it induces translation of specific mRNAs encoding proteins that contribute to the adaptation process, such as the activating transcription factor 4 (ATF4) and ATF5.Citation5,Citation6 Induction of the PERK-eIF2α phosphorylation arm can also lead to the activation of stress-related transcription factors, such as NFκB, by lowering the steady-state levels of its short-lived regulatory IκB inhibitor.Citation7,Citation8 Increased PERK and IRE1 activities, as well as increased calcium levels, have been implicated as mediators of ER-stress-induced autophagy,Citation9,Citation10 which is considered as an important player in life-and-death decisions of the ER-stressed cells. Similarly to ER stress, autophagy, when moderate, increases cellular survival, promoting cancer growth, whereas severe and unbalanced correlates with cell death.Citation9,Citation10
Chronic myeloid leukemia (CML) is a clonal myeloproliferative disease caused by expression of BCR-ABL, a fusion protein with constitutive tyrosine kinase activity.Citation11 CML starts with a chronic phase, but due to genomic instability and activation of additional mechanisms, it progresses to blast crisis, becoming aggressive and increasingly resistant to therapy.Citation12,Citation13 The molecular mechanisms responsible for the progression are largely elusive. A specific inhibitor of the BCR-ABL tyrosine kinase known as imatinib has been effective in the treatment of the chronic phase. However, imatinib responses in chronic phase can lead to the development of drug resistance in some cases, whereas in blast crisis are short-lived and not efficient.Citation14 Despite the advances in CML treatment with new BCR-ABL inhibitors,Citation15 the blastic phase remains almost inevitably fatal.Citation16
Several studies indicate that induction of the UPR promotes tumor cell survival and helps solid tumors to cope with hypoxia and growth factor deprivation.Citation17,Citation18 However, activation of the specific arms of UPR in CML and its impact on cell fate has not been thoroughly investigated. We previously showed that BCR-ABL leads to disturbances in calcium homeostasis in the ER that are associated with increased levels of the ER chaperone Grp78, indicating the presence of an ER stress response.Citation19 Herein, we demonstrate that the PERK-eIF2α phosphorylation arm is upregulated in BCR-ABL-expressing mouse progenitor cell lines, CML cell lines and in CD34+ progenitor cells obtained from CML patients. Analyses of clinical samples indicated a good correlation between increased PERK-eIF2α phosphorylation arm and CML progression and clinical resistance to imatinib treatment. Moreover, we show that the PERK-eIF2α phosphorylation arm is downregulated by imatinib, resulting in an increased sensitivity of BCR-ABL-expressing mouse cells and human CML cells to imatinib-mediated cell death. Thus, the PERK-eIF2α phosphorylation arm may have important implications in the development of CML disease and its treatment with BCR-ABL drug inhibitors.
Results
BCR-ABL expression induces ER stress and upregulates the PERK-eIF2α phosphorylation pathway
We previously showed that BCR-ABL expression in mouse myeloid cells or human CML cell lines increases the expression of ER stress markers.Citation19 To address the potential link between BCR-ABL expression and ER stress response, we investigated the regulation of the PERK-eIF2α phosphorylation arm in immortalized murine 32D myeloid cells expressing either low or high levels of BCR-ABL ().Citation19 We observed that BCR-ABL expression resulted in an increase of PERK phosphorylation at threonine 980 (T980), a residue within the kinase activation loop whose phosphorylation is essential for the autocatalytic activity of the kinase.Citation20 We also noticed that PERK phosphorylation at tyrosine 615 (Y615), a site contributing to the optimal autocatalytic activity of PERK,Citation21 was increased in BCR-ABL-expressing cells. Analysis of total PERK protein by immunoblotting showed the presence of an additional, slower migrating form of PERK in cells with high levels of BCR-ABL (, lane 3). This form corresponds to hyperphosphorylated PERK at serine/threonine and tyrosine residues, as previously described.Citation21 Consistent with the induction of PERK activity, we noticed that the murine cells contained increased levels of phosphorylated eIF2α at S51 that were proportional to BCR-ABL protein levels ().
Figure 1. The PERK-eIF2α phosphorylation pathway is upregulated in mouse and human cell lines expressing BCR-ABL and in CD34+ CML cells. (A) Expression of BCR-ABL, Grp78, PERK, PERK-pY615, PERK-pT980, eIF2α and eIF2α-pS51 in mouse progenitor 32D cells and 32D cells expressing different levels of BCR-ABL. (B) Expression of BCR-ABL, STAT5, STAT5-pY694, Grp78, PERK, PERK-pY615, eIF2α and eIF2α-pS51 in HL60, BV173 and K562 cells or resting and proliferating PBMC from healthy donors. Arrows indicate the lower—unphosphorylated and the higher—hyperphosphorylated PERK. (C) Expression of BCR-ABL, STAT5, STAT5-pY694, Grp78, PERK, PERK-pY615, eIF2α and eIF2α-pS51 proteins in progenitor CD34+ cells isolated from the whole blood of healthy donors (CD34+ HD, lanes 1–4), CML-chronic phase (CD34+ CML-CP, lanes 5–8), CML-accelerated phase (CD34+ CML-AcP, lane 9) or CML-blast crisis patients (CD34+ CML-BC, lanes 10–13). The ratio of phosphorylated STAT5, PERK and eIF2α proteins to actin was calculated based on the densitometry using GeneTools from Syngene.
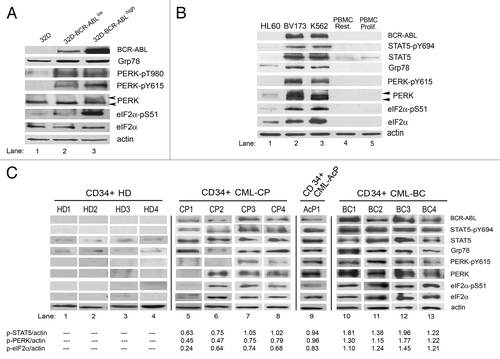
To determine whether our observations with mouse cells are relevant to human CML cells, we examined the activation of the PERK-eIF2α phosphorylation pathway in K562 and BV173 cells derived from CML-blast crisis (BC) patients (). As control, we included the promyelocytic acute leukemia HL60 cells, which do not express BCR-ABL, as well as either resting or PHA-stimulated PBMCs. We observed that Grp78 was upregulated in CML cell lines, indicating the presence of ER stress. We further noticed a substantial increase of PERK and eIF2α proteins which coincided with increased phosphorylation at Y615 and S51, respectively, in CML cells but not in HL60 or PBMCs (). Consistent with our observations in BCR-ABL-transformed 32D cells, PERK was hyperphosphorylated in CML cells, as indicated by the appearance of slow-migrating bands of PERK in the immunoblottings (). Collectively, these data showed the presence of an ER stress response in human CML cells that was associated with increased PERK-eIF2α phosphorylation pathway.
The PERK-eIF2α phosphorylation arm is upregulated in CD34+ progenitor cells from CML patients
To investigate the potential link between the PERK-eIF2α phosphorylation pathway and CML disease, we examined CML progenitor CD34+ cells from peripheral blood of CML patients together with CD34+ cells from healthy donors that were used as control (). We tested four samples from healthy donors—CD34+ HD (lanes 1–4), four samples from patients in chronic phase at the diagnosis before any treatment—CD34+ CML-CP (lanes 5–8), one sample from a patient in accelerated phase—CD34+ CML-AcP (lane 9) and four samples from blast crisis patients that developed resistance to therapy—CD34+ CML-BC (lanes 10–13). summarizes the clinical data for CML patients, with the Sokal score values, type of blast crisis, history of previous treatments and occurrence of clinical resistance. All CP and AcP samples were obtained at the diagnosis stage, and the Sokal score ranged from 0.58 to more than 1.5. All BC patients developed myeloid blast crisis and were clinically resistant to imatinib and other tyrosine kinase inhibitors in two cases.
Table 1. Clinical characterization of samples from CML patients in chronic phase (CP1-CP4), accelerated phase (AcP1) and blast crisis (BC1-BC4)
We observed that CD34+ cells from healthy donors did not show any significant signs of ER stress as determined by Grp78 expression (). Also expression and phosphorylation of PERK and eIF2α proteins were below the detection level in healthy donor cells compared with CML cells. On the other hand, PERK and eIF2α protein expression and phosphorylation levels were increased in CML cells. Cells from patients in the CP displayed different levels of PERK and eIF2α proteins expression and phosphorylation; however, these levels were significantly lower than those observed in BC samples (). In all CML samples obtained from the BC stage of the disease, we noticed a strong upregulation of PERK and eIF2α phosphorylation. Interestingly, we saw a substantial upregulation of PERK and eIF2α phosphorylation in one sample derived from accelerated phase of CML (). When the ratios of phosphorylated STAT5, PERK or eIF2α were normalized to actin levels, we noticed that all ratios were increased in samples with progressed CML disease. Interestingly, all BC samples obtained from patients with resistance to imatinib and other tyrosine kinase inhibitors showed ratios between 1.1–1.9, which were higher than those measured in CP. This data indicated that upregulation of the PERK-eIF2α phosphorylation arm is associated with CML progression as a result of increased BCR-ABL activity.
Induction of cell death by imatinib is associated with suppression of the PERK-eIF2α phosphorylation pathway
We were interested to investigate whether the PERK-eIF2α phosphorylation axis is implicated to BCR-ABL inhibition by imatinib. Thus, we measured the susceptibility of the mouse progenitor cells to apoptosis after treatment with imatinib. Contrary to parental 32D cells, BCR-ABL-expressing 32D cells displayed a dose-dependent increase in apoptosis, as assessed by either annexin V staining () or propidium iodide exclusion viability test (data not shown). We also examined the effects of imatinib treatment on PERK and eIF2α protein expression and phosphorylation levels (). We found that unlike the parental cells, treatment of BCR-ABL-expressing cells with imatinib caused a downregulation of phosphorylated PERK and eIF2α by imatinib in a dose-dependent manner. The downregulation of PERK phosphorylation was associated with a decrease in PERK protein levels after prolonged treatment with high concentrations of imatinib (). Downregulation of PERK activity and protein levels was proportional to eIF2α phosphorylation and protein levels in response to imatinib treatment (). Taken together, these data suggested that imatinib treatment results in the inactivation of the PERK-eIF2α phosphorylation arm in BCR-ABL-expressing cells.
Figure 2. Induction of cell death by imatinib leads to inhibition of the PERK-eIF2α phosphorylation pathway in mouse BCR-ABL-transfected cells. (A) Apoptosis determined by the Annexin V test in cells incubated with imatinib. (B) Expression of PERK, PERK-pY615, eIF2α or eIF2α-pS51 proteins in cells incubated with imatinib. (C) Expression of STAT5, STAT5-pY694, PERK, PERK-pY615, eIF2α or eIF2α-pS51 proteins and PARP cleavage in cells pre-incubated with caspase inhibitor Z-VAD, followed by imatinib. (D) Inhibition of apoptosis by Z-VAD treatment in cells incubated with imatinib at different concentrations. Percentage of apoptotic cells (upper panel) was determined by using the Annexin V test, and percentage of dead cells (lower panel) was determined by propidium iodide exclusion test. The percentage of apoptotic or dead cells is shown as mean ± SEM of three independent experiments. (E) Expression of PERK, PERK-pY615, eIF2α or eIF2α-pS51 proteins in 32D-T315I BCR-ABL cells. The ratio of phosphorylated to total eIF2α protein is indicated (P/T). The ratio was set to 1 for “0” time point (untreated).
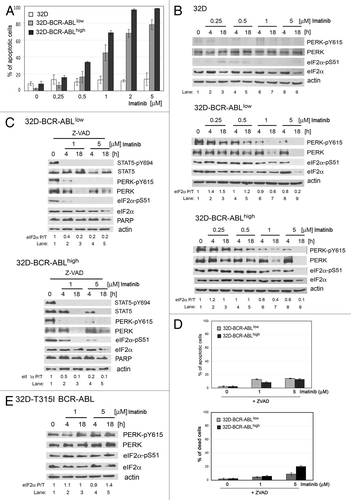
To examine the possibility that downregulation of PERK-eIF2α phosphorylation arm is either a cause or an effect of imatinib-mediated apoptosis, we employed the caspase inhibitor Z-VAD to prevent apoptosis in imatinib-treated cells (). Treatment with Z-VAD did not affect the inhibitory effects of imatinib on BCR-ABL, as shown by immunoblotting analysis of phosphorylated STAT5 (). However, Z-VAD significantly decreased the amount of apoptosis detected in cells expressing BCR-ABL (, upper panel). In addition to Annexin V staining, inhibition of cell death by Z-VAD treatment was confirmed by the propidium iodide exclusion test (, lower panel) as well as by the inhibition of PARP cleavage (). Although Z-VAD treatment significantly protected cells from death, it did not prevent the downregulation of PERK and eIF2α phosphorylation and protein levels, indicating that downregulation of the PERK-eIF2α phosphorylation arm is not an effect of increased apoptosis in imatinib-treated cells (). Furthermore, inhibition of the PERK-eIF2α phosphorylation pathway was not a result of side effects of imatinib, given that 32D cells expressing the imatinib-resistant T315I BCR-ABL mutant were resistant to the downregulation of the PERK-eIF2α phosphorylation arm and apoptosis in response to imatinib ( and data not shown).
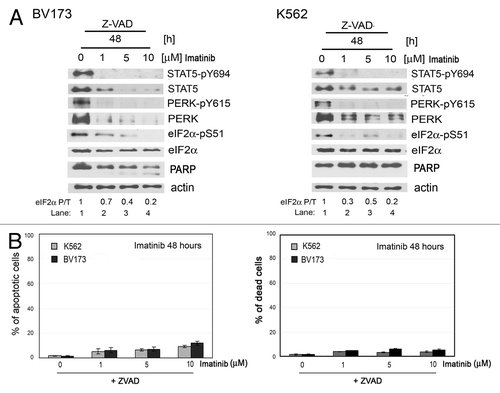
We next determined the relevance of our above observations in the human CML cell lines BV173 and K562. We found that both types of cells were resistant to imatinib treatment for a short period of time and underwent imatinib-induced apoptosis after prolonged treatment (). Short-time imatinib treatment inhibited phosphorylation of STAT5 (, left panels), consistent with recent data supporting the notion that STAT5 is a substrate of BCR-ABL.Citation22 However, PERK Y615 phosphorylation was not affected by the imatinib treatment in these cells, indicating that tyrosine phosphorylation of PERK is rather an autophosphorylation event of activated PERK.Citation21 Although the PERK-eIF2α phosphorylation arm was resistant to shorter treatment with imatinib of BV173 and K562 cells, it became sensitive to prolonged treatment (, right panels). Given that inhibition of apoptosis by Z-VAD treatment was unable to rescue the inhibition of the PERK-eIF2α phosphorylation arm in imatinib-treated BV173 and K562 cells (), we conceivably speculated that downregulation of the arm may be a cause rather than an effect of imatinib-mediated apoptosis.
Figure 3. Inhibition of the PERK-eIF2α phosphorylation pathway correlates with sensitivity to imatinib-induced apoptosis in human K562 and BV173 cells. Apoptosis determined using the Annexin V test in BV173 or K562 cells incubated with imatinib for 18 h (A) or 48 h (B). Whole-cell extracts from BV173 (C) and K562 cells (D) incubated with imatinib were analyzed by immunoblotting using STAT5, STAT5-pY694, PERK, PERK-pY615, eIF2α, eIF2α-pS51 or PARP antibodies.
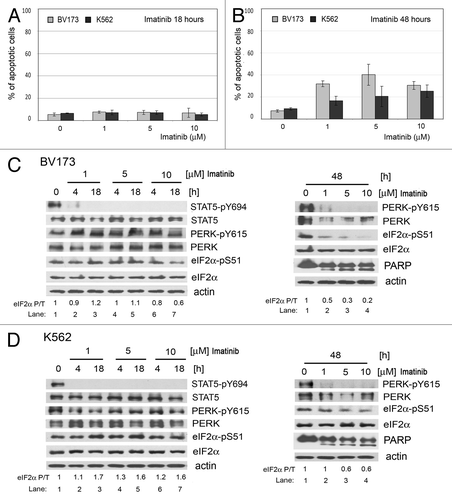
Figure 4. Inhibition of apoptosis by Z-VAD treatment in human K562 and BV173 cells incubated with imatinib. (A) Levels of STAT5-pY694, STAT5, PERK, PERK-pY615, eIF2α or eIF2α-pS51 proteins and PARP cleavage in K562 or BV173 cells pre-incubated with caspase inhibitor Z-VAD, followed by imatinib. The ratio of phosphorylated to total eIF2α protein is indicated (P/T). The ratio was set to 1 for “0” time point (untreated). (B) Percentage of apoptotic cells (left panel) determined by using the Annexin V test and percentage of dead cells (right panel) determined by propidium iodide exclusion test. The percentage of apoptotic or dead cells is shown as mean ± SEM of three independent experiments.
Inhibition of the PERK-eIF2α phosphorylation pathway can sensitize CML cells to imatinib-mediated death
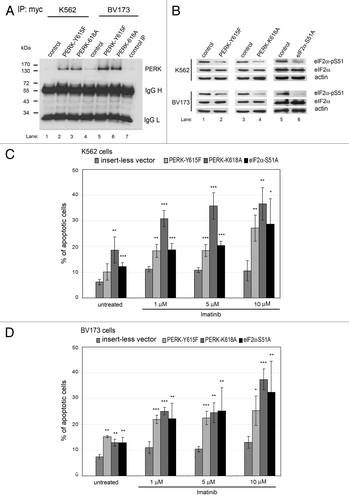
To further address the role of the PERK-eIF2α phosphorylation arm in imatinib sensitivity, we examined whether its inactivation could sensitize cells to imatinib-mediated death. To this end, PERK activity was impaired by the expression of Myc-tagged dominant-negative mutants such as the kinase dead PERK-K618ACitation20 or PERK-Y615F.Citation21 Furthermore, eIF2α phosphorylation was impaired by expression of the eIF2α-S51A mutant, which displays a dominant-negative function.Citation23 Expression of the mutant forms was verified either by immunoprecipitation and immunoblotting analyses of the Myc-tagged PERK proteins () or detection of decreased eIF2α phosphorylation at S51 by immunoblotting (). We found that expression of the mutant forms was associated with increased apoptosis of untreated K562 and BV173 cells, indicating that the PERK-eIF2α phosphorylation arm contributes to cell survival (). We further observed that expression of the mutant proteins increased the sensitivity of K562 and BV173 cells to imatinib-mediated death in a dose-dependent manner (). These data demonstrated that an intact PERK-eIF2α phosphorylation arm has a pro-survival function and mediates resistance of CML cells to imatinib treatment.
Figure 5. Downregulation of the PERK-eIF2α phosphorylation pathway increases sensitivity of human CML cells to imatinib. (A and B) K562 or BV173 cells transfected with the insertless vector or plasmids expressing the PERK-Y615F, the PERK-K618A or the eIF2α-S51A dominant-negative mutants. (A) Expression of mutated Myc-tagged PERK protein analyzed by immunoprecipitation using anti-Myc antibody followed by western blot with anti-PERK antibody detected in PERK mutants. Lane 7 shows the negative IP control. (B) Levels of eIF2α-pS51 or eIF2α proteins in cells expressing mutated forms of PERK or eIF2α proteins. (C and D) The percentage of apoptotic cells in K562 (C) or BV173 (D) cells treated with imatinib. *, p < 0.05; **, p < 0.005; ***, p < 0.0005 vs. cells transfected with the control vector under each condition, by Student’s t-test.
The PERK-eIF2α phosphorylation arm promotes CML cell growth
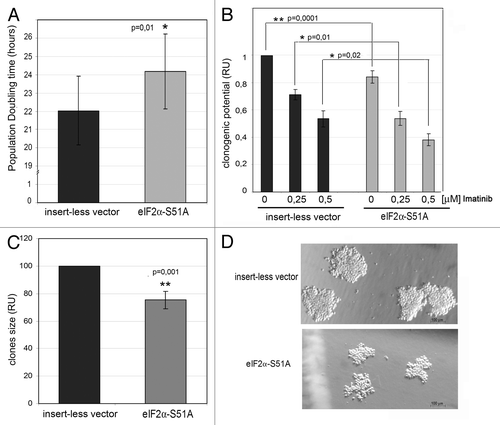
To further understand the function of the PERK-eIF2α phosphorylation arm in CML cells, we examined the effects of the pathway on the proliferative capacity and the clonogenic potential of polyclonal cell lines stably expressing the eIF2α-S51A mutant. First, we determined the population doubling time (PD), which is an indicator of growth potential (). We found that K562 cells expressing eIF2α-S51A had a significantly prolonged PD time compared with control cells, indicating that the PERK-eIF2α phosphorylation pathway facilitates CML cell proliferation. We also found that expression of eIF2α-S51A decreased the clonogenic potential of untreated cells and further decreased the ability of K562 cells to form colonies in the presence of 0.25 or 0.5 μM imatinib (). This result is in line with our observation that inhibition of the PERK-eIF2α phosphorylation pathway increases the sensitivity to imatinib (). Moreover, we observed that cells with impaired eIF2α phosphorylation formed clones of substantially smaller size than cells with an intact phosphorylated eIF2α (). Collectively, these data suggested that the PERK-eIF2α phosphorylation arm supports growth and increases the leukemogenic potential of CML cells.
Figure 6. The PERK-eIF2α phosphorylation pathway mediates the proliferative potential and growth-promoting signaling in human CML cells. (A) Population doubling time (PD) of K562 cells stably expressing control vector or eIF2α-S51A mutant. PD is shown as mean ± SEM from six independent experiments. *, p < 0.05 vs. cells transfected with the control vector, by Student’s t-test. (B–D) Clonal expansion of K562 cells expressing eIF2α-S51A mutant. (B) Clonogenic activity of cells cultured in methylcelulose medium without or with imatinib. Data were calculated relatively to untreated cells expressing the control vector. (C) Clone size (diameter) was calculated relatively to control. *, p < 0.05; **, p < 0.005 vs. cells transfected with the insertless vector, by t-Student test. (D) Picture of clones in control cells and cells expressing eIF2α-S51A mutant.
Discussion
BCR-ABL plays a key role in the activation of survival pathways that contribute to the pathogenesis of CML.Citation24 For example, activation of ERK1/2, Src-family kinase Lyn, aberrantly activated Hedgehog (Hh) or mTOR signaling have been proposed as survival pathways contributing to CML progression and imatinib resistance.Citation25-Citation29 In addition to CML cells, quiescent BCR-ABL-expressing leukemia stem cells (LSCs) also display resistance to imatinib.Citation30-Citation32 Thus, it is important to identify the signaling mechanisms that are responsible for survival as potential targets for therapeutic intervention.Citation32
Our data demonstrate that increased ER stress in BCR-ABL-transformed cells leads to the induction of the PERK-eIF2α phosphorylation pathway. The analysis of acute leukemia HL60 cells, which contain undetectable levels of phosphorylated PERK and eIF2α, indicated that oncogenic transformation of hematopoietic cells may not always be associated with an upregulation of the PERK-eIF2α phosphorylation arm. The mechanisms of upregulation of PERK and eIF2α signaling are not immediately clear but appear to depend on UPR, which is induced due to increased ER stress from BCR-ABL expression. Increased Y615 phosphorylation of PERK, which is required for its maximal activation,Citation21 was not impaired by imatinib, indicating that tyrosine phosphorylation of PERK is rather an autophosphorylation process. Nevertheless, the possibility remains that PERK activation is primed by tyrosine phosphorylation from BCR-ABL, which, in turn, is maintained at elevated levels as a result of autophosphorylation.
We also show that the PERK-eIF2α phosphorylation pathway conveys a cytoprotective effect on CML cells and decreases sensitivity to imatinib. One mechanism by which PERK mediates the cytoprotective effects is the inhibition of oxidative DNA damage.Citation33 PERK was shown to mediate cell survival upon ER stress through induction of the cellular inhibitor of apoptosis proteins cIAP.Citation34 We found that the increased eIF2α phosphorylation supports growth and proliferation of CML cells and protects them from imatinib-mediated apoptosis. This is in line with other studies showing a critical role of eIF2α phosphorylation in the protection of cells from apoptosis induced by hypoxic stress,Citation35,Citation36 chemotherapeutic drugsCitation37-Citation39 or drug inhibitors of glucose metabolism.Citation40 How the PERK-eIF2α phosphorylation arm mediates cytoprotection to imatinib treatment is not clear, but it may be implied by findings linking autophagy to ER stress. Autophagy is a protective mechanism that is essential for CML cell survival, leukemogenesis and resistance to imatinib.Citation41,Citation42 Inasmuch as the PERK-eIF2α phosphorylation arm contributes to induction of autophagy,Citation43 the possibility remains that autophagy is a mechanism utilized by the arm to mediate imatinib resistance in CML cells. It is important to emphasize that PERK activation and eIF2α phosphorylation is one arm of the UPR that mediates resistance to imatinib treatment. Specifically, it has been shown that BCR-ABL expression in mouse 32D cells resulted in the upregulation of the IRE1 and ATF6 members of UPR.Citation44 Thus, effective therapies that bypass imatinib resistance in CML tumors may require simultaneous inactivation of all three arms of UPR.
One of the key observations of our work is that the PERK-eIF2α phosphorylation pathway is detected in human CML samples, and that the levels of PERK and eIF2α activation correlate with stages of the disease and resistance to imatinib. Despite the small sample size, our data show that cells in blast crisis with clinical resistance to imatinib and other tyrosine kinase inhibitors exhibit higher levels and phosphorylation of PERK and eIF2α compared with normal cells. Interestingly, the upregulation was readily observed in accelerated phase tumors, indicating a correlation with disease progression. Our work may have implications in the diagnosis of CML disease given the limitation in diagnostic molecular markers.Citation45 It is important to characterize genes and proteins that distinguish between chronic phase and blast crisis, and identify “molecular signatures” of blast crisis associated with high malignancy and development of resistance.Citation46 Our work may lead to the utilization of phosphorylated eIF2α as reliable biological marker of CML disease development and treatment.
Materials and Methods
Cell culture and treatments
Mouse 32Dc13 cells (CRL-11346) and human K562 cells (CCL-243) were obtained from American Type Culture Collection (ATCC). 32D cells transformed with BCR-ABL and human BV173 cells were kindly provided by Dr. Sharon McKenna.Citation19,Citation47 32D cells expressing BCR-ABL bearing the threonine 315 to isoleucine (T315I) substitution were kindly provided by Prof. T. Skorski.Citation48 Peripheral blood monocytes (PBMCs) from healthy donors were grown in RPMI 1640 with 10% FBS, 2 mM L-glutamine, 1% penicillin/streptomycin in the absence or presence of phytohemagglutinin (PHA). CML CD34+ cells were obtained from bone marrow or blood from patients at the Institute of Hematology and Blood Transfusion in Warsaw (Poland) with patients’ consent and approval of the Ethical Committee. CD34+ cells were separated by gradient centrifugation by using human MACS® CD34+ kit (Miltenyi Biotec) or human CD34+ selection cocktail (StemCell Technologies, Inc.). CD34+ cells from healthy donors were isolated from buffy coats. CD34+ cells were cultured in IMDM medium (Invitrogen) with 10% FBS, 1 ng/ml of granulocyte-macrophage colony-stimulating factor (GM-CSF), 1 ng/ml of stem cell factor (SCF), 2 ng/ml of interleukin-3 (IL-3). Cells were treated with imatinib (a generous gift from the Pharmaceutical Research Institute, Warsaw). Cells were treated with the pan-caspase inhibitor Z-VAD-FMK (Calbiochem) at the final concentration of 40 µM.
Protein extraction, western blot analysis and immunoprecipitations
Cells were lysed in a modified radio-immunoprecipitation (RIPA) buffer as previously described.Citation47 The antibodies used for the western blots were: c-ABL from Calbiochem, phospho-eIF2α (S51) from Invitrogen, BiP/Grp78 from BD Biosciences, eIF2α, phospho-PERK (T980), STAT5, phospho-STAT5 (Y694) and poly (ADP-ribose) polymerase (PARP) from Cell Signaling Technology as well as PERK and phospho-PERK (Y615), as previously described.Citation21 Immunoprecipitations were performed with a mouse monoclonal antibody against the Myc epitope (9E10) (Santa Cruz Biotechnology, Inc.) as described.Citation21
Viability and apoptosis assays
Viability assays were performed by incubating cells with propidium iodide (PI) (Sigma-Aldrich) at the concentration of 50 μg/ml prior to analysis by flow cytometry. Apoptotic cells were stained with Annexin V-PE and 7-AAD and subjected to flow cytometry according to manufacturer's specifications (BD PharMingen).
Transfection assays
Cells (1 × 106) were transfected using the Nucleofector (Nucleofector Technology, Lonza Verviers) according to the manufacturer’s specification. Transfections were performed with 2 µg of the following cDNAs in the pcDNA3.1 vector: eIF2α-S51A,Citation23 PERK-K618A,Citation8,Citation20 or PERK-Y615F.Citation21 As control, 2 µg of insert-less pcDNA 3.1 vector was used. The transfection efficiency was determined by flow cytometry after transfection of cells with the 2 µg of the GFP-expressing vector pEGFP-C1.
Cell growth and clonogenic assays
To determine the population doubling time (PD), cells were cultivated for 48 h (t), and the PD time was calculated from the initial (I) and final cell number (E) according to the following equation: PD = t log 2 / log E - log I.
To determine the clonogenic potential, 5 × 103 cells were plated in cytokine-free methylcellulose (MethoCult H40230, StemCell Rechnologies, Inc.) with or without the addition of imatinib. Colonies containing more than 40 cells were counted after 5 d of culture. The diameters of 150 colonies were measured using Olympus fluorescent microscope and Image Pro Plus program.
Data statistics
All data are shown as mean ± SEM of three independent experiments. Student’s t-test was used to calculate statistical significance; *, p < 0.05; **, p < 0.005; ***, p < 0.0005.
Acknowledgments
We thank Dr. S. McKenna from Cork Cancer Research Centre in Ireland for providing the 32D and BCR-ABL-transfected 32D cell lines; M. Ciezka and W. Dudka-Ruszkowska for help in some of the experiments; Dr. J. Niesiobedzka-Krezel for providing a CML sample. This study was supported by grants from National Science Center (N N301 425938 and 2011/01/B/NZ3/02145 to K.P.; 2011/01/N/NZ4/01707 to M.K.-K. and N N401 594740 to E.G.M.), Ministry of Science and Higher Education (Iuventus Plus IP2010 032870 to P.P.-B.), the Medical University of Warsaw (1M19/NK1W/2009 to T.S. and 1M19/NK1D/2009 to E.G.-M.) and Leukemia and Lymphoma Society of Canada (LLSC) to A.E.K. K.P. is an ISAC Scholar Fellow. P.P. is the recipient of the Montreal Centre for Experimental Therapeutics in Cancer (MCETC) post-doctoral award.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Tassone P, Tagliaferri P, Fulciniti MT, Di Martino MT, Venuta S. Novel therapeutic approaches based on the targeting of microenvironment-derived survival pathways in human cancer: experimental models and translational issues. Curr Pharm Des 2007; 13:487 - 96; http://dx.doi.org/10.2174/138161207780162872; PMID: 17348845
- Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 2003; 4:181 - 91; http://dx.doi.org/10.1038/nrm1052; PMID: 12612637
- Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem 2005; 74:739 - 89; http://dx.doi.org/10.1146/annurev.biochem.73.011303.074134; PMID: 15952902
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8:519 - 29; http://dx.doi.org/10.1038/nrm2199; PMID: 17565364
- Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest 2002; 110:1383 - 8; PMID: 12438433
- Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans 2006; 34:7 - 11; http://dx.doi.org/10.1042/BST0340007; PMID: 16246168
- Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, et al. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol 2004; 24:10161 - 8; http://dx.doi.org/10.1128/MCB.24.23.10161-10168.2004; PMID: 15542827
- Jiang HY, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR, et al. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol Cell Biol 2003; 23:5651 - 63; http://dx.doi.org/10.1128/MCB.23.16.5651-5663.2003; PMID: 12897138
- Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007; 14:230 - 9; http://dx.doi.org/10.1038/sj.cdd.4401984; PMID: 16794605
- Schönthal AH. Endoplasmic reticulum stress and autophagy as targets for cancer therapy. Cancer Lett 2009; 275:163 - 9; http://dx.doi.org/10.1016/j.canlet.2008.07.005; PMID: 18692955
- Kurzrock R, Gutterman JU, Talpaz M. The molecular genetics of Philadelphia chromosome-positive leukemias. N Engl J Med 1988; 319:990 - 8; http://dx.doi.org/10.1056/NEJM198810133191506; PMID: 3047582
- Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer 2005; 5:172 - 83; http://dx.doi.org/10.1038/nrc1567; PMID: 15719031
- Savona M, Talpaz M. Getting to the stem of chronic myeloid leukaemia. Nat Rev Cancer 2008; 8:341 - 50; http://dx.doi.org/10.1038/nrc2368; PMID: 18385684
- Schiffer CA. BCR-ABL tyrosine kinase inhibitors for chronic myelogenous leukemia. N Engl J Med 2007; 357:258 - 65; http://dx.doi.org/10.1056/NEJMct071828; PMID: 17634461
- Quintás-Cardama A, Kantarjian H, Cortes J. Flying under the radar: the new wave of BCR-ABL inhibitors. Nat Rev Drug Discov 2007; 6:834 - 48; http://dx.doi.org/10.1038/nrd2324; PMID: 17853901
- Weisberg E, Manley PW, Cowan-Jacob SW, Hochhaus A, Griffin JD. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat Rev Cancer 2007; 7:345 - 56; http://dx.doi.org/10.1038/nrc2126; PMID: 17457302
- Verfaillie T, Salazar M, Velasco G, Agostinis P.. Linking ER Stress to Autophagy: Potential Implications for Cancer Therapy. Int J Cell Biol 2010; 2010:930509
- Wang G, Yang ZQ, Zhang K. Endoplasmic reticulum stress response in cancer: molecular mechanism and therapeutic potential. Am J Transl Res 2010; 2:65 - 74; PMID: 20182583
- Piwocka K, Vejda S, Cotter TG, O’Sullivan GC, McKenna SL. Bcr-Abl reduces endoplasmic reticulum releasable calcium levels by a Bcl-2-independent mechanism and inhibits calcium-dependent apoptotic signaling. Blood 2006; 107:4003 - 10; http://dx.doi.org/10.1182/blood-2005-04-1523; PMID: 16469868
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999; 397:271 - 4; http://dx.doi.org/10.1038/16729; PMID: 9930704
- Su Q, Wang S, Gao HQ, Kazemi S, Harding HP, Ron D, et al. Modulation of the eukaryotic initiation factor 2 alpha-subunit kinase PERK by tyrosine phosphorylation. J Biol Chem 2008; 283:469 - 75; http://dx.doi.org/10.1074/jbc.M704612200; PMID: 17998206
- Hantschel O, Warsch W, Eckelhart E, Kaupe I, Grebien F, Wagner KU, et al. BCR-ABL uncouples canonical JAK2-STAT5 signaling in chronic myeloid leukemia. Nat Chem Biol 2012; 8:285 - 93; http://dx.doi.org/10.1038/nchembio.775; PMID: 22286129
- Donzé O, Jagus R, Koromilas AE, Hershey JW, Sonenberg N. Abrogation of translation initiation factor eIF-2 phosphorylation causes malignant transformation of NIH 3T3 cells. EMBO J 1995; 14:3828 - 34; PMID: 7641700
- Calabretta B, Perrotti D. The biology of CML blast crisis. Blood 2004; 103:4010 - 22; http://dx.doi.org/10.1182/blood-2003-12-4111; PMID: 14982876
- Donato NJ, Wu JY, Stapley J, Lin H, Arlinghaus R, Aggarwal BB, et al. Imatinib mesylate resistance through BCR-ABL independence in chronic myelogenous leukemia. Cancer Res 2004; 64:672 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-03-1484; PMID: 14744784
- Nambu T, Araki N, Nakagawa A, Kuniyasu A, Kawaguchi T, Hamada A, et al. Contribution of BCR-ABL-independent activation of ERK1/2 to acquired imatinib resistance in K562 chronic myeloid leukemia cells. Cancer Sci 2010; 101:137 - 42; http://dx.doi.org/10.1111/j.1349-7006.2009.01365.x; PMID: 19843070
- Jagani Z, Dorsch M, Warmuth M. Hedgehog pathway activation in chronic myeloid leukemia. Cell Cycle 2010; 9:3449 - 56; http://dx.doi.org/10.4161/cc.9.17.12945; PMID: 20928937
- Vakana E, Platanias LC. AMPK in BCR-ABL expressing leukemias. Regulatory effects and therapeutic implications. Oncotarget 2011; 2:1322 - 8; PMID: 22249159
- Dufies M, Jacquel A, Belhacene N, Robert G, Cluzeau T, Luciano F, et al. Mechanisms of AXL overexpression and function in Imatinib-resistant chronic myeloid leukemia cells. Oncotarget 2011; 2:874 - 85; PMID: 22141136
- Chomel JC, Turhan AG. Chronic myeloid leukemia stem cells in the era of targeted therapies: resistance, persistence and long-term dormancy. Oncotarget 2011; 2:713 - 27; PMID: 21946665
- Graham SM, Jørgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002; 99:319 - 25; http://dx.doi.org/10.1182/blood.V99.1.319; PMID: 11756187
- Chen Y, Peng C, Sullivan C, Li D, Li S. Critical molecular pathways in cancer stem cells of chronic myeloid leukemia. Leukemia 2010; 24:1545 - 54; http://dx.doi.org/10.1038/leu.2010.143; PMID: 20574455
- Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, Koumenis C, et al. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 2010; 29:3881 - 95; http://dx.doi.org/10.1038/onc.2010.153; PMID: 20453876
- Hamanaka RB, Bobrovnikova-Marjon E, Ji X, Liebhaber SA, Diehl JA. PERK-dependent regulation of IAP translation during ER stress. Oncogene 2009; 28:910 - 20; http://dx.doi.org/10.1038/onc.2008.428; PMID: 19029953
- Liu Y, László C, Liu Y, Liu W, Chen X, Evans SC, et al. Regulation of G(1) arrest and apoptosis in hypoxia by PERK and GCN2-mediated eIF2alpha phosphorylation. Neoplasia 2010; 12:61 - 8; PMID: 20072654
- Papadakis AI, Paraskeva E, Peidis P, Muaddi H, Li S, Raptis L, et al. eIF2alpha Kinase PKR modulates the hypoxic response by Stat3-dependent transcriptional suppression of HIF-1alpha. Cancer Res 2010; 70:7820 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-10-0215; PMID: 20924113
- Mounir Z, Krishnamoorthy JL, Wang S, Papadopoulou B, Campbell S, Muller WJ, et al. Akt determines cell fate through inhibition of the PERK-eIF2α phosphorylation pathway. Sci Signal 2011; 4:ra62; http://dx.doi.org/10.1126/scisignal.2001630; PMID: 21954288
- Peidis P, Papadakis AI, Muaddi H, Richard S, Koromilas AE. Doxorubicin bypasses the cytoprotective effects of eIF2α phosphorylation and promotes PKR-mediated cell death. Cell Death Differ 2011; 18:145 - 54; http://dx.doi.org/10.1038/cdd.2010.76; PMID: 20559319
- Peidis P, Papadakis AI, Rajesh K, Koromilas AE. HDAC pharmacological inhibition promotes cell death through the eIF2α kinases PKR and GCN2. Aging (Albany NY) 2010; 2:669 - 77; PMID: 21076179
- Muaddi H, Majumder M, Peidis P, Papadakis AI, Holcik M, Scheuner D, et al. Phosphorylation of eIF2α at serine 51 is an important determinant of cell survival and adaptation to glucose deficiency. Mol Biol Cell 2010; 21:3220 - 31; http://dx.doi.org/10.1091/mbc.E10-01-0023; PMID: 20660158
- Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest 2009; 119:1109 - 23; http://dx.doi.org/10.1172/JCI35660; PMID: 19363292
- Altman BJ, Jacobs SR, Mason EF, Michalek RD, MacIntyre AN, Coloff JL, et al. Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene 2011; 30:1855 - 67; http://dx.doi.org/10.1038/onc.2010.561; PMID: 21151168
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67 - 93; http://dx.doi.org/10.1146/annurev-genet-102808-114910; PMID: 19653858
- Tanimura A, Yujiri T, Tanaka Y, Hatanaka M, Mitani N, Nakamura Y, et al. The anti-apoptotic role of the unfolded protein response in Bcr-Abl-positive leukemia cells. Leuk Res 2009; 33:924 - 8; http://dx.doi.org/10.1016/j.leukres.2009.01.027; PMID: 19237191
- Oehler VG, Yeung KY, Choi YE, Bumgarner RE, Raftery AE, Radich JP. The derivation of diagnostic markers of chronic myeloid leukemia progression from microarray data. Blood 2009; 114:3292 - 8; http://dx.doi.org/10.1182/blood-2009-03-212969; PMID: 19654405
- Zheng C, Li L, Haak M, Brors B, Frank O, Giehl M, et al. Gene expression profiling of CD34+ cells identifies a molecular signature of chronic myeloid leukemia blast crisis. Leukemia 2006; 20:1028 - 34; http://dx.doi.org/10.1038/sj.leu.2404227; PMID: 16617318
- Keeshan K, Mills KI, Cotter TG, McKenna SL. Elevated Bcr-Abl expression levels are sufficient for a haematopoietic cell line to acquire a drug-resistant phenotype. Leukemia 2001; 15:1823 - 33; http://dx.doi.org/10.1038/sj.leu.2402309; PMID: 11753601
- Cramer K, Nieborowska-Skorska M, Koptyra M, Slupianek A, Penserga ET, Eaves CJ, et al. BCR/ABL and other kinases from chronic myeloproliferative disorders stimulate single-strand annealing, an unfaithful DNA double-strand break repair. Cancer Res 2008; 68:6884 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-08-1101; PMID: 18757400