Abstract
The Notch signaling pathway drives proliferation, differentiation, apoptosis, cell fate choices and maintenance of stem cells during embryogenesis and in self-renewing tissues of the adult. In addition, aberrant Notch signaling has been implicated in several tumors, where Notch can function both as an oncogene or a tumor-suppressor gene, depending on the context.
This Extra View aims to review what is currently known about Notch signaling, in particular in gastrointestinal tumors, providing a summary of our data on Notch1 signaling in gastric cancer with results obtained in colorectal cancer (CRC).
We have already reported that the epigenetic regulation of the Notch ligand DLL1 controls Notch1 signaling activation in gastric cancer, and that Notch1 inhibition is associated with the diffuse type of gastric cancer. Here, we describe additional data showing that in CRC cell lines, unlike gastric cancer, DLL1 expression is not regulated by promoter methylation. Moreover, in CRC, Notch1 receptor is not affected by any mutation. These data suggest a different regulation of Notch1 signaling between gastric cancer and CRC.
Introduction
The Notch system is an evolutionarily conserved signaling pathway that regulates many cellular processes, including stem cell maintenance, proliferation, differentiation and apoptosis during development and tumorigenesis.Citation1 Initially cloned in Drosophila in the mid-1980s by the group of Artavanis-Tsakonas,Citation2 in 1991 Notch1 was first implicated in cancer with a constitutive activation in T cell acute lymphocytic leukemia (T-ALL).Citation3 Importantly, the outcome of Notch signaling is dependent on the cellular context and Notch could act both as an oncogene and as a tumor suppressor gene.Citation4
Here we review what is currently known about the Notch signaling, focusing first on the activation and modulation of this system, then on its role in tumors, in particular in gastrointestinal cancers. For clarity, we have summarized results previously obtained by our group on the epigenetic regulation of the Notch ligand DLL1 in gastric cancers and then examined additional data on the lack of methylation of DLL1 in colorectal cancer (CRC) cell lines.
Activation of Notch cascade
In mammals, there are four Notch receptors (Notch 1–4) and five ligands, two of the Jagged family (Jagged1–2) and three of the Delta-like family (DLL1, DLL3, DLL4). Notch signaling activation starts when a membrane bound Notch ligand interacts with a membrane bound Notch receptor on an adjacent cell. This interaction results in a conformational change in Notch receptor, followed by two proteolytic steps. The first cleavage, mediated by a metalloprotease of the ADAM family, occurs extracellularly (at site S2) and makes Notch receptor available for the second cleavage (at site S3) within the transmembrane domain by the γ-secretase complex. In the canonical Notch signaling, following these proteolytic steps, the Notch intracellular domain (NICD) is released in the cytoplasm and translocates into the nucleus where activates the transcription of Notch target genes. NICD binding to the transcriptional repressor CSL (also known as CBF-1, or RBP-jk) converts it into a transcriptional activator recruiting co-activators, such as Mastermind like (MAML) and histone acetyltransferases, such as p300.Citation5 Among the best-known Notch target genes, are the HES (hairy enhancer of split) and Herp/Hey (Hes-related repressor protein with Y-Box) families of basic helix-loop-helix (bHLH) transcriptional repressors, cyclin D1, p21, NRARP (Notch-related ankyrin repeat protein), c-Myc and Deltex. HES and Herp in turn repress the activity of many bHLH transcriptional activators such as Math (mouse homolog of Atonal), Neurogenin and Mash (mouse homolog of Achaete/Scute).Citation6 For instance, in the gut, HES1 mediates the repression of Math1/HATH1.Citation7
Modulation of Notch signaling
Notch signaling can be modulated at different levels by microRNAsCitation8 and post-translational modifications like phosphorylation, ubiquitinylation, glycosylation and fucosylation. Phosphorylation of NICD on serine residues causes the formation of a NICD/Su(H) complex responsible for the intracellular localization of NICD.Citation9 Activation of Notch signaling is shortly terminated by E3 ubiquitin ligases that target Notch to proteosomal degradation.Citation10 Glycosylation by Fringe enzymes on O-linked fucose residues on certain EGF-like repeats inhibits the binding of Jagged ligands potentiating DLL mediated Notch activation.Citation11
Notch signaling in tumorigenesis
A deregulation of Notch signaling is a feature of many tumors and depending on the cellular context Notch can function both as an oncogene or a tumor suppressor. For instance, Notch has an oncogenic role in T-ALL, breast cancer, melanoma, non-small cell lung cancer (NSCLC) and colorectal cancer.Citation12,Citation13 On the contrary, Notch act as a tumor suppressor in the skin, hepatocytes and pancreatic epithelium.Citation12,Citation13
Notch in the gastrointestinal tract
In the intestinal homeostasis, the Notch pathway has a pivotal role for the maintenance of proliferating stem cells and is involved in the control of cell fate decisions between secretory and absorptive lineages.Citation14-Citation16 HES1−/− mice display a relative increase in enteroendrocrine lineages at the expense of enterocytes.Citation7,Citation15 A reciprocal phenotype is observed in Math−/− mice with an increase in absorptive enterocytes.Citation17,Citation18 Accordingly, overexpression of Math1 induces secretory metaplasia with almost complete enterocyte loss.Citation19 Importantly, the goblet cell metaplasia resulting from pharmacological inhibition of Notch signaling with γ-secretase or RBP-jk deletion is blocked in Math1−/− mice suggesting that Notch signaling requires Math1 repression to inhibit secretory cell differentiation.Citation20-Citation22 In addition to Math1 (also called HATH1), other proteins determine the differentiation choice among enteroendocrine, goblet and Paneth cells. In particular, Neurogenin 3 is essential for differentiation toward enteroendocrine cells,Citation23,Citation24 Klf4 (Kruppel-like factor 4) and Spdef (SAM pointed domain containing Ets transcription factor) are required for differentiation of goblet cellsCitation25,Citation26 while Sox9 is essential for Paneth cell differentiation.Citation27,Citation28 Finally, the growth factor-independent 1 (GFI1) transcription factor is expressed in both Paneth and goblet cells.Citation29
Among the five Notch ligands, DLL1 is the most important ligand for Notch1 receptor in the intestinal crypt epithelium and an absence of DLL1 causes an increase in goblet cells.Citation30
Results
DLL1 is epigenetically regulated in gastric cancer
Recently, we demonstrated that DLL1 expression is regulated by promoter hypermethylation in gastric cancer (GC) cell lines.Citation31 Indeed, by bisulfite sequencing, we found a dense promoter methylation in DLL1 non-expressing cell lines (KATOIII, SNU601, SNU719, AGS) and a lack of methylation in DLL1 expressing cell lines (SNU16, MKN1, MKN45, TMK1). Importantly, re-activation of DLL1 expression in SNU719 and AGS, after treatment with the demethylating agent 5-aza-2’deoxycitidine, or overexpression of DLL1 in SNU601, resulted in Notch1 signaling activation with an increase in HES1 and a decrease in HATH1. In addition, in 52 primary GC samples we found that DLL1 promoter hypermethylation was associated to the diffuse histotype. Accordingly, we found a higher level of HATH1 in diffuse and mixed histotypes and a positive correlation between DLL1 and HES1 expression levels in intestinal and mixed types. Intriguingly, lack of DLL1 ligand due to promoter methylation was found only in 50% of diffuse GC. Moreover, we confirmed that DLL1-Notch1 cascade influences the histological differentiation in GC in a mouse model of intestinal gastric cancer, in which we found that DLL1 promoter was unmethylated and DLL1 mRNA was expressed in all the mice.
DLL1 promoter is not methylated in CRC cell lines
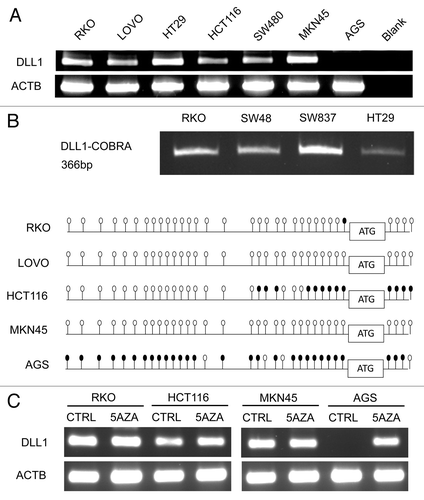
Following our findings on the epigenetic regulation of DLL1 in GC, we explored whether the same regulation is also present in colorectal cancer present in CRC cell lines. For this purpose, we evaluated DLL1 mRNA expression in a panel of CRC cell lines (RKO, LOVO, HT29, HCT116 and SW480) and two GC cell lines (MKN45 DLL1 expressing and AGS DLL1 not expressing were tested as a control). We found that DLL1 was expressed in all the CRC cell lines analyzed and in the GC cell lines as known (). Then, we studied the methylation status of the DLL1 promoter by COBRA and bisulfite sequencing and we found a lack of methylation in CRC cell lines, except for few CGs methylated in HCT116 (). As expected, MKN45 promoter resulted unmethylated while AGS promoter resulted methylated. To further exclude an epigenetic regulation, we treated RKO, HCT116 and the two GC cell lines used as a control with the de-methylating agent 5-aza-2’deoxycitidine (). Only for AGS that has a DLL1 promoter densely methylated, we found a significant increase in DLL1 mRNA expression in the treated cell line compared with the untreated. In line with our methylation results, a very minimal increase in DLL1 mRNA expression after 5aza treatment was found in HCT116. Taken together, these results suggest a different mechanism of regulation of Notch signaling between CRC and GC. To further investigate how Notch signaling is regulated in CRC, we screened a panel of CRC cell lines (RKO, HT29, SW48, CACO-2, HCT116, HCT116 p53−/−, HCT15, SW480, SW837, DLD1) for Notch1 mutations from exon 20 to 34 (the “hot spot” region for mutations in T-ALL) but we didn’t detect any (data not shown).
Figure 1. DLL1 expression and methylation analyses in CRC and GC cell lines (A) DLL1 RNA expression. ACTB, beta-actin; blank, negative RT-PCR control; (B) COBRA (upper panel) and bisulfite sequencing (lower panel) of DLL1 promoter. In COBRA, a single PCR product at 366 bp (base pairs) indicates lack of methylation. In bisulfite sequencing panel, methylation status of each promoter is shown as a row of CpG sites (empty dot, CpG unmethylated; solid dot, CpG methylated); (C) DLL1 mRNA expression after 5-aza treatment; CTRL, not treated; 5AZA, treated with 5-aza-2’deoxycitidine.
Discussion
We have recently shown that promoter hypermethylation of the Notch1 ligand DLL1 regulates its silencing in gastric cancer (GC) cell lines and the repression of Notch1 signaling.Citation31 In addition, in 43 patients with intestinal type of GC, DLL1 expression positively correlated with HES1 while HATH1 expression was associated with the diffuse and mixed histology. Moreover, in 52 GC clinical samples the methylation of DLL1 promoter was associated with the diffuse histotype. Finally, the absence of methylation in DLL1 promoter in intestinal GC was confirmed in a murine model of intestinal carcinogenesis in which DLL1 was expressed and the promoter unmethylated. Taken together these results suggest that Notch1 signaling is activated in intestinal GC where DLL1 is expressed and this expression associates with HES1. Conversely, the methylation of DLL1 promoter in diffuse GC, together with the high level of HATH1 (that is negatively regulated by HES1-Notch1 signaling), suggests that Notch1 signaling is switched off in diffuse GC. Intriguingly, DLL1 methylation was found only in 50% of diffuse GC, even though this histotype was associated with high HATH1 expression. These findings suggest that changes in DLL1 expression are only partially regulated by DLL1 promoter methylation and HATH1 expression could be modulated independently from canonical Notch1 signaling. Indeed, in the intestine, there is a complex signaling network among Notch and many other pathways such as Wnt, Bmp and Sonic Hedgehog.Citation32 For instance, the activation of Wnt/β-catenin leads to HATH1 degradation in CRC cells,Citation33-Citation35 while the activation of Notch1 signaling downregulates Wnt/β-catenin targets genes through chromatin modification.Citation36
In this Extra View, we reported new data on the absence of DLL1 methylation in CRC cell lines. Importantly, DLL1 promoter was unmethylated also in RKO and SW48 that display a widespread methylation due to the CpG island methylator phenotype (CIMP). To explain the discrepancy in DLL1 methylation between CRC and diffuse gastric cancer we should take into account the different histology of these two cancers. It’s known that the absence of DLL1 mimics the inactivation of Notch1 and causes an increase in mucus secreting goblet cells.Citation30 In line with this, diffuse type of gastric adenocarcinoma frequently displays signet ring cells rich in mucin, in particular in familial syndromes,Citation37 while signet ring carcinoma is a rare entity in CRC.Citation38
It’s known that loss of E-cadherin is crucial in diffuse gastric cancersCitation39,Citation40 and E-cadherin promoter methylation has been reported to be the second hit both in sporadic and hereditary diffuse gastric cancers.Citation41,Citation42 As described above, there is an interplay between Wnt/β-catenin and Notch signaling in CRC: it is possible that E-cadherin could interfere with Notch signaling in gastric cancer. In fact, in other systems, it has been shown that E-cadherin loss is associated with increase of Notch activityCitation43 and that Notch can work as an E-cadherin repressor.Citation44,Citation45 However, one should keep in mind that the Notch signaling pathway can alternatively work as either tumor suppressor or oncogene depending on the specific cell subtypes. Furthermore, even though we didn’t analyze E-cadherin promoter status in our patients, Notch signaling loss in diffuse gastric cancer could be a contributing feature to this specific histotype along with loss of E-cadherin. In addition, E-cadherin promoter methylation is extremely frequent in cancers arising in the settings of ulcerative colitis, an inflammatory disease that increases the risk of colon cancer.Citation46 Thus, future studies should be targeted in order to find important information on the interplay between E-Cadherin/Wnt and Notch signaling in inflammatory-driven colon cancers.
Material and Methods
Cell cultures and treatments
The human colorectal cancer (CRC) cell lines RKO, SW480, SW48, SW837, HCT116, LOVO and HT29 were obtained from ATCC. The human gastric cancer (GC) cell lines AGS and MKN45 were a kind gift of Dr. Antonia R. Sepulveda. Cell lines were cultured in the appropriate media (IMDM for CRC cell lines and RPMI for GC cell lines) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM glutamine (Life Technologies). Cells were maintained at 37°C in a 5% CO2 incubator.
5-aza-2′deoxycitidine (5AZA) was purchased from Sigma-Aldrich and the treatment was performed on RKO, HCT116, MKN45 and AGS cell lines at 1–5 µM for 96 h.
RNA extraction and RT-PCR
RNA extraction from cell lines was performed with TRIzol (Life Technologies), and 2 µg of RNA were retrotranscribed using random hexamers and MMLV reverse transcriptase (Life Technologies). RT-PCR was performed using the HotstarTaq Master Mix Kit (Qiagen). Primers sequences are the following: DLL1 Forward 5′-TATCCGCTATCCAGGCTGTC-3′ and Reverse 5′-GGTGGGCAGGTACAGGAGTA-3′; β-actin Forward 5′- TCACACTGGCATCGTGATGGACTC-3′ and Reverse 5′- TCCTGCTTGCTGATCCACATCTGC-3′. PCR products were separated on a 2% agarose gel, stained with 0.5 µg/ml ethidium bromide and the image was acquired under UV illumination (Gel Logic Imaging System).
Methylation assays
The methylation status of the DLL1 promoter was determined by bisulfite sequencing or combined bisulfite restriction assay (COBRA). One μg of DNA was treated with sodium bisulfite with the Epitect Bisulfite Kit (Qiagen), following the manufacturer’s protocol. Modified DNA was used as a template for PCR reactions. For both COBRA and bisulfite sequencing, a 366-bp fragment of the DLL1 gene located in the 5′UTR at -329 from the transcriptional start codon was amplified using primers DLL1-F (5′-AGGAAGTYGGYGATTTTTATTTT-3′) and DLL1-R (5′-AAAACACCRCCAAAACCAACRC-3′). PCR was performed using HotstarTaq Master Mix Kit for 45 cycles as follow: 94°C for 30 sec (denaturation) and 72°C for 30 sec (elongation). The annealing temperature was reduced by 2°C every 15 cycles starting from 55°C. For COBRA, PCR product (5–10 μL) was digested with 10 to 20 units of TaqI at 37°C for 2 h. If DLL1 was methylated, the PCR product was cleaved into five fragments of 78, 175, 113, 288, 253 bp. Digested products were analyzed by agarose electrophoresis (3%), and visualized by staining with ethidium bromide. For the bisulfite sequencing, 2 μl of PCR products were ligated into the TOPO vector (Life Technologies), and, after transformation, clones were sequenced in both directions with M13 primers.
| Abbreviations: | ||
| GC | = | gastric cancer |
| DLL1 | = | delta-like 1 |
| NICD | = | Notch intracellular domain |
| 5AZA | = | 5-aza-2’deoxycitidine |
| HES1 | = | hairy/enhancer of split |
| HATH1 | = | atonal |
Acknowledgments
This work was supported by an Italian Association for Cancer Research (AIRC) investigator grant 10216 (to L.R.).
References
- Borggrefe T, Liefke R. Fine-tuning of the intracellular canonical Notch signaling pathway. Cell Cycle 2012; 11:264 - 76; http://dx.doi.org/10.4161/cc.11.2.18995; PMID: 22223095
- Artavanis-Tsakonas S, Muskavitch MA, Yedvobnick B. Molecular cloning of Notch, a locus affecting neurogenesis in Drosophila melanogaster. Proc Natl Acad Sci USA 1983; 80:1977 - 81; http://dx.doi.org/10.1073/pnas.80.7.1977; PMID: 6403942
- Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD, et al. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991; 66:649 - 61; http://dx.doi.org/10.1016/0092-8674(91)90111-B; PMID: 1831692
- Koch U, Radtke F. Notch and cancer: a double-edged sword. Cell Mol Life Sci 2007; 64:2746 - 62; http://dx.doi.org/10.1007/s00018-007-7164-1; PMID: 17687513
- Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 2009; 137:216 - 33; http://dx.doi.org/10.1016/j.cell.2009.03.045; PMID: 19379690
- Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol 2003; 194:237 - 55; http://dx.doi.org/10.1002/jcp.10208; PMID: 12548545
- Jensen J, Pedersen EE, Galante P, Hald J, Heller RS, Ishibashi M, et al. Control of endodermal endocrine development by Hes-1. Nat Genet 2000; 24:36 - 44; http://dx.doi.org/10.1038/71657; PMID: 10615124
- Wang Z, Li Y, Kong D, Ahmad A, Banerjee S, Sarkar FH. Cross-talk between miRNA and Notch signaling pathways in tumor development and progression. Cancer Lett 2010; 292:141 - 8; http://dx.doi.org/10.1016/j.canlet.2009.11.012; PMID: 20022691
- Kidd S, Lieber T, Young MW. Ligand-induced cleavage and regulation of nuclear entry of Notch in Drosophila melanogaster embryos. Genes Dev 1998; 12:3728 - 40; http://dx.doi.org/10.1101/gad.12.23.3728; PMID: 9851979
- Lai EC. Protein degradation: four E3s for the notch pathway. Curr Biol 2002; 12:R74 - 8; http://dx.doi.org/10.1016/S0960-9822(01)00679-0; PMID: 11818085
- Haines N, Irvine KD. Glycosylation regulates Notch signalling. Nat Rev Mol Cell Biol 2003; 4:786 - 97; PMID: 14570055
- Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer 2011; 11:338 - 51; http://dx.doi.org/10.1038/nrc3035; PMID: 21508972
- Lobry C, Oh P, Aifantis I. Oncogenic and tumor suppressor functions of Notch in cancer: it’s NOTCH what you think. J Exp Med 2011; 208:1931 - 5; http://dx.doi.org/10.1084/jem.20111855; PMID: 21948802
- Fre S, Pallavi SK, Huyghe M, Laé M, Janssen KP, Robine S, et al. Notch and Wnt signals cooperatively control cell proliferation and tumorigenesis in the intestine. Proc Natl Acad Sci USA 2009; 106:6309 - 14; http://dx.doi.org/10.1073/pnas.0900427106; PMID: 19251639
- van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005; 435:959 - 63; http://dx.doi.org/10.1038/nature03659; PMID: 15959515
- VanDussen KL, Carulli AJ, Keeley TM, Patel SR, Puthoff BJ, Magness ST, et al. Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development 2012; 139:488 - 97; http://dx.doi.org/10.1242/dev.070763; PMID: 22190634
- Yang Q, Bermingham NA, Finegold MJ, Zoghbi HY. Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science 2001; 294:2155 - 8; http://dx.doi.org/10.1126/science.1065718; PMID: 11739954
- Shroyer NF, Helmrath MA, Wang VY, Antalffy B, Henning SJ, Zoghbi HY. Intestine-specific ablation of mouse atonal homolog 1 (Math1) reveals a role in cellular homeostasis. Gastroenterology 2007; 132:2478 - 88; http://dx.doi.org/10.1053/j.gastro.2007.03.047; PMID: 17570220
- VanDussen KL, Samuelson LC. Mouse atonal homolog 1 directs intestinal progenitors to secretory cell rather than absorptive cell fate. Dev Biol 2010; 346:215 - 23; http://dx.doi.org/10.1016/j.ydbio.2010.07.026; PMID: 20691176
- Kazanjian A, Noah T, Brown D, Burkart J, Shroyer NF. Atonal homolog 1 is required for growth and differentiation effects of notch/gamma-secretase inhibitors on normal and cancerous intestinal epithelial cells. Gastroenterology 2010; 139:28 e1-6 918 - 28, 928, e1-6; http://dx.doi.org/10.1053/j.gastro.2010.05.081; PMID: 20621629
- van Es JH, de Geest N, van de Born M, Clevers H, Hassan BA. Intestinal stem cells lacking the Math1 tumour suppressor are refractory to Notch inhibitors. Nat Commun 2010; 1:18; http://dx.doi.org/10.1038/ncomms1017; PMID: 20975679
- Kim TH, Shivdasani RA. Genetic evidence that intestinal Notch functions vary regionally and operate through a common mechanism of Math1 repression. J Biol Chem 2011; 286:11427 - 33; http://dx.doi.org/10.1074/jbc.M110.188797; PMID: 21282114
- Jenny M, Uhl C, Roche C, Duluc I, Guillermin V, Guillemot F, et al. Neurogenin3 is differentially required for endocrine cell fate specification in the intestinal and gastric epithelium. EMBO J 2002; 21:6338 - 47; http://dx.doi.org/10.1093/emboj/cdf649; PMID: 12456641
- Mellitzer G, Beucher A, Lobstein V, Michel P, Robine S, Kedinger M, et al. Loss of enteroendocrine cells in mice alters lipid absorption and glucose homeostasis and impairs postnatal survival. J Clin Invest 2010; 120:1708 - 21; http://dx.doi.org/10.1172/JCI40794; PMID: 20364088
- Katz JP, Perreault N, Goldstein BG, Lee CS, Labosky PA, Yang VW, et al. The zinc-finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon. Development 2002; 129:2619 - 28; PMID: 12015290
- Noah TK, Kazanjian A, Whitsett J, Shroyer NF. SAM pointed domain ETS factor (SPDEF) regulates terminal differentiation and maturation of intestinal goblet cells. Exp Cell Res 2010; 316:452 - 65; http://dx.doi.org/10.1016/j.yexcr.2009.09.020; PMID: 19786015
- Mori-Akiyama Y, van den Born M, van Es JH, Hamilton SR, Adams HP, Zhang J, et al. SOX9 is required for the differentiation of paneth cells in the intestinal epithelium. Gastroenterology 2007; 133:539 - 46; http://dx.doi.org/10.1053/j.gastro.2007.05.020; PMID: 17681175
- Bastide P, Darido C, Pannequin J, Kist R, Robine S, Marty-Double C, et al. Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J Cell Biol 2007; 178:635 - 48; http://dx.doi.org/10.1083/jcb.200704152; PMID: 17698607
- Bjerknes M, Cheng H. Cell Lineage metastability in Gfi1-deficient mouse intestinal epithelium. Dev Biol 2010; 345:49 - 63; http://dx.doi.org/10.1016/j.ydbio.2010.06.021; PMID: 20599897
- Pellegrinet L, Rodilla V, Liu Z, Chen S, Koch U, Espinosa L, et al. Dll1- and dll4-mediated notch signaling are required for homeostasis of intestinal stem cells. Gastroenterology 2011; 140:e1-7 1230 - 40; http://dx.doi.org/10.1053/j.gastro.2011.01.005; PMID: 21238454
- Piazzi G, Fini L, Selgrad M, Garcia M, Daoud Y, Wex T, et al. Epigenetic regulation of Delta-Like1 controls Notch1 activation in gastric cancer. Oncotarget 2011; 2:1291 - 301; PMID: 22249198
- Sancho E, Batlle E, Clevers H. Signaling pathways in intestinal development and cancer. Annu Rev Cell Dev Biol 2004; 20:695 - 723; http://dx.doi.org/10.1146/annurev.cellbio.20.010403.092805; PMID: 15473857
- Peignon G, Durand A, Cacheux W, Ayrault O, Terris B, Laurent-Puig P, et al. Complex interplay between β-catenin signalling and Notch effectors in intestinal tumorigenesis. Gut 2011; 60:166 - 76; http://dx.doi.org/10.1136/gut.2009.204719; PMID: 21205878
- Tsuchiya K, Nakamura T, Okamoto R, Kanai T, Watanabe M. Reciprocal targeting of Hath1 and beta-catenin by Wnt glycogen synthase kinase 3beta in human colon cancer. Gastroenterology 2007; 132:208 - 20; http://dx.doi.org/10.1053/j.gastro.2006.10.031; PMID: 17241872
- Aragaki M, Tsuchiya K, Okamoto R, Yoshioka S, Nakamura T, Sakamoto N, et al. Proteasomal degradation of Atoh1 by aberrant Wnt signaling maintains the undifferentiated state of colon cancer. Biochem Biophys Res Commun 2008; 368:923 - 9; http://dx.doi.org/10.1016/j.bbrc.2008.02.011; PMID: 18275842
- Kim HA, Koo BK, Cho JH, Kim YY, Seong J, Chang HJ, et al. Notch1 counteracts WNT/β-catenin signaling through chromatin modification in colorectal cancer. J Clin Invest 2012; 122:3248 - 59; http://dx.doi.org/10.1172/JCI61216; PMID: 22863622
- Charlton A, Blair V, Shaw D, Parry S, Guilford P, Martin IG. Hereditary diffuse gastric cancer: predominance of multiple foci of signet ring cell carcinoma in distal stomach and transitional zone. Gut 2004; 53:814 - 20; http://dx.doi.org/10.1136/gut.2002.010447; PMID: 15138207
- Psathakis D, Schiedeck TH, Krug F, Oevermann E, Kujath P, Bruch HP. Ordinary colorectal adenocarcinoma vs. primary colorectal signet-ring cell carcinoma: study matched for age, gender, grade, and stage. Dis Colon Rectum 1999; 42:1618 - 25; http://dx.doi.org/10.1007/BF02236218; PMID: 10613484
- Becker KF, Atkinson MJ, Reich U, Becker I, Nekarda H, Siewert JR, et al. E-cadherin gene mutations provide clues to diffuse type gastric carcinomas. Cancer Res 1994; 54:3845 - 52; PMID: 8033105
- Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, et al. E-cadherin germline mutations in familial gastric cancer. Nature 1998; 392:402 - 5; http://dx.doi.org/10.1038/32918; PMID: 9537325
- Machado JC, Oliveira C, Carvalho R, Soares P, Berx G, Caldas C, et al. E-cadherin gene (CDH1) promoter methylation as the second hit in sporadic diffuse gastric carcinoma. Oncogene 2001; 20:1525 - 8; http://dx.doi.org/10.1038/sj.onc.1204234; PMID: 11313896
- Grady WM, Willis J, Guilford PJ, Dunbier AK, Toro TT, Lynch H, et al. Methylation of the CDH1 promoter as the second genetic hit in hereditary diffuse gastric cancer. Nat Genet 2000; 26:16 - 7; http://dx.doi.org/10.1038/79120; PMID: 10973239
- Ferreira AC, Suriano G, Mendes N, Gomes B, Wen X, Carneiro F, et al. E-cadherin impairment increases cell survival through Notch-dependent upregulation of Bcl-2. Hum Mol Genet 2012; 21:334 - 43; http://dx.doi.org/10.1093/hmg/ddr469; PMID: 21989054
- Leong KG, Niessen K, Kulic I, Raouf A, Eaves C, Pollet I, et al. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J Exp Med 2007; 204:2935 - 48; http://dx.doi.org/10.1084/jem.20071082; PMID: 17984306
- Stylianou S, Clarke RB, Brennan K. Aberrant activation of notch signaling in human breast cancer. Cancer Res 2006; 66:1517 - 25; http://dx.doi.org/10.1158/0008-5472.CAN-05-3054; PMID: 16452208
- Azarschab P, Porschen R, Gregor M, Blin N, Holzmann K. Epigenetic control of the E-cadherin gene (CDH1) by CpG methylation in colectomy samples of patients with ulcerative colitis. Genes Chromosomes Cancer 2002; 35:121 - 6; http://dx.doi.org/10.1002/gcc.10101; PMID: 12203775