It is easy to conceive that in a toxic environment most cells would abandon the division process. In the past decades, it has indeed become clear that cells have developed complex cell cycle checkpoint mechanisms, which slow-down or stop cells from proliferating under challenging conditions. More recently, however, is the discovery and molecular characterization of signaling cascades capable of integrating classical stress-activated responses and cell cycle checkpoints. A report published in a recent issue of Cell CycleCitation1 sheds light on the increasing complexity of such pathways. The authors found that the stress-activated protein kinases p38 and JNK cooperate with Chk1 to block progression into mitosis under conditions that alter DNA replication (i.e., treatments of cells with antiproliferative drugs such as hydroxyurea, aphidicolin, camptothecin or etoposide). By using a combination of pharmacological and genetic tools, together with precise protocols of cell cycle synchronization, the study shows compelling evidence that in murine NIH3T3 and embryonic fibroblasts, hydroxyurea (used at concentrations capable of abrogating DNA synthesis) added during progression through S-phase first induces an early acute activation of Chk1 that is immediately followed by a phasic activation of both p38 and JNK. Remarkably, p38 and JNK activities are triggered completely independent of ATM-ATR and Chk1 activation, since their activities were similarly induced in the presence of caffeine and UCN-01, respectively.
Additionally, activation of p38 under DNA replication blockade seems to be mediated by MKK3 and MKK6 (usual kinases turning-on p38 signaling), while interestingly, JNK activation seems to be exclusively caused by MKK4 (one of the regular kinases switching-on the JNK pathway) but not MKK7. Furthermore, downstream of p38 signaling, the authors unveiled activation of MK2 and MK3, kinases classically activated by p38 under many stressors. Strikingly, activation of MKK3/6-p38-MK2/3 and MKK4-JNK cascades in the presence of hydroxyurea is required to suppress entry into mitosis as measured by reduced (Serine 10)-histone H3 phospho-signal and impaired (Threonine 14 and Tyrosine 15)-CDK1 dephosphorylation and activation. The effect on CDK1 activity is likely mediated by inhibitory regulation of members of the Cdc25 family of phosphatases (), as previously reported by others.Citation2-Citation4 Finally, the authors establish that at least p38α and p38β are involved in this mechanism; however, it is still unclear whether JNK1 and JNK2 both play significant roles in the process. Also, it remains to be identified which kinase(s) is (are) responsible for the activation of the respective p38 and JNK MKKs, under conditions blocking DNA synthesis/replication.
Figure 1. A proposed model of signaling circuitry integrating stress-response and cell cycle checkpoint pathways. Refer to text and references for details.
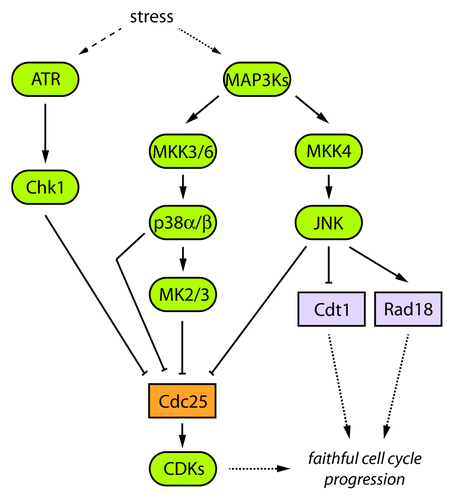
On the other hand, some progress has been made in the identification of substrates of the stress-activated protein kinases p38 and JNK implicated in DNA replication. Two recent studies found that Cdt1 is phosphorylated, probably at several residues, by both p38 and JNK, in the presence of genotoxicCitation5 (UV-C) and non-genotoxicCitation6 (including sorbitol and anisomycin) stressors. Cdt1 is a key DNA replication licensing factor that contributes with Cdc6 to ensure proper loading of the MCM complex onto chromatin to form the pre-replication complex. Notably, stress-mediated phosphorylation of Cdt1 blocks its degradation during S-phase,Citation5,Citation6 leads to the dissociation of the histone acetylase HBO1/KAT7 from replication originsCitation6 and compromises the ability of Cdt1 to instigate loading of the MCM complex,Citation5 therefore blocking initiation of DNA replication.Citation5,Citation6
Moreover, JNK was also found to phosphorylate the RING-finger type E3 ubiquitin ligase Rad18, involved in postreplication repair of damaged DNA (UV-C irradiation). JNK-mediated Rad18 phosphorylation appears to facilitate recruitment of the translesion synthesis DNA polymerase Polη to stalled replication forks.Citation7 Polη is then presumably capable of DNA synthesis over the damaged DNA, later allowing DNA replication by conventional DNA polymerases to occur. This intriguing observation rather suggests a role for JNK in DNA damage response tolerance pathways.
Based on those and other studies describing a direct control of cell cycle regulators by stress-response pathways, at least in the case of JNK, it is possible to advocate a general function in cellular genome integrity maintenance, through the regulation of substrates such as Cdt1 and Rad18, or histone H3 and Cdh1/FZR1, under toxicCitation5-Citation7 or unperturbedCitation8,Citation9 conditions, respectively. Finally, according to our current understanding of the stress-activated signaling pathways, it is noteworthy that the analytical dynamics, subcellular localization and temporal occurrence of the stress input (directly associated with cell cycle checkpoints or not) would likely determine not only the strength, duration and diffusion of the stress response but more importantly its final functional outcome.Citation1 In practical terms, the modulability of the stress-response could decide which exact pathways are activated and which substrates need to be accordingly modified. Given the growing involvement of stress responses in the control of cell cycle (checkpoint) mechanisms, we would speculate that in coming years, the cell cycle-related phosphoproteome governed by canonical stress kinases will continue to augment.
| Abbreviations: | ||
| JNK | = | c-Jun NH2-terminal kinase |
| Chk1 | = | checkpoint kinase 1 |
| DNA | = | deoxyribonucleic acid |
| ATM | = | ataxia telangiectasia mutated protein kinase |
| ATR | = | ataxia telangiectasia and Rad3-related protein kinase |
| MKK | = | (MAPKK), mitogen-activated protein kinase kinase |
| MK | = | (MAPKAPK), MAPK-activated protein kinase |
| CDK | = | cyclin-dependent kinase |
| UVC | = | ultraviolet C |
| HBO1 | = | histone acetyltransferase binding to ORC 1 |
| MCM | = | mini-chromosome maintenance |
| Cdh1/FZR1 | = | cell division cycle 20 homolog 1/fizzy-related 1 |
References
- Llopis A, et al. Cell Cycle 2012; 11; this issue http://dx.doi.org/10.4161/cc.21917; PMID: 22935704
- Kittipatarin C, et al. Cell Cycle 2006; 5:907 - 12; http://dx.doi.org/10.4161/cc.5.9.2693; PMID: 16628013
- Uchida S, et al. Cancer Res 2009; 69:6438 - 44; http://dx.doi.org/10.1158/0008-5472.CAN-09-0869; PMID: 19638579
- Gutierrez GJ, et al. J Biol Chem 2010; 285:14217 - 28; http://dx.doi.org/10.1074/jbc.M110.121848; PMID: 20220133
- Chandrasekaran S, et al. Mol Cell Biol 2011; 31:4405 - 16; http://dx.doi.org/10.1128/MCB.06163-11; PMID: 21930785
- Miotto B, et al. Mol Cell 2011; 44:62 - 71; http://dx.doi.org/10.1016/j.molcel.2011.06.021; PMID: 21856198
- Barkley LR, et al. Mol Biol Cell 2012; 23:1943 - 54; http://dx.doi.org/10.1091/mbc.E11-10-0829; PMID: 22456510
- Lee K, et al. Cell Cycle 2008; 7:216 - 21; http://dx.doi.org/10.4161/cc.7.2.5155; PMID: 18256527
- Gutierrez GJ, et al. Nat Cell Biol 2010; 12:686 - 95; http://dx.doi.org/10.1038/ncb2071; PMID: 20581839