Abstract
The incidence of esophageal adenocarcinoma (EAC) is rapidly rising in the western world and accounts for 2% of all cancer-related deaths. The precursor lesion for EAC is Barrett esophagus (BE), which is strongly associated with gastresophageal reflux disease. A major limitation to the study of EAC has been the absence of tractable and genetically modifiable preclinical models of BE. A mouse model of BE and EAC that resembles human disease could provide novel insights into the origins and molecular pathogenesis of BE. In addition, validated animal models could help stratify BE patients given the limited predictive power of current standard endoscopic measures and clinical assessment. Here, we review the findings from recently developed mouse models of BE and EAC and their impact on clinical decision making, surveillance programs and therapeutic options. The data, taken together, suggest potential origins of BE from the gastric cardia, a role of bile acid and hypergatrinemia for carcinogenesis, a growing importance for columnar-like epithelium and a critical role for Notch signaling.
Introduction
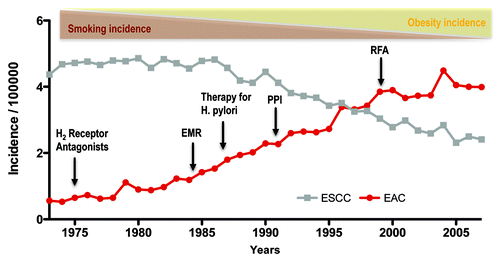
There has been a considerable amount of controversy and debate on the topic of Barrett esophagus (BE) regarding clinical surveillance strategies, useful biomarkers and the origin of this disease.Citation1,Citation2 Here, we aim to summarize the current knowledge and discuss recent insights derived from a novel genetic mouse model of inflammation-dependent esophageal metaplasia.Citation3 Barrett esophagus (BE) is a premalignant condition of the distal esophagus defined by replacement of the normal squamous epithelium in the esophagus by columnar epithelium, typically with intestinal metaplasia.Citation4 Barrett esophagus represents the initial step in the histopathologic progression that can lead to low-grade dysplasia, high-grade dysplasia and esophageal adenocarcinoma (EAC). The incidence of EAC, which comprises both esophageal and gastresophageal junction cancers, has increased at a rate of 4–10% annually in Western countries, an increase greater than that for any other cancer.Citation5 This increase has occurred despite the introduction of powerful acid-suppressant medications (proton pump inhibitors or PPIs) to treat GERD, and has been associated with a rapid decline in the prevalence of Helicobacter pylori in the United States ().
Figure 1. The increasing incidence of esophageal adenocarcinoma (EAC) between 1975 and 2005 and associated factors. During this period of time, esophageal squamous cell carcinoma (ESCC) has declined in incidence, as has its major risk factor, tobacco use. EAC incidence has risen concomitantly with obesity, which is one risk factor for the disease. Despite the advent of medical therapies such as acid inhibition through H2 receptor antagonists and proton pump inhibitors (PPIs), and the treatment of H pylori, EAC has continued to rise in incidence. Techniques such as endoscopic mucosal resection (EMR) and RFA (radiofrequency ablation) have also failed to stem the rise in EAC incidence. Data from the Surveillance Epidemiology and End Results (SEER) database of the National Cancer Institute.
Risk factors for esophageal adenocarcinoma include white race, older age, male sex, gastro-esophageal reflux disease (GERD), smoking and obesity. BE is the precursor of esophageal adenocarcinoma and the most important risk factor. In developed countries, substantial resources are expended on surveillance of BE, with the goal of early detection of high-grade dysplasia or esophageal adenocarcinoma. However, recent studies demonstrating rates of progression lower than previously reported raise questions regarding the cost effectiveness and overall utility of endoscopic surveillance as currently employed. The rate of progression from non-dysplastic BE to EAC had been previously accepted as ~0.5% per year.Citation6 However, two recent, large population-based studies reported rates of progression from non-dysplastic BE to cancer of 0.10–0.13% per year. These figures correspond with a relative risk of EAC of ~11 for a patient with BE, a substantial drop from the 30- to 40-fold increased risk estimated in early reports.Citation7,Citation8 Nevertheless, surveillance strategies could be improved by the identification of additional risk factors, or biomarkers could be found to target a higher risk population. Research on validated preclinical models could assist in this search by providing new insight into the biology of inflammation-driven metaplasia, and the factors that lead to the development of BE and EAC. Mouse models of Barrett-like metaplasia have provided further clarification of the mechanisms by which bile acid and inflammation induce metaplasia, the molecular pathways that drive proliferation and expansion of the columnar epithelial lineage and the progenitor cells that represent the origins of BE and EAC. Greater knowledge and understanding of the cell of origin of BE, and the molecular pathways that promote and trigger carcinogenesis, are likely to be critical in stratifying BE patients and identifying the subset that is at greatest risk for progression to EAC.Citation9
Modeling Barrett Esophagus in the Mouse
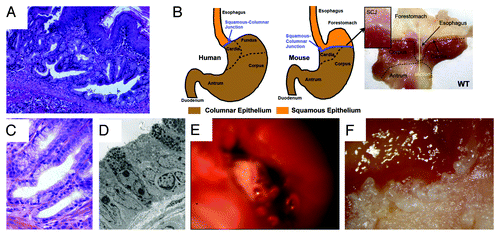
Despite the fact that EAC is the most rapidly increasing cancer in the western world, and BE as broadly defined is the predominant precursor lesion for EAC, there have been a relatively small number of basic research studies or preclinical models that have been able to address important questions in the field, or that have been useful to clinicians managing these patients. A major challenge in the field has been to identify suitable preclinical models whereby esophageal metaplasia resembling Barrett esophagus precedes the development of neoplasia. Until recently, the best animal model used to study BE has been a rat surgical model, in which an esophagojejunostomy is used to induce gastroduodenal reflux.Citation10 However, this is a model that has been difficult to reproduce in mice. We recently generated a novel transgenic mouse model for BE and EAC that has provided fundamental insights into the early pathogenesis of BE, and offers a molecular basis for an emerging paradigm shift regarding the cell of origin of BE and EAC.Citation3 We established a mouse line that carried the EBV-L2-IL-1β transgene, where IL-1β was overexpressed in the esophageal and squamous forestomach mucosa (). The mice exhibited esophagitis, and with no additional intervention, the mice progressed to BE by 12 mo and spontaneously to adenocarcinoma with older age. However, with the addition of bile acids to the drinking water (0.2% deoxycholic acid), the mice developed accelerated BE and earlier onset tumorigenesis. Furthermore, with the addition of both bile acids and nitrosamine (N-methyl-N-nitrosourea) in the drinking water, the mice developed markedly accelerated BE and tumors. The tumors could be identified in mice through a novel endoscopic technique and also through PET scanning. The L2-IL-1β mouse model of BE and EAC was validated through electron microscopy (EM) studies, which showed ultrastructural similarities between human and murine lesions through microarray profile comparing human BE to murine lesions and through microarray analysis by LCM of dysplastic lesions. Finally, similar to the notion proposed in the original paper by Dr. Norman Barrett himself,Citation11 the murine model of BE and EAC has suggested that the metaplastic lesions originate from progenitor cells in the gastric cardia,Citation11,Citation12 which, over time, appear to migrate proximally into the squamous esophagus and are strongly associated with the development of dysplasia in both human and murine BE. In particular, using Lgr5-CreERT mice, we were able to demonstrate that the metaplasia can arise from an Lgr5+ cardia stem cell. Lgr5 and other progenitor markers are absent from normal murine and human squamous mucosa but are markedly upregulated in Barrett esophagus in both the mouse and human models. We also observed upregulation of other progenitor markers, including K19, CCK2R and Dclk1.
Figure 2. The L2-IL-1β mouse model of Barrett esophagus (BE) and esophageal adenocarcinoma (EAC). (A) Histological appearance of BE lesions, consisting of columnar mucosa and mucus-producing cells at the squamocolumnar junction (SCJ) in the L2-IL-1β mouse. (B) Anatomical differences between human and mouse stomach. The SCJ is distal to the esophago-gastric junction and comprises the border between the squamous forestomach and the columnar-lined stomach in the mouse. In the figure on the far right, the mouse stomach has been opened along the greater curvature to demonstrate the location of the SCJ. The vertical line demonstrates the location of the sagittal section seen in . (with kind permission from Cancer Cell / Elsevier, Quante, Bhagat et al. 2012) (C) Human Barrett esophagus, containing columnar cells and goblet cells. (D) Electron microscopic appearance of mouse BE lesions demonstrating columnar appearance, mucin granules, and surface microvilli. (E) Tumor at the SCJ seen during upper endoscopy of an L2-IL-1β mouse. (F) Macroscopic appearance of tumor at the SCJ in the L2-IL-1β mouse.
Nevertheless, there are some limitations to both the mouse and rat models, in that the presence of the squamous forestomach distinguishes the upper GI tract of rodents from that in the human. However, the squamocolumnar junction (SCJ) of the mouse still comprises the junction of the esophagus and gastric cardia, as in the human. In our mouse model, the main pathologic changes occur at the SCJ adjacent to the esophagus, and the tumors consistently arise in the distal esophagus. However, these anatomical differences between mice and humans do need to be taken into consideration when extrapolating findings from the mouse model to human patients. Another mouse model of BE was recently described by Frank McKeon’s groupCitation13 in p63-deficient neonatal mice (since p63−/− mice die several weeks after birth), which exhibit a Barrett-like lesion characterized by “well-developed columnar epithelium” with “positive staining with Alcian blue and periodic acid-Schiff (PAS),” similar in many respects to the metaplasia in our model. However, this particular model was a developmental study; it was not physiologic, with no modulation by bile acids or inflammatory stimuli, and there was no progression to dysplasia. Therefore, the study could only partly address the origins of metaplasia in the adult animal. However, taken together, the two models point to the origin of metaplasia from cells present at the squamous-columnar junction, and they suggest that at least some BE does not arise from the squamous epithelium. These insights from mouse models into the origin of BE are clearly more than of minor academic interest, since the findings are likely to shape future screening and therapeutic approaches, which may include targeting stem cells of BE in the gastric cardia, as a recent review by Frank McKeon’s group also suggests.Citation2
Importance of Inflammation and Bile Acids
BE and EAC are strongly associated with gastresophageal reflux disease (GERD), and the development of BE has been primarily attributed to chronic GERD. Approximately 40% of US adults report heartburn symptoms at least monthly; one-third of these patients have esophagitis, and 10% have BE. GERD is thought to induce chronic esophageal inflammation, known as reflux esophagitis, a risk factor for both BE and EAC. Thus, chronic inflammation is thought to be one of the primary mediators of both BE and EAC. The link between inflammation and cancer is now well-established, and while the mechanism is still not completely understood, it is clear that inflammatory cells release a variety of mediators that establish an inflammatory and pro-carcinogenic microenvironment.Citation14,Citation15 Chronic inflammation, likely induced by reflux of both gastric acid and bile acids, promotes the proliferation and survival of malignant cells by subverting innate and adaptive immune responses and altering responses to hormones and chemotherapeutic agents. In particular, elevated levels of IL-6, a downstream target of NFκB, have been identified as an important mediator of tumorigenesis in mouse models of colon and liver cancer, and are potentially responsible for gender disparities in cancer.Citation16,Citation17 A number of clinical studies have suggested that genetic polymorphisms in pro-inflammatory cytokines are associated with cancer.Citation18,Citation19 IL-1β, along with IL-8, has been shown to be consistently upregulated in BE, particularly at the SCJ,Citation20 pointing to the relevance of our mouse model. In the L2-IL-1β mouse model, high levels of IL-1b induce chronic inflammation and spontaneous carcinogenesis in an IL-6-dependent manner, indicating that these two cytokines (IL-1β and IL-6), which are often upregulated in preneoplastic tissues and the tumor microenvironment, are important factors in the development of BE and EAC in humans as well.Citation3 Not surprisingly, an abrogation of esophageal inflammation is considered an important treatment goal in patients with esophagitis and BE.
Nevertheless, esophagitis and Barrett metaplasia arise in the setting of reflux of both gastric contents (acid) and duodenal contents (bile acids) into the esophagus. The role of bile reflux has been established from surgical models in rats, where esophagojejunostomy and esophagogastroduodenostomy results in a mixed bile and gastric refluxate that leads to intestinal metaplasia, with many similarities to human BE, including early induction of Cdx1 and Cdx2 expression and the development of a columnar-lined epithelium containing intestinal mucin-secreting goblet cells,Citation21-Citation25 and esophageal adenocarcinoma.Citation26,Citation27 Bile acids, particularly unconjugated bile acids such as deoxycholate that induce DNA damage, are one component of gastroduodenal reflux that has been strongly linked to the development of BE, as well as to other cancers (e.g., colorectal cancer).Citation25,Citation28,Citation29 Bile acids have been shown to activate both survival and anti-apoptotic pathways in cultured cells, and unconjugated bile acids are increased in the refluxate in patients on a high-fat diet and in patients taking proton pump inhibitors.Citation30 The exposure to bile acids has been reported to lead to promoter demethylation and activation of the Cdx2 promoter in esophageal cells.Citation25,Citation31 The L2-IL-1b mouse model demonstrates that bile acids accelerate the development of BE and EAC, but only in the setting of an existing inflammatory condition in the esophagus (e.g., in L2-IL-1β mice), analogous to reflux-induced esophagitis in humans. Interestingly, a number of recent studies have now shown that feeding 0.2% unconjugated bile acids to mice induces mild esophagitis and metaplasia over time,Citation32 although we were not able to reproduce this phenotype in our WT mice.Citation3 In the inflammatory L2-IL-1β mouse, bile acids induced an early amplification of a TFF2-expressing columnar-type metaplasia in the distal esophagus, and increased Cdx2 gene expression was found in inflamed esophageal mucosa before other intestinal markers were increased, similar to earlier studies that suggested that columnar cell differentiation is also an early event in the formation of BE.Citation33 Importantly, in the L2-IL-1β mouse model, chronic exposure to unconjugated bile acids rather than gastric acid contributed heavily to the development of metaplasia and initiation of carcinogenesis. In summary, the data suggest that the combination of inflammation and bile acid exposure is critical to the induction of intestinal metaplasia of the esophagus and plays an important role during esophageal carcinogenesis. Further studies are needed to determine if bile acids or gastric acid are more important for induction of chronic inflammation and cancer promotion, since clarification of this issue would likely guide future therapeutic and chemoprevention strategies in patients with BE.
Proton Pump Inhibitor Therapy and Hypergastrinemia in BE
Omeprazole was introduced into the United States in 1989, and the use of proton pump inhibitors (PPIs) for the treatment of GERD has since grown tremendously over the past 20 years (currently 5% of the U.S. population takes PPIs). PPIs have numerous beneficial effects, including symptom control, reduction of inflammation and healing of esophageal ulceration. While gastric acid is believed to be an important etiological factor in the pathogenesis of BE, there has been a dramatic increase in the incidence of BE and EAC despite the widespread use of PPIs (). Currently PPIs have become the mainstay of therapy in BE; between 95–98% of patients with BE are prescribed these drugs.Citation34,Citation35 However, roughly 30–50% of BE patients diagnosed in the clinical setting do not have reflux,Citation36,Citation37 and population-based studies also suggest that roughly one-half of all BE patients do not have GERDCitation38 (Rex, 2003 #27637). Use of PPIs have been shown to normalize esophageal pH, lower cell proliferation and promote cell differentiation,Citation39 but long-term PPI treatment does not reduce the length of the BE segment.Citation40 Studies on the beneficial effects of PPIs have been conflicting. A few studies suggest that there may be an inverse association between the use of PPI and risk of EAC or HGD in individuals with BE.Citation41,Citation42 In addition, in one study, a delay in using a PPI for 2 years or more after diagnosis with BE was associated with an increased risk of developing LGD and an increased risk of developing HGD or EAC.Citation43 However, other studies have suggested that treatment with PPI or other acid-suppressing drugs was associated with an increased risk of EAC.Citation44,Citation45 While PPIs may reduce the rate of progression in some patients with BE, they also induce significant hypergastrinemia in a subset of patients by reducing acid-inhibitory feedback pathways. Gastrin plays an important role in the regulation of epithelial proliferation and differentiation of the gastrointestinal tract through binding of gastrin peptides to the CCK-2/gastrin receptor. An important study by Haigh et al. showed that the CCK-2 receptor is expressed at low levels in normal esophageal squamous mucosa and markedly increased in reflux esophagitis, Barrett metaplasia and the majority of EACs with a functional importance to the development of BE.Citation46 In theory, hypergastrinemia secondary to PPI treatment could lead to increased Barrett proliferation and amplification of metaplastic clones within the esophagus. While epidemiologic studies have provided conflicting data on the effect of PPIs on EAC risk, none of these studies stratified patients on the basis of serum gastrin levels. While the role of PPIs in the development of BE has been controversial, elevated serum gastrin was associated with a significantly increased odds of high-grade dysplasia or adenocarcinomaCitation47 and correlated with proliferation (Green DA, et al., 2011). A number of highly specific gastrin/CCK2R antagonists have been developed in recent years, but none have yet been investigated clinically in BE. Our group has previously shown that a specific and active CCK2R antagonist, YF476, can reduce proliferation and inhibit Helicobacter-dependent gastric neoplasia.Citation48 In addition, we showed that a closely related molecule, YM022, was able to block colonic proliferation, expansion of colonic stem/progenitor cells and inhibit AOM-dependent colonic neoplasia.Citation49 Thus, one can postulate that hypergastrinemia could play a role in BE progression, although further studies are needed to examine this notion in a preclinical model. Additional prospective studies are needed in human BE patients to confirm this association between serum gastrin levels and progression to dysplasia. Most GI specialty societies recommend PPIs for the management of reflux symptoms in the setting of Barrett esophagus, yet stop short of advocating PPIs solely for chemoprevention in asymptomatic BE patients,Citation50,Citation51 as two recent reviews have confirmed that the available data are insufficient to draw any definite conclusions or support a chemopreventive effect.Citation52,Citation53
Cell of Origin of Barrett Esophagus
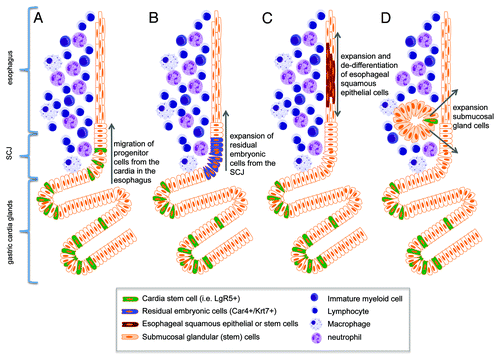
The incidence of BE and EAC are increasing rapidly, but the reasons for this trend as well as the source of the neoplastic lesions remain poorly understood.Citation54 Given the stable nature of BE—which most often does not progress to cancer for decades, if at all—the development of BE probably reflects the expansion of a novel or altered stem cell population that is long-lived, distinct from normal squamous cells and can acquire the genetic changes necessary for malignant transformation. Thus, one of the major questions in the field of BE/EAC, and one that has been debated for decades, is the cell of origin for BE.Citation55 It seems reasonable to assume that EAC arises from the same progenitors that give rise to BE, making the identification of the progenitors responsible for BE a high priority, but there has been little consensus on which set of stem cells represents the most likely candidates. One possibility is that the development of BE involves the reprogramming of normal squamous stem cells into intestinal epithelial stem cells. However, another possibility is that BE develops from a novel stem cell recruited into the squamous esophagus, such as a gastric cardia stem cell, a submucosal stem cell (squamous gland duct cells)Citation70 or even a circulating (e.g., bone marrow-derived) stem cell.Citation22,Citation56 The finding that Barrett heterogeneity results from multiple independent stem/progenitor clones, would tend to argue against a transdifferentiation model.Citation57 This question has been difficult to address in part due to the absence of useful in vitro or in vivo models, such as a tractable animal model of BE that would allow definitive lineage tracing ().
Figure 3. Schematic diagram of hypothesized cells of origin for BE. (A) Lgr5+ stem cells migrate proximally from the cardia into the esophagus in response to proinflammatory stimuli (Quante, Bhagat et al. 2012). (B) Car4+/Krt7+ residual embryonic cells expand proximally from the SCJ into the esophagus (Wang, Ouyang et al. 2011). (C) Esophageal squamous epithelial cells de-differentiate and expand to form squamous metaplasia. (D) Stem cells of the submucosal esophageal glands expand and give rise to BE (Leedham SJ et al., 2008).
In most tissues, stem cells within a niche are assumed to be present in relatively small numbers. These cells are thought to remain largely quiescent or undergo division at a relatively slow rate,Citation58 such that they are generally negative for most proliferation markers. The very proximal stomach, or gastric cardia, while difficult to define anatomically, represents a zone of 4–5 glands units just below the SCJ that shows a paucity of parietal and zymogenic cells and resembles more closely the gastric antrum or the intestine/colon. The absence of parietal cells and the abundance of progenitor markers in this area suggests that the gastric cardia is a transitional epithelium. Stem cells in the cardia have not been intensively investigated, but stem cells have been shown to exist at the base of the antral glands and in the crypts of the small intestine and colon.Citation59 In the gastric corpus, stem cells reside in the gastric isthmus, where they give rise to two sets of progenitors that undergo bidirectional migration.Citation60 Recently, a single marker, Lgr5/GPR49, a leucine-rich orphan G protein-coupled receptor, was shown to specifically label stem cells in the small intestinal and colon crypts, the so-called crypt based columnar (CBC) cells.Citation61,Citation62 Activation of Wnt signaling in Lgr5+ cells was able to induce intestinal neoplasia.Citation62 Furthermore, isolation of single Lgr5+ cells showed that at least some of these cells were able to produce intestinal glands in vitro.Citation63 Lgr5 has been identified as a valid marker for stem cells at the base of the antral glands and the gastric cardia, but is reportedly not expressed in the gastric corpus.Citation64
In the L2-IL-1β mouse model, we demonstrated that Lgr5-labeled cells functioned as stem cells in the gastric cardia (Quante M et al., 2011), as previously shown.Citation64 Interestingly, in lineage tracing experiments in the IL-1β mouse model, we observed labeled BE metaplasia, indicating that Lgr5+ cells within the cardia may function as cells of origin for BE and dysplasia. Furthermore, in mice and humans we observed increased Krt19 expression, which is a known marker for surface mucus and gastric progenitor cells,Citation65,Citation66 as well as increased expression of TFF2, which labels an early progenitor cell in the secretory lineage in the stomach.Citation60 Lgr5, Krt19 and TFF2 are not normally expressed in the esophageal squamous epithelium. These data suggest that BE (and EAC) most likely do not arise from differentiated cells of the squamous epithelium of the esophagus, but rather migrate up from the stomach to form a columnar-lined epithelium with intestinal differentiation. Given the fact that de-differentiation of any differentiated cell has not been achieved in normal human or mouse tissue, except in the case of reprogramming using exogenous transcription factors,Citation67 we argue that the stem cell population within the gastric cardia, which is by origin and definition primed to generate columnar cells, is the cell of origin for columnar-lined esophagus (CLE) and intestinal metaplasia in BE. This notion is supported by a recent study that also suggests that BE arises from cells at the SCJ; in this study, p63-knockout mice were shown to develop a Barrett-like metaplasia in the squamous forestomach, and retrograde growth of a population of Car4-expressing cells located at the SCJ was proposed as the origin of the metaplasia, although no formal lineage tracing was performed. The authors similarly concluded that the metaplasia did not originate from transdifferentiation of squamous epithelial cells, but instead appeared to be arising from cells at the cardia, in this case, remnants from embryonic development.Citation13
The issue of the cell of origin for BE and EAC is closely related with the questions of clonality and of clonal malignant progression of BE to EAC. Clonal progression of BE metaplasia has been previously studied using flow-purified analysis of a series of whole biopsies and assessment of the mutational profile of relevant genes, including CDKN2A (P16) and TP53.Citation68,Citation69 Using these methods, it has been demonstrated that an entire Barrett segment can be clonally derived, with rapid spread of mutations through the epithelium. However, a separate analysis at a crypt-by-crypt level showed that BE metaplasia was genetically heterogeneous, containing many apparently independent clones.Citation70 Analysis for deficiency of cytochrome c oxidase (CCO; a mitochondrially encoded enzyme) allows for the clear identification of a clonal lineage in human tissue, since loss of CCO is typically associated with mtDNA mutations. In this study, patches of CCO-deficient cells that share mtDNA mutations revealed clonal expansion of independent stem cell populations. Adjacent metaplastic BE glands were clearly derived from distinct clones,Citation71 indicating the presence of multiple stem cells, which would be consistent with migrating stem cell populations from the cardia (). The finding by the same group that squamous and columnar cells rarely originate from the same population of stem cells was an intriguing observation that requires further study.
Other putative progenitor markers have been reported to be upregulated in Barrett esophagus. Doublecortin and CaM Kinase-Like-1 (Dclk1 or DCAMKL1) is a microtubule-associated-kinase that has been postulated to be expressed in gut epithelial progenitors.Citation72 In our L2-IL-1β mouse model and in human BE, we found an accumulation of Dclk1+ cells, and this was confirmed in a study by another group, which reported a progressive increase of Dclk1 expression in BE from dysplasia to EAC.Citation73 Dclk1+ cells are highly abundant in the gastric cardia, particularly in areas immediately adjacent to the SCJ. However, formal lineage tracing for Dclk1 has not been reported, and the function of these cells remains to be defined.
In summary, data from the L2-IL-1β mouse model suggest that a progenitor/stem cell population within the gastric cardia increases during the development of BE. Lgr5+ cells, in particular, possess the ability to differentiate into a secretory cell lineage and migrate into the esophagus to give rise to Barrett metaplasia and dysplasia. These findings have significant potential to impact prevention and surveillance strategies of BE patients. If findings from the mouse model are confirmed in humans, then greater attention will need to be given during endoscopic evaluations to the gastric cardia, and the carditis that is associated with reflux esophagitis will likely be given much greater scrutiny. Moreover, the current findings from the mouse model, which suggest a lineage relationship between the gastric cardia and BE metaplasia, may enable us to broaden the definition of BE histologically. Indeed, cardiac mucosa seems to be an abnormal metaplastic epithelium that expands into the esophagus in response to injury or inflammatory signals. If a histologic rather than anatomic definition were to be used for junctional tumors, it seems apparent that the majority of adenocarcinomas presently classified as “esophagogastric” or “gastric cardiac” in western countries are indeed identical to the adenocarcinoma of the esophagus.
Notch Signaling in Barrett Esophagus
The importance of classical intestinal (goblet cell) metaplasia has been much debated by pathologists in the field,Citation74 and the recent discussions regarding the US definition of BE was largely in response to the observations by a few pathologists regarding the “well-defined risk of neoplasia in patients with esophageal columnar metaplasia but without goblet cells.”Citation74 While BE with prominent goblet cell metaplasia has been better studied than patients with columnar lined esophagus (CLE) alone or CLE with rare goblet cells, data from the L2-IL-1β mouse model argue that nongoblet cell metaplasia might be a greater risk factor for cancer. Indeed, the development of IM in the mouse occurs in a low Notch signaling environment, while maintenance of the CLE phenotype and progression to dysplasia occurs in a high Notch signaling environment. As has been shown for the intestine, differentiation of the migrating cardia stem/progenitor cells into goblet cells is associated with inhibition of Notch signaling. A recent study from the NetherlandsCitation75 demonstrated in a rat BE model that Notch inhibition resulted in both a reduction in cellular proliferation and increased goblet cell differentiation, again suggesting that the two phenomena are inversely correlated, and that pharmacological induction of goblet cell metaplasia could be a viable strategy for prevention of EAC in BE patients. We demonstrated a similar effect in our mouse model—an increase in intestinal gene expression and decreased proliferation with Notch inhibitors. Indeed, we provide important evidence pointing to the utility of Notch1 expression and activated Notch signaling as a biomarker for the activated cardia stem/progenitors that migrate into the esophageal squamous epithelium in the setting of chronic inflammation. The Notch pathway regulates binary cell-lineage specification and differentiation through cell-cell communication and is critically important during early development, suggesting, as a conclusion of the results from the rat and mouse models, that proliferation and progression of BE is promoted by Notch signaling, and that Notch inhibition can induce goblet cell differentiation and prevent transformation of BE metaplasia to EAC.
Definition of Barrett Esophagus
Early studies from the 1950s that first described Barrett esophagus initially mentioned the proximal stomach, or cardia, as a potential source of Barrett esophagus.Citation76 Barrett himself described the ulcerated columnar lining of his patient as “histologically gastric in type”Citation77 and assumed that BE resulted from proximal migration of stomach epithelium just below the GE junction. However, in the 1980s, the prevailing hypothesis formed that reflux-induced damage to the esophageal squamous epithelium leads to reprogramming or transdifferentiation of multipotent stem cells in the basal layer of the squamous epithelium. In addition, the notion developed that intestinal (goblet cell) metaplasia, the most common type of BE, was the epithelial cell type most associated with cancer development. Consequently, researchers in the US chose to define BE by the presence of intestinal metaplasia (IM), and a number of gastroenterology organizations (e.g., the ACG) adopted this narrow definition.
However, prior work points to a more diverse spectrum of esophageal metaplasia that can occur in association with chronic reflux esophagitis. In 1976, Paull et al. described three types of columnar epithelia in the esophagus: a cardia-type, a gastric fundic-type and IM, which was also referred to as specialized columnar epithelium.Citation12 As noted above, for the last several decades, specialized columnar epithelium or IM was considered to be mandatory for the diagnosis of BE in the United States. However, gastric cardia-type epithelium or CLE also represents an abnormal metaplastic epithelium that develops as a consequence of GERD. It is noteworthy that BE and EAC are, in the vast majority of cases, located just above the GEJ and, thus, always arise contiguous with the gastric cardia. Indeed, junctional or cardia cancers appear biologically identical to EAC and are difficult to distinguish at a clinical and pathological level. The notion of a gastric cardiac origin for BE/EAC has been supported by histopathological studies of patients following surgical esophagogastrostomy, where cardiac type mucosa regenerated at the anastomosis, followed later by Barrett-like mucosa in association with reflux, suggesting that BE was derived from cardiac and/or fundic mucosa.Citation78 Indeed, gastric cardiac mucosa has been found in association with esophageal cancer in 100% of cases in a large series of 131 consecutive patients.Citation79 The trigger for activating progenitors in the gastric cardia may be a disorder called carditis, chronic inflammation of the proximal stomach associated with GERD. In GERD patients, “carditis” is highly localized to a region immediately adjacent to the SCJ, and Lembo et al. suggested that cytokines upregulated in esophagitis may interact with the cardia columnar cells, leading to histologic changes.Citation80 In many cases of BE, however, both goblet cell and gastric-type metaplasia are present and coexist. Nevertheless, data from the mouse model and from clinical studies raise the interesting possibility that non-goblet cell metaplasia might represent more of a risk for EAC than classic goblet cell metaplasia. European societies such as the BSG include cardia-type epithelium for the diagnosis of BE, based on a belief that cardia metaplasia is at risk for progression, and a recent AGA Institute technical review now concurs that any epithelium that can progress such as columnar epithelium should be considered BE.Citation50,Citation51
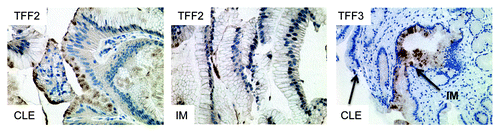
In the L2-IL-1β mouse model, we only rarely observed true goblet cells within the secretory cell lineage at the SCJ in the distal mouse esophagus; nevertheless, 20–30% of the mice developed dysplasia and cancer, indicating that the cancer most likely does not arise from goblet cells, but instead originates from the heterogeneous population of columnar cells within Barrett metaplasia. While IM is present in BE in the vast majority of cases, it is not present in every case, as noted by GatenbyCitation81 and Takubo.Citation82 Moreover, columnar-like epithelium seems to precede the development of IM, and progression to IM is associated with the extent of metaplasia and the duration of the disease.Citation81 We were able to confirm these earlier findings in a review of published studies, which showed no significant difference (p = 0.88) between CLE and IM as adjacent tissues associated with EAC ().Citation3 Interestingly, in the above study, investigators observed that the rate of progression to the next more differentiated step (IM, LGD, HGD and EAC) was increased in CLE compared with IM, supporting our concept of stepwise progression, where the cardia progenitor first differentiates into CLE and only later differentiates into IM or EAC. In this regard it seems of interest for risk evaluation that TFF2 might mark a progenitor cell in the columnar-lined epithelium that finally differentiates into TFF3+ IM in mice and in humans (). In addition, we have performed our own evaluation on a German cohort of 31 resection samples with EAC.Citation83 In histopathological analysis of 31 single 2–4 cm specimens harboring esophageal mucosa and EAC, we found that in six cases, EAC was adjacent to IM; in 12 cases, EAC was adjacent to cardia-type tissue; and in 28 cases, the EAC was adjacent to CLE (). Thus, there is strong evidence that EAC can arise in CLE and does not require IM as a direct precursor lesion. These histopathological observations, along with the results from the novel L2-IL-1β BE mouse model, will hopefully inspire further prospective studies that will determine the cancer risk of “goblet cell poor” vs. “goblet cell rich” metaplasia.
Figure 4. (A) Pooled analysis of studies that characterized the mucosa adjacent to esophageal adenocarcinoma in patients as intestinal metaplasia (IM, in %) or columnar-lined esophagus (CLE, %). In these nine studies comprising 1328 patients, there was no significant difference between the presence of IM or CLE as the tissue adjacent to EAC (p = 0.88). (with kind permission from Cancer Cell / Elsevier, Quante, Bhagat et al. 2012) (B) Histopathologic evaluation of the mucosa adjacent to EAC in 31 specimens in a German cohort. CLE was most commonly seen, compared with IM, cardia-type tissue and squamous mucosa (SQ) (*p < 0.05).
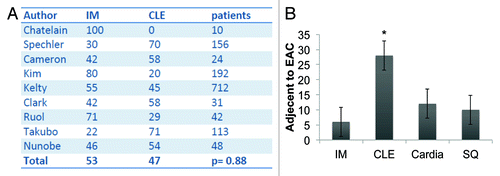
Figure 5. Expression of TFF2 and TFF3 in human columnar-lined epithelium (CLE) and intestinal metaplasia (IM). TFF2 marks an early precursor cell in the columnar lineage; staining is seen in CLE, but (B) with the development of IM, TFF2 staining decreases. (C) Conversely, TFF3 staining is not seen in CLE but increases with the development of IM.
Therapeutic Conclusions
Despite numerous advances over the years in the field of Barrett esophagus and esophageal adenocarcinoma, we continue to lack a clear understanding of the origins of BE and its progression to cancer. Studying the origins of BE in humans has been particularly challenging given the low population prevalence, lack of identified precursors and its apparent rapid development and subsequent static nature. All of our knowledge related to progression has relied on the presence of intestinal metaplasia. However, as clearly demonstrated by recent data on rates of progression,Citation7,Citation8 IM is a very crude marker of EAC risk. Basing broad clinical strategies solely on the presence or absence of IM has led to cost-ineffective management and little to no appreciable impact on mortality. Therefore, arguably the most important initial goal from a clinical standpoint is the identification of truly “at-risk” tissue for the development of EAC. The L2-IL-1β mouse model now provides the opportunity to employ a rational bench-to-bedside approach to address these clinically important gaps in our knowledge.
For example, Notch signaling in the mouse is a key driver of the development of CLE and subsequent progression to cancer. Notch inhibition results in decreased proliferation, conversion to a goblet cell-rich phenotype and reduced progression to EAC.Citation3,Citation75 Therefore, does increased Notch signaling in humans result in goblet-cell poor columnar-lined epithelium that is actually at high risk for progression? Could the application of topical Notch inhibitors transform a high-risk BE patient into a goblet cell-rich, low-risk patient?
Once we identify patients who are at high risk for progression, the subsequent focus would be to devise patient-specific targeted strategies and interventions to maximize cost-effective EAC prevention. Experiments in the L2-IL-1β mouse show that expansion of Cck2r+ progenitor cell lineages occurs prior to established BE,Citation3 suggesting that gastrin could potentially have a direct influence on the development of BE. Gastrin also has numerous effects in BE that could promote progression to EAC,Citation46,Citation84,Citation85 and there is an association between hypergastrinemia and advanced neoplasia in patients with BE.Citation47 Cck2r inhibition in the L2-IL-1β mouse markedly reduces proliferation, and there is now an ongoing study evaluating the effects of a Cck2r antagonist in human patients with BE (NCT01298999). Do patients with high circulating gastrin levels have an increased risk of progression? And can Cck2r inhibition effectively reduce the risk of EAC, particularly in hypergastrinemic patients?
Endoscopic ablative techniques have come to the forefront in the management of patients who develop high-grade dysplasia and early adenocarcinoma. While early results have demonstrated high rates of BE eradication with ablation,Citation86,Citation87 longer-term follow-up data are emerging to indicate that recurrence of BE as well as dysplasia and EAC occur at a clinically significant rate.Citation88,Citation89 Recurrence of BE after ablation, however, may serve as a good human model to study the origins of BE and to validate findings from the L2-IL-1β mouse model. These studies could, in turn, lead to the development of adjuvant treatments to improve durability of treatment. For example, can therapeutic targeting of intestinal stem cells after ablation reduce the risk of recurrence?
As we apply lessons learned from the mouse model to humans, we will begin to understand the origins of Barrett esophagus, its progression to cancer and factors that influence these processes. Hopefully, this will lead to the accurate identification of high-risk patient subgroups as well as the development of risk-stratified clinical algorithms that include patient-specific and biologically based chemopreventive strategies.
References
- Kahrilas PJ. The problems with surveillance of Barrett’s esophagus. N Engl J Med 2011; 365:1437 - 8; http://dx.doi.org/10.1056/NEJMe1108435; PMID: 21995392
- Xian W, Ho KY, Crum CP, McKeon F. Cellular origin of Barrett’s esophagus: controversy and therapeutic implications. Gastroenterology 2012; 142:1424 - 30; http://dx.doi.org/10.1053/j.gastro.2012.04.028; PMID: 22537611
- Quante M, Bhagat G, Abrams JA, Marache F, Good P, Lee MD, et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell 2012; 21:36 - 51; http://dx.doi.org/10.1016/j.ccr.2011.12.004; PMID: 22264787
- Spechler SJ, Fitzgerald RC, Prasad GA, Wang KK. History, molecular mechanisms, and endoscopic treatment of Barrett’s esophagus. Gastroenterology 2010; 138:854 - 69; http://dx.doi.org/10.1053/j.gastro.2010.01.002; PMID: 20080098
- Everhart JE, Ruhl CE. Burden of digestive diseases in the United States part I: overall and upper gastrointestinal diseases. Gastroenterology 2009; 136:376 - 86; http://dx.doi.org/10.1053/j.gastro.2008.12.015; PMID: 19124023
- Shaheen NJ, Crosby MA, Bozymski EM, Sandler RS. Is there publication bias in the reporting of cancer risk in Barrett’s esophagus?. Gastroenterology 2000; 119:333 - 8; http://dx.doi.org/10.1053/gast.2000.9302; PMID: 10930368
- Hvid-Jensen F, Pedersen L, Drewes AM, Sørensen HT, Funch-Jensen P. Incidence of adenocarcinoma among patients with Barrett’s esophagus. N Engl J Med 2011; 365:1375 - 83; http://dx.doi.org/10.1056/NEJMoa1103042; PMID: 21995385
- Bhat S, Coleman HG, Yousef F, Johnston BT, McManus DT, Gavin AT, et al. Risk of malignant progression in Barrett’s esophagus patients: results from a large population-based study. J Natl Cancer Inst 2011; 103:1049 - 57; http://dx.doi.org/10.1093/jnci/djr203; PMID: 21680910
- Moyes LH, Going JJ. Still waiting for predictive biomarkers in Barrett’s oesophagus. J Clin Pathol 2011; 64:742 - 50; http://dx.doi.org/10.1136/jclinpath-2011-200084; PMID: 21606229
- Fein M, Peters JH, Chandrasoma P, Ireland AP, Oberg S, Ritter MP, et al. Duodenoesophageal reflux induces esophageal adenocarcinoma without exogenous carcinogen. J Gastrointest Surg 1998; 2:260 - 8; http://dx.doi.org/10.1016/S1091-255X(98)80021-8; PMID: 9841983
- Barrett NR. Chronic peptic ulcer of the oesophagus and ‘oesophagitis’. Br J Surg 1950; 38:175 - 82; http://dx.doi.org/10.1002/bjs.18003815005; PMID: 14791960
- Paull A, Trier JS, Dalton MD, Camp RC, Loeb P, Goyal RK. The histologic spectrum of Barrett’s esophagus. N Engl J Med 1976; 295:476 - 80; http://dx.doi.org/10.1056/NEJM197608262950904; PMID: 940579
- Wang X, Ouyang H, Yamamoto Y, Kumar PA, Wei TS, Dagher R, et al. Residual embryonic cells as precursors of a Barrett’s-like metaplasia. Cell 2011; 145:1023 - 35; http://dx.doi.org/10.1016/j.cell.2011.05.026; PMID: 21703447
- Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell 2006; 124:823 - 35; http://dx.doi.org/10.1016/j.cell.2006.02.016; PMID: 16497591
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature 2008; 454:436 - 44; http://dx.doi.org/10.1038/nature07205; PMID: 18650914
- Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007; 317:121 - 4; http://dx.doi.org/10.1126/science.1140485; PMID: 17615358
- Grivennikov S, Karin M. Autocrine IL-6 signaling: a key event in tumorigenesis?. Cancer Cell 2008; 13:7 - 9; http://dx.doi.org/10.1016/j.ccr.2007.12.020; PMID: 18167335
- Bidwell J, Keen L, Gallagher G, Kimberly R, Huizinga T, McDermott MF, et al. Cytokine gene polymorphism in human disease: on-line databases. Genes Immun 1999; 1:3 - 19; http://dx.doi.org/10.1038/sj.gene.6363645; PMID: 11197303
- Howell WM, Calder PC, Grimble RF. Gene polymorphisms, inflammatory diseases and cancer. Proc Nutr Soc 2002; 61:447 - 56; http://dx.doi.org/10.1079/PNS2002186; PMID: 12691174
- Fitzgerald RC, Abdalla S, Onwuegbusi BA, Sirieix P, Saeed IT, Burnham WR, et al. Inflammatory gradient in Barrett’s oesophagus: implications for disease complications. Gut 2002; 51:316 - 22; http://dx.doi.org/10.1136/gut.51.3.316; PMID: 12171950
- Tatsuta T, Mukaisho K, Sugihara H, Miwa K, Tani T, Hattori T. Expression of Cdx2 in early GRCL of Barrett’s esophagus induced in rats by duodenal reflux. Dig Dis Sci 2005; 50:425 - 31; http://dx.doi.org/10.1007/s10620-005-2452-9; PMID: 15810620
- Sarosi G, Brown G, Jaiswal K, Feagins LA, Lee E, Crook TW, et al. Bone marrow progenitor cells contribute to esophageal regeneration and metaplasia in a rat model of Barrett’s esophagus. Dis Esophagus 2008; 21:43 - 50; PMID: 18197938
- Theisen J, Peters JH, Fein M, Hughes M, Hagen JA, Demeester SR, et al. The mutagenic potential of duodenoesophageal reflux. Ann Surg 2005; 241:63 - 8; PMID: 15621992
- Cheng P, Gong J, Wang T, Jie C, Liu GS, Zhang R. Gene expression in Barrett’s esophagus and reflux esophagitis induced by gastroduodenoesophageal reflux in rats. World J Gastroenterol 2005; 11:3277 - 80; PMID: 15929182
- Kazumori H, Ishihara S, Rumi MA, Kadowaki Y, Kinoshita Y. Bile acids directly augment caudal related homeobox gene Cdx2 expression in oesophageal keratinocytes in Barrett’s epithelium. Gut 2006; 55:16 - 25; http://dx.doi.org/10.1136/gut.2005.066209; PMID: 16118348
- Szentpáli K, Széll M, Paszt A, Wolfárd A, Dobozy A, Németh I, et al. Simultaneous adeno- and squamous cell carcinoma with different phenotypic profiles in a rat model of chronic gastroesophageal reflux. Dis Esophagus 2007; 20:305 - 10; http://dx.doi.org/10.1111/j.1442-2050.2007.00682.x; PMID: 17617879
- Buskens CJ, Hulscher JB, van Gulik TM, Ten Kate FJ, van Lanschot JJ. Histopathologic evaluation of an animal model for Barrett’s esophagus and adenocarcinoma of the distal esophagus. J Surg Res 2006; 135:337 - 44; http://dx.doi.org/10.1016/j.jss.2006.04.023; PMID: 16926029
- Kaur BS, Triadafilopoulos G. Acid- and bile-induced PGE(2) release and hyperproliferation in Barrett’s esophagus are COX-2 and PKC-epsilon dependent. Am J Physiol Gastrointest Liver Physiol 2002; 283:G327 - 34; PMID: 12121879
- Souza RF, Krishnan K, Spechler SJ. Acid, bile, and CDX: the ABCs of making Barrett’s metaplasia. Am J Physiol Gastrointest Liver Physiol 2008; 295:G211 - 8; http://dx.doi.org/10.1152/ajpgi.90250.2008; PMID: 18556417
- Theisen J, Nehra D, Citron D, Johansson J, Hagen JA, Crookes PF, et al. Suppression of gastric acid secretion in patients with gastroesophageal reflux disease results in gastric bacterial overgrowth and deconjugation of bile acids. J Gastrointest Surg 2000; 4:50 - 4; http://dx.doi.org/10.1016/S1091-255X(00)80032-3; PMID: 10631362
- Jankowski JA, Wright NA, Meltzer SJ, Triadafilopoulos G, Geboes K, Casson AG, et al. Molecular evolution of the metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am J Pathol 1999; 154:965 - 73; http://dx.doi.org/10.1016/S0002-9440(10)65346-1; PMID: 10233832
- Guy NC, Garewal H, Holubec H, Bernstein H, Payne CM, Bernstein C, et al. A novel dietary-related model of esophagitis and Barrett’s esophagus, a premalignant lesion. Nutr Cancer 2007; 59:217 - 27; http://dx.doi.org/10.1080/01635580701499529; PMID: 18001217
- Eda A, Osawa H, Satoh K, Yanaka I, Kihira K, Ishino Y, et al. Aberrant expression of CDX2 in Barrett’s epithelium and inflammatory esophageal mucosa. J Gastroenterol 2003; 38:14 - 22; http://dx.doi.org/10.1007/s005350300001; PMID: 12560917
- Wani S, Falk G, Hall M, Gaddam S, Wang A, Gupta N, Singh M, Singh V, Chuang KY, Boolchand V, Gavini H, Kuczynski J, Sud P, Reddymasu S, Bansal A, Rastogi A, Mathur SC, Young P, Cash B, Lieberman DA, Sampliner RE, Sharma P. Patients with nondysplastic Barrett's esophagus have low risks for developing dysplasia or esophageal adenocarcinoma. Clin Gastroenterol Hepatol 2011; 9:220 - 7; http://dx.doi.org/10.1016/j.cgh.2010.11.008
- Nguyen DM, Richardson P, El-Serag HB. Medications (NSAIDs, statins, proton pump inhibitors) and the risk of esophageal adenocarcinoma in patients with Barrett’s esophagus. Gastroenterology 2010; 138:2260 - 6; http://dx.doi.org/10.1053/j.gastro.2010.02.045; PMID: 20188100
- Voutilainen M, Sipponen P, Mecklin JP, Juhola M, Färkkilä M. Gastroesophageal reflux disease: prevalence, clinical, endoscopic and histopathological findings in 1,128 consecutive patients referred for endoscopy due to dyspeptic and reflux symptoms. Digestion 2000; 61:6 - 13; http://dx.doi.org/10.1159/000007730; PMID: 10671769
- Abrams JA, Fields S, Lightdale CJ, Neugut AI. Racial and ethnic disparities in the prevalence of Barrett’s esophagus among patients who undergo upper endoscopy. Clin Gastroenterol Hepatol 2008; 6:30 - 4; http://dx.doi.org/10.1016/j.cgh.2007.10.006; PMID: 18063419
- Ronkainen J, Aro P, Storskrubb T, Johansson SE, Lind T, Bolling-Sternevald E, et al. Prevalence of Barrett’s esophagus in the general population: an endoscopic study. Gastroenterology 2005; 129:1825 - 31; http://dx.doi.org/10.1053/j.gastro.2005.08.053; PMID: 16344051
- Ouatu-Lascar R, Fitzgerald RC, Triadafilopoulos G. Differentiation and proliferation in Barrett’s esophagus and the effects of acid suppression. Gastroenterology 1999; 117:327 - 35; http://dx.doi.org/10.1053/gast.1999.0029900327; PMID: 10419913
- Cooper BT, Chapman W, Neumann CS, Gearty JC. Continuous treatment of Barrett’s oesophagus patients with proton pump inhibitors up to 13 years: observations on regression and cancer incidence. Aliment Pharmacol Ther 2006; 23:727 - 33; http://dx.doi.org/10.1111/j.1365-2036.2006.02825.x; PMID: 16556174
- El-Serag HB, Aguirre TV, Davis S, Kuebeler M, Bhattacharyya A, Sampliner RE. Proton pump inhibitors are associated with reduced incidence of dysplasia in Barrett’s esophagus. Am J Gastroenterol 2004; 99:1877 - 83; http://dx.doi.org/10.1111/j.1572-0241.2004.30228.x; PMID: 15447744
- El-Serag H. The association between obesity and GERD: a review of the epidemiological evidence. Dig Dis Sci 2008; 53:2307 - 12; http://dx.doi.org/10.1007/s10620-008-0413-9; PMID: 18651221
- Hillman LC, Chiragakis L, Shadbolt B, Kaye GL, Clarke AC. Effect of proton pump inhibitors on markers of risk for high-grade dysplasia and oesophageal cancer in Barrett’s oesophagus. Aliment Pharmacol Ther 2008; 27:321 - 6; http://dx.doi.org/10.1111/j.1365-2036.2007.03579.x; PMID: 18047565
- Lagergren J, Bergström R, Lindgren A, Nyrén O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N Engl J Med 1999; 340:825 - 31; http://dx.doi.org/10.1056/NEJM199903183401101; PMID: 10080844
- García Rodríguez LA, Lagergren J, Lindblad M. Gastric acid suppression and risk of oesophageal and gastric adenocarcinoma: a nested case control study in the UK. Gut 2006; 55:1538 - 44; http://dx.doi.org/10.1136/gut.2005.086579; PMID: 16785284
- Haigh CR, Attwood SE, Thompson DG, Jankowski JA, Kirton CM, Pritchard DM, et al. Gastrin induces proliferation in Barrett’s metaplasia through activation of the CCK2 receptor. Gastroenterology 2003; 124:615 - 25; http://dx.doi.org/10.1053/gast.2003.50091; PMID: 12612900
- Wang JS, Varro A, Lightdale CJ, Lertkowit N, Slack KN, Fingerhood ML, et al. Elevated serum gastrin is associated with a history of advanced neoplasia in Barrett’s esophagus. Am J Gastroenterol 2010; 105:1039 - 45; http://dx.doi.org/10.1038/ajg.2009.629; PMID: 19904251
- Takaishi S, Cui G, Frederick DM, Carlson JE, Houghton J, Varro A, et al. Synergistic inhibitory effects of gastrin and histamine receptor antagonists on Helicobacter-induced gastric cancer. Gastroenterology 2005; 128:1965 - 83; http://dx.doi.org/10.1053/j.gastro.2005.03.027; PMID: 15940630
- Jin G, Ramanathan V, Quante M, Baik GH, Yang X, Wang SS, et al. Inactivating cholecystokinin-2 receptor inhibits progastrin-dependent colonic crypt fission, proliferation, and colorectal cancer in mice. J Clin Invest 2009; 119:2691 - 701; PMID: 19652364
- Spechler SJ, Sharma P, Souza RF, Inadomi JM, Shaheen NJ, American Gastroenterological Association. American Gastroenterological Association medical position statement on the management of Barrett’s esophagus. Gastroenterology 2011; 140:1084 - 91; http://dx.doi.org/10.1053/j.gastro.2011.01.031; PMID: 21376940
- Spechler SJ, Sharma P, Souza RF, Inadomi JM, Shaheen NJ, American Gastroenterological Association. American Gastroenterological Association technical review on the management of Barrett’s esophagus. Gastroenterology 2011; 140:e18 - 52, quiz e13; http://dx.doi.org/10.1053/j.gastro.2011.01.031; PMID: 21376939
- Winberg H, Lindblad M, Lagergren J, Dahlstrand H. Risk factors and chemoprevention in Barrett’s esophagus--an update. Scand J Gastroenterol 2012; 47:397 - 406; http://dx.doi.org/10.3109/00365521.2012.667145; PMID: 22428928
- Kastelein F, Spaander MC, Biermann K, Vucelic B, Kuipers EJ, Bruno MJ. Role of acid suppression in the development and progression of dysplasia in patients with Barrett’s esophagus. Dig Dis 2011; 29:499 - 506; http://dx.doi.org/10.1159/000331513; PMID: 22095018
- Corley DA, Kubo A, Levin TR, Block G, Habel L, Rumore G, et al. Race, ethnicity, sex and temporal differences in Barrett’s oesophagus diagnosis: a large community-based study, 1994-2006. Gut 2009; 58:182 - 8; http://dx.doi.org/10.1136/gut.2008.163360; PMID: 18978173
- Barbera M, Fitzgerald RC. Cellular origin of Barrett’s metaplasia and oesophageal stem cells. Biochem Soc Trans 2010; 38:370 - 3; http://dx.doi.org/10.1042/BST0380370; PMID: 20298185
- Hutchinson L, Stenstrom B, Chen D, Piperdi B, Levey S, Lyle S, et al. Human Barrett's adenocarcinoma of the esophagus, associated myofibroblasts, and endothelium can arise from bone marrow-derived cells after allogeneic stem cell transplant. Stem Cells Dev 2011; 20:11 - 7; http://dx.doi.org/10.1089/scd.2010.0139; PMID: 20677919
- Leedham SJPS, Preston SL, McDonald SA, Elia G, Bhandari P, Poller D, et al. Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett’s oesophagus. Gut 2008; 57:1041 - 8; http://dx.doi.org/10.1136/gut.2007.143339; PMID: 18305067
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001; 414:105 - 11; http://dx.doi.org/10.1038/35102167; PMID: 11689955
- Quante M, Wang TC. Stem cells in gastroenterology and hepatology. Nat Rev Gastroenterol Hepatol 2009; 6:724 - 37; http://dx.doi.org/10.1038/nrgastro.2009.195; PMID: 19884893
- Quante M, Marrache F, Goldenring JR, Wang TC. TFF2 mRNA transcript expression marks a gland progenitor cell of the gastric oxyntic mucosa. Gastroenterology 2010; 139:2018 - 27, e2; http://dx.doi.org/10.1053/j.gastro.2010.08.003; PMID: 20708616
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007; 449:1003 - 7; http://dx.doi.org/10.1038/nature06196; PMID: 17934449
- Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009; 457:608 - 11; http://dx.doi.org/10.1038/nature07602; PMID: 19092804
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009; 459:262 - 5; http://dx.doi.org/10.1038/nature07935; PMID: 19329995
- Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH, et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 2010; 6:25 - 36; http://dx.doi.org/10.1016/j.stem.2009.11.013; PMID: 20085740
- Means AL, Xu Y, Zhao A, Ray KC, Gu GA. A CK19(CreERT) knockin mouse line allows for conditional DNA recombination in epithelial cells in multiple endodermal organs. Genesis 2008; 46:318 - 23; http://dx.doi.org/10.1002/dvg.20397; PMID: 18543299
- Brembeck FH, Moffett J, Wang TC, Rustgi AK. The keratin 19 promoter is potent for cell-specific targeting of genes in transgenic mice. Gastroenterology 2001; 120:1720 - 8; http://dx.doi.org/10.1053/gast.2001.24846; PMID: 11375953
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126:663 - 76; http://dx.doi.org/10.1016/j.cell.2006.07.024; PMID: 16904174
- Wong DJ, Paulson TG, Prevo LJ, Galipeau PC, Longton G, Blount PL, et al. p16(INK4a) lesions are common, early abnormalities that undergo clonal expansion in Barrett’s metaplastic epithelium. Cancer Res 2001; 61:8284 - 9; PMID: 11719461
- Barrett MT, Sanchez CA, Prevo LJ, Wong DJ, Galipeau PC, Paulson TG, et al. Evolution of neoplastic cell lineages in Barrett oesophagus. Nat Genet 1999; 22:106 - 9; http://dx.doi.org/10.1038/8816; PMID: 10319873
- Leedham SJ, Preston SL, McDonald SA, Elia G, Bhandari P, Poller D, et al. Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett’s oesophagus. Gut 2008; 57:1041 - 8; http://dx.doi.org/10.1136/gut.2007.143339; PMID: 18305067
- Nicholson AM, Graham TA, Simpson A, Humphries A, Burch N, Rodriguez-Justo M, et al. Barrett's metaplasia glands are clonal, contain multiple stem cells and share a common squamous progenitor. Gut 2011; PMID: 22200839
- Giannakis M, Stappenbeck TS, Mills JC, Leip DG, Lovett M, Clifton SW, et al. Molecular properties of adult mouse gastric and intestinal epithelial progenitors in their niches. J Biol Chem 2006; 281:11292 - 300; http://dx.doi.org/10.1074/jbc.M512118200; PMID: 16464855
- Vega KJ, May R, Sureban SM, Lightfoot SA, Qu D, Reed A, et al. Identification of the putative intestinal stem cell marker doublecortin and CaM kinase-like-1 in Barrett's esophagus and esophageal adenocarcinoma. J Gastroenterol Hepatol 2012; 27:773 - 80; http://dx.doi.org/10.1111/j.1440-1746.2011.06928.x; PMID: 21916995
- Riddell RH, Odze RD. Definition of Barrett’s esophagus: time for a rethink--is intestinal metaplasia dead?. Am J Gastroenterol 2009; 104:2588 - 94; http://dx.doi.org/10.1038/ajg.2009.390; PMID: 19623166
- Menke V, van Es JH, de Lau W, van den Born M, Kuipers EJ, Siersema PD, et al. Conversion of metaplastic Barrett’s epithelium into post-mitotic goblet cells by gamma-secretase inhibition. Dis Model Mech 2010; 3:104 - 10; http://dx.doi.org/10.1242/dmm.003012; PMID: 20075383
- Allison PR, Johnstone AS. The oesophagus lined with gastric mucous membrane. Thorax 1953; 8:87 - 101; http://dx.doi.org/10.1136/thx.8.2.87; PMID: 13077502
- Barrett NR. Chronic peptic ulcer of the oesophagus and ‘oesophagitis’. Br J Surg 1950; 38:175 - 82; http://dx.doi.org/10.1002/bjs.18003815005; PMID: 14791960
- Hamilton SR, Yardley JH. Regnerative of cardiac type mucosa and acquisition of Barrett mucosa after esophagogastrostomy. Gastroenterology 1977; 72:669 - 75; PMID: 838221
- Nakanishi Y, Saka M, Eguchi T, Sekine S, Taniguchi H, Shimoda T. Distribution and significance of the oesophageal and gastric cardiac mucosae: a study of 131 operation specimens. Histopathology 2007; 51:515 - 9; http://dx.doi.org/10.1111/j.1365-2559.2007.02793.x; PMID: 17711448
- Lembo TIA, Ippoliti AF, Ramers C, Weinstein WM. Inflammation of the gastro-oesophageal junction (carditis) in patients with symptomatic gastro-oesophageal reflux disease: a prospective study. Gut 1999; 45:484 - 8; http://dx.doi.org/10.1136/gut.45.4.484; PMID: 10486352
- Gatenby PA, Ramus JR, Caygill CP, Shepherd NA, Watson A. Relevance of the detection of intestinal metaplasia in non-dysplastic columnar-lined oesophagus. Scand J Gastroenterol 2008; 43:524 - 30; http://dx.doi.org/10.1080/00365520701879831; PMID: 18415743
- Takubo K, Vieth M, Aida J, Sawabe M, Kumagai Y, Hoshihara Y, et al. Differences in the definitions used for esophageal and gastric diseases in different countries: endoscopic definition of the esophagogastric junction, the precursor of Barrett’s adenocarcinoma, the definition of Barrett’s esophagus, and histologic criteria for mucosal adenocarcinoma or high-grade dysplasia. Digestion 2009; 80:248 - 57; http://dx.doi.org/10.1159/000235923; PMID: 19828957
- Langer R, Von Rahden BH, Nahrig J, Von Weyhern C, Reiter R, Feith M, et al. Prognostic significance of expression patterns of c-erbB-2, p53, p16INK4A, p27KIP1, cyclin D1 and epidermal growth factor receptor in oesophageal adenocarcinoma: a tissue microarray study. J Clin Pathol 2006; 59:631 - 4; http://dx.doi.org/10.1136/jcp.2005.034298; PMID: 16731604
- Harris JC, Clarke PA, Awan A, Jankowski J, Watson SA. An antiapoptotic role for gastrin and the gastrin/CCK-2 receptor in Barrett’s esophagus. Cancer Res 2004; 64:1915 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-03-2713; PMID: 15026323
- Abdalla SI, Lao-Sirieix P, Novelli MR, Lovat LB, Sanderson IR, Fitzgerald RC. Gastrin-induced cyclooxygenase-2 expression in Barrett’s carcinogenesis. Clin Cancer Res 2004; 10:4784 - 92; http://dx.doi.org/10.1158/1078-0432.CCR-04-0015; PMID: 15269153
- Shaheen NJ, Greenwald BD, Peery AF, Dumot JA, Nishioka NS, Wolfsen HC, et al. Safety and efficacy of endoscopic spray cryotherapy for Barrett’s esophagus with high-grade dysplasia. Gastrointest Endosc 2010; 71:680 - 5; http://dx.doi.org/10.1016/j.gie.2010.01.018; PMID: 20363409
- Shaheen NJ, Sharma P, Overholt BF, Wolfsen HC, Sampliner RE, Wang KK, et al. Radiofrequency ablation in Barrett’s esophagus with dysplasia. N Engl J Med 2009; 360:2277 - 88; http://dx.doi.org/10.1056/NEJMoa0808145; PMID: 19474425
- Vaccaro BJ, Gonzalez S, Poneros JM, Stevens PD, Capiak KM, Lightdale CJ, et al. Detection of intestinal metaplasia after successful eradication of Barrett’s Esophagus with radiofrequency ablation. Dig Dis Sci 2011; 56:1996 - 2000; http://dx.doi.org/10.1007/s10620-011-1680-4; PMID: 21468652
- Halsey KD, Chang JW, Waldt A, Greenwald BD. Recurrent disease following endoscopic ablation of Barrett’s high-grade dysplasia with spray cryotherapy. Endoscopy 2011; 43:844 - 8; http://dx.doi.org/10.1055/s-0030-1256649; PMID: 21826629