Abstract
Fifteen years ago, we reported that proto-oncogene MYC promoted differentiation of human epidermal stem cells, a finding that was surprising to the MYC and the skin research communities. MYC was one of the first human oncogenes identified, and it had been strongly associated with proliferation. However, it was later shown that MYC could induce apoptosis under low survival conditions. Currently, the notion that MYC promotes epidermal differentiation is widely accepted, but the cell cycle mechanisms that elicit this function remain unresolved. We have recently reported that keratinocytes respond to cell cycle deregulation and DNA damage by triggering terminal differentiation. This mechanism might constitute a homeostatic protection face to cell cycle insults. Here, I discuss recent and not-so-recent evidence suggesting the existence of a largely unexplored oncogene-induced differentiation response (OID) analogous to oncogene-induced apoptosis (OIA) or senescence (OIS). In addition, I propose a model for the role of the cell cycle in skin homeostasis maintenance and for the dual role of MYC in differentiation.
Myc and Differentiation
The proto-oncogene MYC is amplified in a variety of human malignancies. MYC regulates transcription; it was initially shown to drive proliferation, and its diverse functions have been thoroughly reviewed elsewhere (e.g., refs. Citation1 and Citation2). MYC acts on the cell cycle in multiple ways.Citation3 Notably, it induces the expression of positive regulators of S phase such as Cyclin D or Cyclin E, and it downregulates cell cycle inhibitors such as p15INK4, p27KIP1 or p21CIP1.
The function of MYC in the cell cycle could readily account for its oncogenic capacity, but matters became more complex when it was shown to drive apoptosis under growth-restrictive conditions.Citation4-Citation6 MYC-induced apoptosis was proposed as a compensatory mechanism to limit MYC-induced tumorigenesis. Therefore, MYC has a dual effect on cell multiplication, positive and negative, by driving proliferation and apoptosis, the final outcome depending on the balance between one and another, between cell survival factors and pro-apoptotic factors.
The role of MYC in differentiation is even more intriguing. Because MYC promotes cell proliferation, it was thought to be incompatible with cell differentiation. Indeed, MYC has been found to inhibit differentiation of a variety of cell types when ectopically expressedCitation7 and is critical in the maintenance of pluripotency,Citation8 but it has also been shown to drive differentiation (reviewed in ref. Citation2).
The epidermis of the skin is a paradigm of MYC-induced differentiation.Citation9-Citation11 Epidermis is a self-renewing stratified epithelium. Keratinocytes proliferate within the basal layer, and after a transient phase of rapid proliferation, they initiate post-mitotic terminal differentiation and migrate throughout the suprabasal layers toward the surface of the skin. The epidermis is the most frequent target of cancer.Citation12 Yet, considering that the skin is continuously exposed to mutagenic hazard, mainly solar UV radiation (UV) and the human papilloma virus (HPV), the clinical manifestations are relatively scarce.
The epidermis must have powerful mechanisms ensuring that the balance between cell multiplication and cell differentiation is maintained. What are these mechanisms? Evidence of apoptosis has been shown in severely sun-damaged skin, and DNA repair must have an active role in maintaining genome integrity.Citation13,Citation14 However, what tells epidermis how many cells are being shed from the surface of the skin, for the basal layer to produce a similar number of cells? In the case of benign hyperproliferative conditions, how does the epidermis manage to maintain its equilibrium while avoiding tumorigenesis? The study of the cell cycle mechanisms promoting epidermal differentiation downstream of MYC might provide some clues. Herein, I shall examine some recent and old evidence for an oncogene-induced differentiation response in the epidermis and beyond.
The Epidermis Cell Cycle Paradoxes
Epidermal cell cycle progression and terminal differentiation have been considered incompatible processes. Keratinocytes are thought to arrest cell cycle and cell growth in the onset of initiation of terminal differentiation (). This model, however, does not explain two key observations:
Figure 1. The traditional keratinocyte cell cycle model. Keratinocytes are thought to undergo cell growth and cell cycle arrest in G0 before terminal differentiation (A). However, the most frequent hyperproliferative skin conditions include thickening of differentiating strata (acanthosis or hyperkeratosis) (B). The G0 model would predict that a hyperproliferative stimulus would block differentiation, and this would easily result in a tumor (C). Red are cells with the capacity to divide; green are post-mitotic terminally differentiating cells.
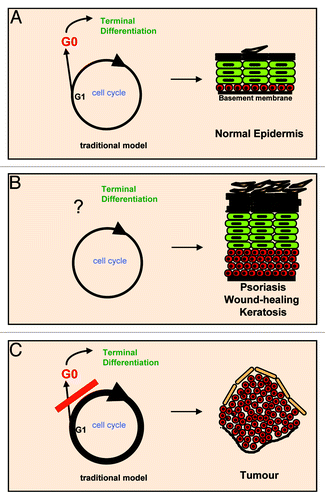
(1) Keratinocytes become larger as they differentiate,Citation15 thus indicating that cellular growth continues.
(2) Benign hyperproliferative skin conditions such as psoriasis, wound-healing or keratosis involve extension of proliferative layers (hyperplasia) and thickening of squamous differentiated layers (acanthosis or hyperkeratosis). Yet, the balance between proliferation and differentiation is somehow maintained in these lesions, with accumulation of both proliferative and differentiating cells. As a result, the structure of the tissue remains, even if its properties are altered ().
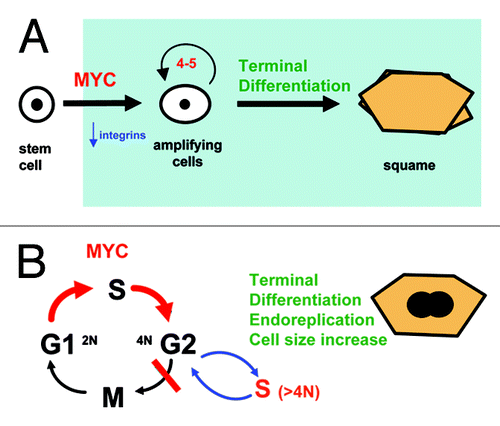
If keratinocytes had to undergo cell cycle arrest prior to differentiation, then cell cycle hyperactivation would hamper differentiation (). This was our rationale when we expressed MYC ectopically in human primary keratinocytes. However, this is not what we observed. We were aiming for transcription factors with a role in the commitment to differentiation. To our surprise, overexpression of wild-type MYC or conditional activation of MYCER (fusion protein with the binding domain of estrogen receptor) promoted epidermal differentiation.Citation10 MYC was driving stem cells into the differentiation pathway, involving downregulation of cell adhesion molecules to the basement membrane (integrins) and a step of rapid proliferation. These events normally precede keratinocyte terminal differentiation and stratification ().
Figure 2. MYC drives epidermal differentiation and endoreplication. (A) Overactivation of MYC in human keratinocytes downregulates cell adhesion integrins and drives stem cells into active cell cycle and clonal expansion, thus committing them to terminal differentiation.Citation10 The function of MYC in epidermis becomes clearer if the keratinocyte clonal expansion phase is considered as part of the differentiation program (light blue). (B) By driving the keratinocyte cell cycle but not cell division, overactivation of MYC induces cell size increase and endoreplication, part of the normal differentiation program.Citation19,Citation23,Citation29,Citation46,Citation51
Our experiments on human primary keratinocytes in vitro were supported by studies on tissue-specific transgenic mice. Overexpression of MYC under various different basal or suprabasal promoters in mouse epidermis did not cause apparent apoptosis, but drove stem cells into differentiation, thereby depleting the stem cell compartment.Citation9,Citation11,Citation16 Regardless of whether or not MYC caused hyperplasia in mouse skin, terminal differentiation prevailed over proliferation, as we had observed in human cells in vitro. Consistently, MYC function in mouse epidermis also involves downregulation of cell adhesion molecules.Citation17,Citation18
The fact that active MYC was compatible with terminal differentiation was in contradiction with the notion that cell cycle progression and differentiation were incompatible. How could we explain these observations? We previously showed that human primary keratinocytes continue cell cycle progression and DNA replication during terminal differentiation in vitro.Citation19 Terminal differentiation blocks cytokinesis, and cells undergo successive cycles of DNA replication and growth in the absence of cell division (). This phenomenon is referred to as endoreplication.Citation20 Interestingly, endoreplication allows cells to increase in size, and thus it can explain how keratinocytes become larger as they differentiate. Moreover, endoreplication has a well-known role in plant epidermis.Citation21
Endoreplication reconciles MYC activation of the cell cycle with epidermal differentiation. Does endoreplication have a physiological role in the epidermis? To answer this question we chose two different approaches: (1) inactivation of MYC in mouse epidermis and (2) determining whether endoreplication takes place in human epidermis.
We took advantage of the tissue-specific knockout technology to inactivate MYC in mouse epidermis by use of the recombinase CRE upon the promoter of the keratinocyte-specific keratin K5.Citation22 Mice with complete loss of MYC in the epidermis were viable, and adults had a differentiated skin, showing that keratinocytes do not need MYC to divide and differentiate.Citation23 However, MYC-deficient epidermis contained smaller keratinocytes and a smaller proportion of polyploid cells than their normal littermates. As a consequence, MYC-deficient skin was impaired in plasticity and integrity. The skin ripped off in areas of mechanical tension, and wound healing was inefficient. MYC was dispensable for the division of stem cells, but it was requisite for their capacity to amplify and undergo rapid proliferation. Therefore, MYC was required for normal epidermal growth. This was consistent with reports showing a primary role for MYC in protein and ribosome biosynthesis.Citation24,Citation25 Also consistently, partial deletion of MYC in epidermis was reported to confer resistance to experimental skin carcinogenesis.Citation26
Impairment of endoreplication and cellular growth by MYC inactivation did not prevent mouse epidermis from differentiation, but it affected stem cell renewal and skin regeneration. We have made similar observations after inactivating MYC in the liverCitation27 or in the mammary gland.Citation28
Human Epidermal Endoreplication
Does endoreplication take place in human skin? Despite that cell cycle regulation has been extensively studied in mammalian cells in the last decades, its relationship with differentiation in human epidermis is still unclear. We have performed extensive studies of cell cycle dynamics, DNA replication and nuclear DNA content in normal human epidermis of skin from various body sites by different means. The results consistently showed that cell cycle progression, DNA replication and differentiation coexist in suprabasal layers of the epidermis.Citation29 The very first surprise was the striking accumulation of Cyclin E in differentiating layers. The second surprise was that most cells expressing mitotic Cyclin A or Cyclin B were within the first suprabal layers. The third surprise was to see that suprabasal layers were more active on DNA replication than the basal layer.
However surprising human epidermal endoreplication might seem, a careful study of the epidermal cell cycle behavior does not provide evidence for a G0/G1 arrest in differentiating cells, but rather against. For instance:
(1) Inhibition of the keratinocyte cell cycle in G1 did not efficiently induce differentiation and in some cases (such as the overexpression of the cdk inhibitor p21CIP1), it even attenuated differentiation (refs. Citation19, Citation30 and Citation31 and references therein).
(2) Primary keratinocytes differentiate from any phase of the cell cycle, and differentiating cells do not accumulate in G0/G1 but in G2/M.Citation19,Citation32
(3) As it occurs in natural hyperproliferative benign skin disorders and in MYC epidermal transgenic mice, a variety of transgenic mouse lines overexpressing a cell cycle molecule in the epidermis displayed hyperplasia with hyperkeratosis (accumulation of differentiated layers); these include E2F,Citation33 Cyclin D1,Citation34,Citation35 MDM2,Citation36 cdk4Citation37 or cdk2.Citation38 Some of the authors of these works stated their surprise by the persistence of differentiation.
Therefore, a large body of evidence does not reconcile well with a cell cycle arrest model for keratinocyte differentiation.
The endoreplication model can explain these apparent paradoxes. In our studies, not only basal keratinocytes continued on cycle as they stratified into suprabasal layers, but it was the more actively Cycling cells that initiated terminal differentiation. Even more strikingly, most basal cells detaching from the basement membrane, sometimes referred to as “mushroom cells” due to the stalk they still have within the basal layer,Citation39 were undergoing mitosis. Although, as it would be expected, some cells within the basal layer were on cycle, most of the cells expressing mitotic Cyclins were migrating into the suprabasal layers. Patches of Cyclin A-expressing cells, including metaphasic figures, were initiating post-mitotic differentiation as they begun to stratify ().
Figure 3. Actively Cycling clones initiate terminal differentiation and migration. (A) Stem cells (SC; yellow) divide in the basal layer (1); some daughter cells stay as stem cells; others activate the cell cycle and enter a phase of clonal expansion or amplification (2; CAC; red, Cyclin A positive); as the cell cycle accelerates, the group of cells blocks mitosis, initiates terminal differentiation (keratins K1 and K10, green) and migrates into suprabasal layers (3). Some cells still divide their nucleus (endomitosis); subsequently, the nuclei undergo re-replication. Top micrographs correspond to immunofluorescent detection of Cyclin A (red), keratin K1 (green) DNA (blue) on different areas of a microsection from normal skin. BM, basement membrane; Bar: 40 µm. For more details and cell cycle markers see ref. Citation29. (B) By linking terminal differentiation to cell cycle hyperactivation, epidermis can coordinate proliferation with differentiation and maintain tissue structure upon hyperproliferative stimuli. Cell cycle hyperactivation then simply increases the epidermal turnover. Red are cells with the capacity to divide; green are post-mitotic terminally differentiating cells.
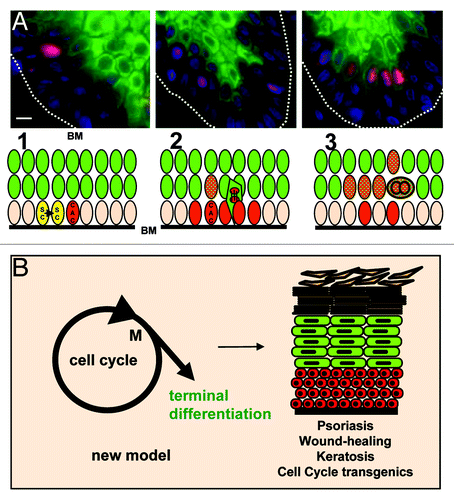
In a steady-state epidermis, suprabasal keratinocytes undergo terminal differentiation, and there is wide consensus that they can no longer proliferate. Indeed, it has been shown that proliferation is maintained by cell adhesion integrins through interaction with the basement membrane.Citation40 In addition, the epidermal suprabasal cytoskeleton is formed by keratins K1 and K10, which suppress proliferation when ectopically expressed in dividing cells.Citation41-Citation43 Therefore, strong evidence indicates that keratinocytes cease cell division as they detach from the basement membrane within the basal layer. In our analyses, most cells expressing mitotic Cyclins within the basal and peribasal layers were also expressing the post-mitotic keratin cytoskeleton K1/K10 (). Those cells continued DNA replication, increased in ploidy and contained duplicated centrosomes.Citation29,Citation44
A Cell Cycle-Differentiation Checkpoint
Endoreplication per se is interesting, and very enlightening reviews cover this topic.Citation20,Citation21,Citation45 Endoreplication has likely important functions in epidermis such as cell enlargement, increase of genes copy number, limiting the number of cell divisions and others. The issue of this essay is how epidermis coordinates cell cycle with differentiation. Endoreplication is consequence of a mitosis-defective cell cycle, and, thus, it reveals that the key control in keratinocyte proliferation might not be G1/S but G2/M. This not only may change the way we see skin homeostasis, but also the understanding of skin epithelial carcinogenesis. If the critical checkpoint is at mitosis, then oncogenic alterations driving cell cycle entry would not be tumorigenic, since the mitotic control would block cell division and trigger differentiation (). This would be a simple way to explain why overexpression of oncogenes or G1/S regulators in epidermis results in hyperplasia with hyperkeratosis and not in tumorigenesis.
We hypothesized that cell cycle hyperactivation and deregulation might trigger differentiation as an anti-oncogenic self-defense mechanism. To demonstrate this we needed to find two connections:
(1) That a keratinocyte mitosis block triggers epidermal terminal differentiation.
(2) That cell cycle hyperactivation in keratinocytes results in a mitosis block.
(1) Aiming to answer this question, we blocked primary keratinocytes in G2/M with various different chemical or molecular agents, including nocodazole, cdk chemical inhibitors and a topoisomerase inhibitor (ICRF193), or bleomycin, that causes DNA double-strand breaks. In addition, we made use of inhibitors of Aurora B and Polo-like kinases, components of the mitosis spindle checkpoint. Finally, we knocked-down the endogenous mitosis kinase cdk1 or Aurora B kinase by specific shRNAs or siRNAs. All these treatments blocked keratinocyte mitosis yet allowed DNA replication, raised the polyploidy index and strikingly induced terminal differentiation.Citation19,Citation29,Citation46
(2) Activation of MYCER in keratinocytes rapidly induces cell cycle activation prior to the increase of post-mitotic terminal differentiation.Citation46 However, MYC transcription factor elicits pleiotropic cellular effects, and we needed a direct and clean activation of the cell cycle that pushed DNA replication. To this end we constructed a retroviral vector carrying a GFP form of Cyclin E. Cyclin E accumulates during epidermal differentiation,Citation29 and it is widely involved in animal and plant endoreplication.Citation20,Citation21,Citation45 Overexpression of Cyclin E in keratinocytes has had revealing consequences.
Human primary keratinocytes overexpressing Cyclin E-GFP, as expected, had a higher index of cdk2 activity, Cycling cells and DNA replication.Citation46 Interestingly, Cyclin E-GFP also caused increased polyploidy and multinucleate cells, consequences of mitosis failure. What was more striking, the keratinocyte clonogenic potential was reduced, and the proportion of large, differentiating cells increased very significantly. Ectopic Cyclin E in keratinocytes caused no senescence or apoptosis, but terminal differentiation. By accelerating the cell cycle, Cyclin E also accelerated the differentiation program.
Missing Links
We have shown both that a mitosis block triggers keratinocyte differentiation, and that cell cycle hyperactivation blocks the keratinocyte mitosis. This suggests an exciting and simple model for the epidermal maintenance of tissue structure upon cell cycle stimuli, where differentiation is linked to cell cycle deregulation (). However, the first missing link is a mechanism that can translate cell cycle hyperactivation into a mitosis block.
As discussed, we had shown that treatment of primary keratinocytes with chemicals causing genotoxic insult triggered differentiation. In addition, it was proposed that MYC and other oncogenes might cause genomic instability by provoking DNA damage,Citation47,Citation48 and this is likely mediated by “replication stress.” Replication stress can be caused by accelerated DNA replication (hyper-replication), due to errors of chain synthesis or to depletion of cellular nucleotide pools.Citation49,Citation50 DNA damage can trigger cell cycle checkpoints leading to apoptosis or senescence. Our rationale was therefore that Cyclin E might push the keratinocyte cell cycle, inducing too many errors, which eventually would trigger the mitosis checkpoints. Consistently, overexpression of Cyclin E in keratinocytes caused accumulated DNA damage as measured by phosphorylation of γ-H2AX, an early marker of DNA repair. Moreover, Cyclin E induced the p53/p21 pathway, a typical response to DNA damage. Interestingly, activation of conditional MYC in primary keratinocytes, that induces p53 and p21, also upregulates Cyclin E.Citation46,Citation51 Moreover, knocking-down Cyclin E by specific shRNAs attenuated MYC-induced differentiation, suggesting that this process is mediated by cell cycle hyperactivation. Our model is therefore that the deregulation of the keratinocyte cell cycle produces DNA damage up to a threshold when the cell cannot repair it. This would alarm the cell cycle checkpoints that block mitosis. The mitosis block would then trigger terminal differentiation ().
Figure 4. Molecular control of epidermal oncogene-induced differentiation. By deregulating the cell cycle, overexpression of Cyclin E in keratinocytes causes DNA damage (possibly by replication stress); this induces DNA damage responses including the mitosis checkpoints that block cell division; this triggers cell size increase, terminal differentiation and DNA re-replication; transient p21CIP1 is able to inhibit both cdk2 and cdk1, but the high amounts of Cyclin E may make less efficient the inhibition of S phase-cdk2.Citation46
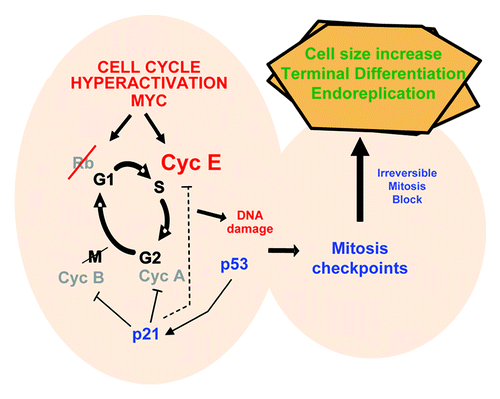
The next missing link is the mechanism by which a mitosis block switches on terminal differentiation. Here we need to speculate further. Cell adhesion to the basement membrane via integrins is known to critically control keratinocyte proliferation and differentiation. Twice less the amount of integrin molecules on the surface can suffice for a daughter of a stem cell to enter differentiation.Citation52 Elegant models for a link between cell size and signaling have been proposed.Citation53,Citation54 As a result of a sustained mitosis block, the cell enlarges, and the increased size might make cell adhesion molecules relatively less important (). Cells might lose adherence and be pushed by neighbor more strongly adherent cells. A similar phenomenon may act in some processes of anoikis (loss of anchorage-induced apoptosis). This provides a mechanism by which mitosis checkpoints may control epidermal cell fate, but it is merely hypothetical and difficult to prove. In keratinocytes, the model would be in agreement with studies on mouse epidermis, suggesting that asymmetric cell division might promote keratinocyte stratification and differentiation.Citation55
Figure 5. Proposed models for the control of epidermal cell fate by mitosis and the dual role of MYC in differentiation. (A) Cell adhesion integrins maintain keratinocyte proliferation, and their inhibition triggers differentiation;Citation40 a sustained mitosis block in keratinocytes would increase their cell volume, and this may result in loss of adherence via integrins; they would then be pushed to stratify by neighboring, more strongly adherent cells. (B) In systems where overactivation of MYC has no direct action on mitosis, cell fate would result in proliferation when cell division is allowed, but it would result in apoptosis or differentiation, depending on the cell type and context, when cell division is blocked. Similarly, MYC may inhibit differentiation processes that require cell growth arrest (G0-), but it may stimulate differentiation processes that involve cellular growth or cell size increase (Growth-).
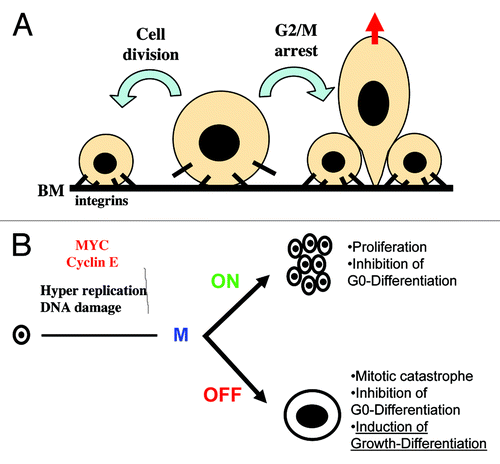
Molecular Switch
Our results indicate that as keratinocytes undergo rapid proliferation, they become uninhibited, and the cell cycle is deregulated. The mitosis block would prevent cells from uncontrolled proliferation. Accumulation of Cyclin E or other cell cycle activators might limit the number of cell divisions that keratinocytes can undergo before they enter terminal differentiation. While the cell cycle accelerates, Cyclin E accumulates; Rb is downregulated; mitotic Cyclins A and B and kinase cdk1 are inhibited ( and refs. Citation29 and Citation46 and references therein). All these changes occur as proliferative keratinocytes irreversibly commit to terminal differentiation. Concomitantly, cdk inhibitor p21CIP1 (p21) is transiently induced.Citation30,Citation31,Citation56 P21 has been shown to bind cdk2 complexes and block cells in G1 or to bind cdk1 complexes and block cells in G2/M, which allows DNA re-replication and cell size increase.Citation57-Citation59 In keratinocytes, transient binding of p21 to cdk1 peaks by the time when they irreversibly lose their capacity to proliferate.Citation46 P21 is also transiently expressed in post-mitotic peribasal cells of skin (ref. Citation31 and references therein). However, constitutive overexpression of p21 in keratinocytes inhibited both the cell cycle and differentiation.Citation30,Citation31
In endoreplicating systems, the induction of p21 might need to be transient in order for cdk2 to continue DNA replication once mitosis is irreversibly blocked. We have studied this issue in human leukemia myeloid cells K562. Upon certain stimuli, these cells undergo megakaryocytic differentiation involving endoreplication. Interestingly, overexpression of p21 induces differentiation in a proportion of cells that become polyploid.Citation60 However, the expression of p21 needs to be transient to allow full extent of polyploidy and differentiation.Citation61 The cell cycle analyses during p21-induced megakaryocytic differentiation and keratinocyte differentiation provides revealing similarities.Citation46,Citation61 In differentiating K562, S phase Cyclins are also increased, while DNA replication continues, mitotic Cyclins are inhibited and p21 is transiently induced. In both cell systems Cyclin E induces endoreplication.Citation46,Citation62
As stated before, keratinocytes are not the only primary system in which MYC stimulates differentiation (reviewed in ref. Citation2). MYC tends to stimulate differentiation when the normal process is concomitant with active cell growth or rapid proliferation, as it is the case of keratinocytes or some hematopoietic lineages. In K562 cells, which lack functional p53, ectopic MYC appears to favor mainly apoptosis. However, when p21 is transiently expressed in the presence of exogenous MYC, it protects K562 cells from apoptosis, and instead, they undergo megakaryocytic differentiation, increased cell size and polyploidization.Citation61
Therefore, MYC is capable to induce differentiation in certain circumstances or cell lineages. Why does MYC in some cases inhibit and in others stimulate differentiation? Our results in keratinocytes suggest the existence of a mitosis-differentiation checkpoint.Citation29,Citation46 We and others have suggested that at least in some cell lineages, the consequences of MYC action on proliferation might depend on whether mitosis is active or inactive.Citation23,Citation63 When cell division is impaired, the stimulus to grow is sustained, and the cell is protected from mitotic catastrophe (apoptosis), then the cell continues to grow. Consistently, we previously proposed that keratinocyte differentiation may share pathways with apoptosis.Citation64,Citation65). Therefore, it is tempting to speculate that in self-renewal systems, MYC, by pushing cellular growth, blocks processes of differentiation that require cell growth arrest (small size), such as erythrocyte differentiation (reviewed in ref. Citation7), whereas it stimulates processes of differentiation that involve active cell cycle and cellular growth (cell size increase; ). MYC might even have different effects on differentiation within the same cell lineage depending on whether the stage at which it is activated requires or not cellular growth. For instance, in embryo or neonatal liver, MYC induced neoplasia, whereas in adult liver, it induced cell size increase and polyploidy.Citation66
Although, in some cases, the inhibitory effect of MYC on differentiation was independent on the cell cycle,Citation7 it might drive cells out of G0 in a G1 growth-active state. Cell cycle-independent effects of MYC on differentiation can, in some cells, be driven by its post-translational modifications, such as protein cleavage and re-localization in muscle differentiation,Citation67 or phosphorylation in retinoic acid modulation of leukemia cells.Citation68
Paradoxical effects on cell fate depending on the state of the cell cycle, reminiscent of those of MYC but in the opposite sense, have been observed for the p53 pathway. Whereas, by halting the cell cycle, p53 is able to inhibit osteoblast or thymocyte maturation,Citation69-Citation71 it induces markers of erythrocyte differentiation in K562 cells.Citation72 Consistently, overexpression of cdk2 inhibitor p27KIP1 in these cells also induces erythrocyte differentiation.Citation60 p53 can also elicit alternative effects on cellular senescence depending on the crosstalk with the mTOR growth-transduction pathway through the cell cycle-inhibitor p21CIP1 in fibrosarcoma cells.Citation73,Citation74
Oncogene-Induced Differentiation
In self-renewing tissues involving continuous cell proliferation and differentiation, it is critical that the number of cells differentiating equals the number of cells produced. Upon a stimulus to grow such as the affluence of blood serum after injury or potentially oncogenic mutations, the balance must be maintained. When the cell cycle accelerates, a brake system must be established. By linking cell cycle hyperactivation to differentiation through DNA damage, both processes are chained. The outcome is orchestrating proliferation with differentiation. Within these lines, it is interesting to note that columns of stratifying cells harboring p53 mutations are present in normal skin.Citation75 Therefore, although p53 accumulation has been associated with apoptosis after severe damageCitation13 in steady-state epidermis, damaged keratinocytes might be “expelled” from the tissue by means of differentiation.
Might this mechanism exist in other human differentiating tissues? Although it is nowadays well-accepted that oncogenes can induce apoptosis (OIA) or senescence (OIS) to counterbalance their effect on cell multiplication, the existence of an oncogene-induced differentiation response (OID) has been largely unexplored.
The finding that oncogenes can cause DNA damage provides a link between their activity and differentiation through the cell cycle checkpoints. Some recent and old works have found further evidence for such kind of checkpoints. Very recently, a lymphoid differentiation response to DNA damage limiting self-renewal has been reported in hematopoietic cells,Citation76 and a G2-mitosis checkpoint has been proposed to control trophoblast differentiation into polyploid giant cells.Citation77 Previously, a negative relationship was established between genotoxic insult and myogenic differentiation.Citation78,Citation79 Much earlier, oncogenic forms of the Ras signal transduction family were shown to trigger neuron differentiation of PC12 cells (e.g., ref. Citation80). Although KRas is frequently activated in colorectal carcinoma, in some cases oncogenic Ras has also been associated with colon differentiation.Citation81-Citation83 As mentioned above, osteoblast differentiationCitation69 and thymocyte maturationCitation70,Citation71 might also depend on cell cycle progression. Indeed, there are parallels between the proliferative phase involved in T lymphocyte maturation and the rapid proliferative phase preceding epidermal differentiation (). If this proliferative phase is considered as part of the epidermal differentiation program or keratinocyte maturation, the model becomes simpler. Due to cell cycle-differentiationcheckpoints, by accelerating the cell cycle single oncogenes may just increase the epidermal turnover without disrupting tissue structure. Additional alterations in these checkpoints would be required for cancer development and would favor selection of MYC-overexpressing cells.Citation84
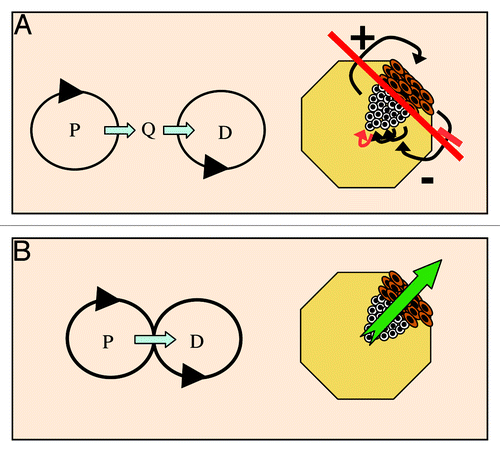
A rapid analysis of the terms “oncogene-induced differentiation” or “differentiation checkpoint” in two important web scientific databases show that the notion has been scarcely employed (). Yet, the coordination between proliferation and differentiation is essential to tissue homeostasis, development and cancer (). The question now is whether cell cycle-induced differentiation checkpoints have still been poorly documented because they are restricted to a few cell systems, or because they have so far received little attention.
Table 1. Presence in scientific literature
Figure 6. Linking proliferation with differentiation orchestrates homeostasis. (A) In developing or self-renewing cell systems such as epidermis, a quiescence phase (Q) between proliferation (P) and differentiation (D) would need to be controlled by a complex dialog of soluble and non-soluble factors (arrows); any alteration of one of the connexions (red) would alter the system (long red line). (B) Linking differentiation to cell cycle hyperactivation through a mitosis block would automatically coordinate cell numbers. The evidence for such a link in human epidermis: cell growth, DNA replication and differentiation coexist; there is a G2/M accumulation in differentiation; there is a link between G2/M and the start of differentiation; a mitosis block triggers differentiation; cell cycle hyperactivation triggers a mitosis block and differentiation.Citation29,Citation46 White are proliferative cells; brown are differentiating cells.
In summary, an oncogene- or cell cycle-induced differentiation checkpoint analogous to the alarms that result in apoptosis or senescence might trigger terminal differentiation in some cell systems as an automatic homeostasis-maintenance mechanism.
| Abbreviations: | ||
| UV | = | ultraviolet radiation |
| HPV | = | human papilloma virus |
| cdk | = | Cyclin-dependent kinase |
| OID | = | oncogene-induced differentiation |
| MYCER | = | fusion protein made of MYC and a mutant ligand binding domain of estrogen receptor |
Acknowledgments
I apologize to all those authors whose works have importantly contributed to the topics discussed and I did not cite. This essay was not intended as a comprehensive review but to propose a model and I have discussed some examples that support the model. I wish to thank Jean Jacques Guilhou, Javier León, the INSERM (France) and the ISCIII (Spain) for fundamental support and all those who contributed to our findings within my group, especially Jeniffer Zanet and Ana Freije. I also thank James Elder, Ana Freije, Agustí Toll and Philippe Pasero for critical reading of the manuscript and Diana Solinís for revising the English writing. This work was completed with funds from the national Instituto de Salud Carlos III (ISCIII, Spain; FIS-PI08/0890, FIS-PI11/02070 and Programa Regiones Emergentes-EMER) and from a cooperative program Universidad de Cantabria-Sistema Cántabro de Salud (UC-SCS; Cantabria, Spain).
References
- Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer 2008; 8:976 - 90; http://dx.doi.org/10.1038/nrc2231; PMID: 19029958
- Eilers M, Eisenman RN. Myc’s broad reach. Genes Dev 2008; 22:2755 - 66; http://dx.doi.org/10.1101/gad.1712408; PMID: 18923074
- Blagosklonny MV, Pardee AB. The restriction point of the cell cycle. Cell Cycle 2002; 1:103 - 10; http://dx.doi.org/10.4161/cc.1.2.108; PMID: 12429916
- Askew DS, Ashmun RA, Simmons BC, Cleveland JL. Constitutive c-myc expression in an IL-3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene 1991; 6:1915 - 22; PMID: 1923514
- Shi Y, Glynn JM, Guilbert LJ, Cotter TG, Bissonnette RP, Green DR. Role for c-myc in activation-induced apoptotic cell death in T cell hybridomas. Science 1992; 257:212 - 4; http://dx.doi.org/10.1126/science.1378649; PMID: 1378649
- Evan GI, Littlewood TD. The role of c-myc in cell growth. Curr Opin Genet Dev 1993; 3:44 - 9; http://dx.doi.org/10.1016/S0959-437X(05)80339-9; PMID: 8453273
- León J, Ferrandiz N, Acosta JC, Delgado MD. Inhibition of cell differentiation: a critical mechanism for MYC-mediated carcinogenesis?. Cell Cycle 2009; 8:1148 - 57; http://dx.doi.org/10.4161/cc.8.8.8126; PMID: 19282668
- Smith KN, Lim JM, Wells L, Dalton S. Myc orchestrates a regulatory network required for the establishment and maintenance of pluripotency. Cell Cycle 2011; 10:592 - 7; http://dx.doi.org/10.4161/cc.10.4.14792; PMID: 21293186
- Waikel RL, Kawachi Y, Waikel PA, Wang XJ, Roop DR. Deregulated expression of c-Myc depletes epidermal stem cells. Nat Genet 2001; 28:165 - 8; http://dx.doi.org/10.1038/88889; PMID: 11381265
- Gandarillas A, Watt FM. c-Myc promotes differentiation of human epidermal stem cells. Genes Dev 1997; 11:2869 - 82; http://dx.doi.org/10.1101/gad.11.21.2869; PMID: 9353256
- Arnold I, Watt FM. c-Myc activation in transgenic mouse epidermis results in mobilization of stem cells and differentiation of their progeny. Curr Biol 2001; 11:558 - 68; http://dx.doi.org/10.1016/S0960-9822(01)00154-3; PMID: 11369200
- Böni R, Schuster C, Nehrhoff B, Burg G. Epidemiology of skin cancer. Neuro Endocrinol Lett 2002; 23:Suppl 2 48 - 51; PMID: 12163848
- Brash DE, Ziegler A, Jonason AS, Simon JA, Kunala S, Leffell DJ. Sunlight and sunburn in human skin cancer: p53, apoptosis, and tumor promotion. J Investig Dermatol Symp Proc 1996; 1:136 - 42; PMID: 9627707
- Kulms D, Schwarz T. Molecular mechanisms of UV-induced apoptosis. Photodermatol Photoimmunol Photomed 2000; 16:195 - 201; http://dx.doi.org/10.1034/j.1600-0781.2000.160501.x; PMID: 11068857
- Banks-Schlegel S, Green H. Involucrin synthesis and tissue assembly by keratinocytes in natural and cultured human epithelia. J Cell Biol 1981; 90:732 - 7; http://dx.doi.org/10.1083/jcb.90.3.732; PMID: 6895225
- Flores I, Murphy DJ, Swigart LB, Knies U, Evan GI. Defining the temporal requirements for Myc in the progression and maintenance of skin neoplasia. Oncogene 2004; 23:5923 - 30; http://dx.doi.org/10.1038/sj.onc.1207796; PMID: 15208685
- Frye M, Gardner C, Li ER, Arnold I, Watt FM. Evidence that Myc activation depletes the epidermal stem cell compartment by modulating adhesive interactions with the local microenvironment. Development 2003; 130:2793 - 808; http://dx.doi.org/10.1242/dev.00462; PMID: 12736221
- Gebhardt A, Frye M, Herold S, Benitah SA, Braun K, Samans B, et al. Myc regulates keratinocyte adhesion and differentiation via complex formation with Miz1. J Cell Biol 2006; 172:139 - 49; http://dx.doi.org/10.1083/jcb.200506057; PMID: 16391002
- Gandarillas A, Davies D, Blanchard JM. Normal and c-Myc-promoted human keratinocyte differentiation both occur via a novel cell cycle involving cellular growth and endoreplication. Oncogene 2000; 19:3278 - 89; http://dx.doi.org/10.1038/sj.onc.1203630; PMID: 10918584
- Edgar BA, Orr-Weaver TL. Endoreplication cell cycles: more for less. Cell 2001; 105:297 - 306; http://dx.doi.org/10.1016/S0092-8674(01)00334-8; PMID: 11348589
- De Veylder L, Larkin JC, Schnittger A. Molecular control and function of endoreplication in development and physiology. Trends Plant Sci 2011; 16:624 - 34; http://dx.doi.org/10.1016/j.tplants.2011.07.001; PMID: 21889902
- Ramirez A, Page A, Gandarillas A, Zanet J, Pibre S, Vidal M, et al. A keratin K5Cre transgenic line appropriate for tissue-specific or generalized Cre-mediated recombination. Genesis 2004; 39:52 - 7; http://dx.doi.org/10.1002/gene.20025; PMID: 15124227
- Zanet J, Pibre S, Jacquet C, Ramirez A, de Alborán IM, Gandarillas A. Endogenous Myc controls mammalian epidermal cell size, hyperproliferation, endoreplication and stem cell amplification. J Cell Sci 2005; 118:1693 - 704; http://dx.doi.org/10.1242/jcs.02298; PMID: 15797928
- Levens D. Disentangling the MYC web. Proc Natl Acad Sci USA 2002; 99:5757 - 9; http://dx.doi.org/10.1073/pnas.102173199; PMID: 11983876
- Gomez-Roman N, Grandori C, Eisenman RN, White RJ. Direct activation of RNA polymerase III transcription by c-Myc. Nature 2003; 421:290 - 4; http://dx.doi.org/10.1038/nature01327; PMID: 12529648
- Oskarsson T, Essers MA, Dubois N, Offner S, Dubey C, Roger C, et al. Skin epidermis lacking the c-Myc gene is resistant to Ras-driven tumorigenesis but can reacquire sensitivity upon additional loss of the p21Cip1 gene. Genes Dev 2006; 20:2024 - 9; http://dx.doi.org/10.1101/gad.381206; PMID: 16882980
- Baena E, Gandarillas A, Vallespinós M, Zanet J, Bachs O, Redondo C, et al. c-Myc regulates cell size and ploidy but is not essential for postnatal proliferation in liver. Proc Natl Acad Sci USA 2005; 102:7286 - 91; http://dx.doi.org/10.1073/pnas.0409260102; PMID: 15857952
- Moumen M, Chiche A, Deugnier MA, Petit V, Gandarillas A, Glukhova MA, et al. The proto-oncogene Myc is essential for mammary stem cell function. Stem Cells 2012; 30:1246 - 54; http://dx.doi.org/10.1002/stem.1090; PMID: 22438054
- Zanet J, Freije A, Ruiz M, Coulon V, Sanz JR, Chiesa J, et al. A mitosis block links active cell cycle with human epidermal differentiation and results in endoreplication. PLoS One 2010; 5:e15701; http://dx.doi.org/10.1371/journal.pone.0015701; PMID: 21187932
- Harvat BL, Wang A, Seth P, Jetten AM. Up-regulation of p27Kip1, p21WAF1/Cip1 and p16Ink4a is associated with, but not sufficient for, induction of squamous differentiation. J Cell Sci 1998; 111:1185 - 96; PMID: 9547295
- Di Cunto F, Topley G, Calautti E, Hsiao J, Ong L, Seth PK, et al. Inhibitory function of p21Cip1/WAF1 in differentiation of primary mouse keratinocytes independent of cell cycle control. Science 1998; 280:1069 - 72; http://dx.doi.org/10.1126/science.280.5366.1069; PMID: 9582119
- Hauser PJ, Agrawal D, Pledger WJ. Primary keratinocytes have an adhesion dependent S phase checkpoint that is absent in immortalized cell lines. Oncogene 1998; 17:3083 - 92; http://dx.doi.org/10.1038/sj.onc.1202235; PMID: 9872324
- Pierce AM, Gimenez-Conti IB, Schneider-Broussard R, Martinez LA, Conti CJ, Johnson DG. Increased E2F1 activity induces skin tumors in mice heterozygous and nullizygous for p53. Proc Natl Acad Sci USA 1998; 95:8858 - 63; http://dx.doi.org/10.1073/pnas.95.15.8858; PMID: 9671769
- Robles AI, Larcher F, Whalin RB, Murillas R, Richie E, Gimenez-Conti IB, et al. Expression of Cyclin D1 in epithelial tissues of transgenic mice results in epidermal hyperproliferation and severe thymic hyperplasia. Proc Natl Acad Sci USA 1996; 93:7634 - 8; http://dx.doi.org/10.1073/pnas.93.15.7634; PMID: 8755527
- Rodriguez-Puebla ML, LaCava M, Miliani De Marval PL, Jorcano JL, Richie ER, Conti CJ. Cyclin D2 overexpression in transgenic mice induces thymic and epidermal hyperplasia whereas Cyclin D3 expression results only in epidermal hyperplasia. Am J Pathol 2000; 157:1039 - 50; http://dx.doi.org/10.1016/S0002-9440(10)64616-0; PMID: 10980142
- Alkhalaf M, Ganguli G, Messaddeq N, Le Meur M, Wasylyk B. MDM2 overexpression generates a skin phenotype in both wild type and p53 null mice. Oncogene 1999; 18:1419 - 34; http://dx.doi.org/10.1038/sj.onc.1202448; PMID: 10050879
- Miliani de Marval PL, Gimenez-Conti IB, LaCava M, Martinez LA, Conti CJ, Rodriguez-Puebla ML. Transgenic expression of Cyclin-dependent kinase 4 results in epidermal hyperplasia, hypertrophy, and severe dermal fibrosis. Am J Pathol 2001; 159:369 - 79; http://dx.doi.org/10.1016/S0002-9440(10)61703-8; PMID: 11438484
- Macias E, Miliani de Marval PL, Senderowicz A, Cullen J, Rodriguez-Puebla ML. Expression of CDK4 or CDK2 in mouse oral cavity is retained in adult pituitary with distinct effects on tumorigenesis. Cancer Res 2008; 68:162 - 71; http://dx.doi.org/10.1158/0008-5472.CAN-07-2461; PMID: 18172308
- Régnier M, Vaigot P, Darmon M, Pruniéras M. Onset of epidermal differentiation in rapidly proliferating basal keratinocytes. J Invest Dermatol 1986; 87:472 - 6; http://dx.doi.org/10.1111/1523-1747.ep12455517; PMID: 2428884
- Watt FM, Lo Celso C, Silva-Vargas V. Epidermal stem cells: an update. Curr Opin Genet Dev 2006; 16:518 - 24; http://dx.doi.org/10.1016/j.gde.2006.08.006; PMID: 16919447
- Kartasova T, Roop DR, Yuspa SH. Relationship between the expression of differentiation-specific keratins 1 and 10 and cell proliferation in epidermal tumors. Mol Carcinog 1992; 6:18 - 25; http://dx.doi.org/10.1002/mc.2940060105; PMID: 1380247
- Paramio JM, Casanova ML, Segrelles C, Mittnacht S, Lane EB, Jorcano JL. Modulation of cell proliferation by cytokeratins K10 and K16. Mol Cell Biol 1999; 19:3086 - 94; PMID: 10082575
- Santos M, Bravo A, López C, Paramio JM, Jorcano JL. Severe abnormalities in the oral mucosa induced by suprabasal expression of epidermal keratin K10 in transgenic mice. J Biol Chem 2002; 277:35371 - 7; http://dx.doi.org/10.1074/jbc.M205143200; PMID: 12119299
- Rosa-Garrido M, Ceballos L, Alonso-Lecue P, Abraira C, Delgado MD, Gandarillas A. A cell cycle role for the epigenetic factor CTCF-L/BORIS. PLoS One 2012; 7:e39371; http://dx.doi.org/10.1371/journal.pone.0039371; PMID: 22724006
- Lilly MA, Duronio RJ. New insights into cell cycle control from the Drosophila endocycle. Oncogene 2005; 24:2765 - 75; http://dx.doi.org/10.1038/sj.onc.1208610; PMID: 15838513
- Freije A, Ceballos L, Coisy M, Barnes L, Rosa M, De Diego E, et al. Cyclin E drives human keratinocyte growth into differentiation. Oncogene 2012; http://dx.doi.org/10.1038/onc.2012.22; PMID: 22349815
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science 2008; 319:1352 - 5; http://dx.doi.org/10.1126/science.1140735; PMID: 18323444
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006; 444:638 - 42; http://dx.doi.org/10.1038/nature05327; PMID: 17136094
- Osborn AJ, Elledge SJ, Zou L. Checking on the fork: the DNA-replication stress-response pathway. Trends Cell Biol 2002; 12:509 - 16; http://dx.doi.org/10.1016/S0962-8924(02)02380-2; PMID: 12446112
- Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011; 145:435 - 46; http://dx.doi.org/10.1016/j.cell.2011.03.044; PMID: 21529715
- Dazard JE, Piette J, Basset-Seguin N, Blanchard JM, Gandarillas A. Switch from p53 to MDM2 as differentiating human keratinocytes lose their proliferative potential and increase in cellular size. Oncogene 2000; 19:3693 - 705; http://dx.doi.org/10.1038/sj.onc.1203695; PMID: 10949923
- Jones PH, Harper S, Watt FM. Stem cell patterning and fate in human epidermis. Cell 1995; 80:83 - 93; http://dx.doi.org/10.1016/0092-8674(95)90453-0; PMID: 7813021
- Meyers J, Craig J, Odde DJ. Potential for control of signaling pathways via cell size and shape. Curr Biol 2006; 16:1685 - 93; http://dx.doi.org/10.1016/j.cub.2006.07.056; PMID: 16950104
- Moseley JB, Mayeux A, Paoletti A, Nurse P. A spatial gradient coordinates cell size and mitotic entry in fission yeast. Nature 2009; 459:857 - 60; http://dx.doi.org/10.1038/nature08074; PMID: 19474789
- Lechler T, Fuchs E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature 2005; 437:275 - 80; http://dx.doi.org/10.1038/nature03922; PMID: 16094321
- Hauser P, Ma L, Agrawal D, Haura E, Cress WD, Pledger WJ. Efficient down-regulation of Cyclin A-associated activity and expression in suspended primary keratinocytes requires p21(Cip1). Mol Cancer Res 2004; 2:96 - 104; PMID: 14985466
- Niculescu AB 3rd, Chen X, Smeets M, Hengst L, Prives C, Reed SI. Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol 1998; 18:629 - 43; PMID: 9418909
- Medema RH, Klompmaker R, Smits VA, Rijksen G. p21waf1 can block cells at two points in the cell cycle, but does not interfere with processive DNA-replication or stress-activated kinases. Oncogene 1998; 16:431 - 41; http://dx.doi.org/10.1038/sj.onc.1201558; PMID: 9484832
- Ullah Z, Lee CY, Depamphilis ML. Cip/Kip Cyclin-dependent protein kinase inhibitors and the road to polyploidy. Cell Div 2009; 4:10; http://dx.doi.org/10.1186/1747-1028-4-10; PMID: 19490616
- Muñoz-Alonso MJ, Acosta JC, Richard C, Delgado MD, Sedivy J, León J. p21Cip1 and p27Kip1 induce distinct cell cycle effects and differentiation programs in myeloid leukemia cells. J Biol Chem 2005; 280:18120 - 9; http://dx.doi.org/10.1074/jbc.M500758200; PMID: 15746092
- Muñoz-Alonso MJ, Ceballos L, Bretones G, Frade P, León J, Gandarillas A. MYC accelerates p21CIP-induced megakaryocytic differentiation involving early mitosis arrest in leukemia cells. J Cell Physiol 2012; 227:2069 - 78; http://dx.doi.org/10.1002/jcp.22935; PMID: 21769863
- García P, Frampton J, Ballester A, Calés C. Ectopic expression of Cyclin E allows non-endomitotic megakaryoblastic K562 cells to establish re-replication cycles. Oncogene 2000; 19:1820 - 33; http://dx.doi.org/10.1038/sj.onc.1203494; PMID: 10777216
- Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P. Drosophila myc regulates cellular growth during development. Cell 1999; 98:779 - 90; http://dx.doi.org/10.1016/S0092-8674(00)81512-3; PMID: 10499795
- Polakowska RR, Piacentini M, Bartlett R, Goldsmith LA, Haake AR. Apoptosis in human skin development: morphogenesis, periderm, and stem cells. Dev Dyn 1994; 199:176 - 88; http://dx.doi.org/10.1002/aja.1001990303; PMID: 7517223
- Gandarillas A. Epidermal differentiation, apoptosis, and senescence: common pathways?. Exp Gerontol 2000; 35:53 - 62; http://dx.doi.org/10.1016/S0531-5565(99)00088-1; PMID: 10705039
- Beer S, Zetterberg A, Ihrie RA, McTaggart RA, Yang Q, Bradon N, et al. Developmental context determines latency of MYC-induced tumorigenesis. PLoS Biol 2004; 2:e332; http://dx.doi.org/10.1371/journal.pbio.0020332; PMID: 15455033
- Conacci-Sorrell M, Ngouenet C, Eisenman RN. Myc-nick: a cytoplasmic cleavage product of Myc that promotes alpha-tubulin acetylation and cell differentiation. Cell 2010; 142:480 - 93; http://dx.doi.org/10.1016/j.cell.2010.06.037; PMID: 20691906
- Uribesalgo I, Benitah SA, Di Croce L. From oncogene to tumor suppressor: the dual role of Myc in leukemia. Cell Cycle 2012; 11:1757 - 64; http://dx.doi.org/10.4161/cc.19883; PMID: 22510570
- Wang X, Kua HY, Hu Y, Guo K, Zeng Q, Wu Q, et al. p53 functions as a negative regulator of osteoblastogenesis, osteoblast-dependent osteoclastogenesis, and bone remodeling. J Cell Biol 2006; 172:115 - 25; http://dx.doi.org/10.1083/jcb.200507106; PMID: 16380437
- Jiang D, Lenardo MJ, Zúñiga-Pflücker JC. p53 prevents maturation to the CD4+CD8+ stage of thymocyte differentiation in the absence of T cell receptor rearrangement. J Exp Med 1996; 183:1923 - 8; http://dx.doi.org/10.1084/jem.183.4.1923; PMID: 8666950
- Sulic S, Panic L, Barkic M, Mercep M, Uzelac M, Volarevic S. Inactivation of S6 ribosomal protein gene in T lymphocytes activates a p53-dependent checkpoint response. Genes Dev 2005; 19:3070 - 82; http://dx.doi.org/10.1101/gad.359305; PMID: 16357222
- Feinstein E, Gale RP, Reed J, Canaani E. Expression of the normal p53 gene induces differentiation of K562 cells. Oncogene 1992; 7:1853 - 7; PMID: 1501893
- Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci USA 2010; 107:9660 - 4; http://dx.doi.org/10.1073/pnas.1002298107; PMID: 20457898
- Galluzzi L, Kepp O, Kroemer G. TP53 and MTOR crosstalk to regulate cellular senescence. Aging (Albany NY) 2010; 2:535 - 7; PMID: 20876940
- Ren Z, Ponten F, Nister M, Ponten J. Reconstruction of the two-dimensional distribution of p53 positive staining patches in sun-exposed morphologically normal skin. Int J Oncol 1997; 11:111 - 5; PMID: 21528187
- Wang J, Sun Q, Morita Y, Jiang H, Gross A, Lechel A, et al. A differentiation checkpoint limits hematopoietic stem cell self-renewal in response to DNA damage. Cell 2012; 148:1001 - 14; http://dx.doi.org/10.1016/j.cell.2012.01.040; PMID: 22385964
- Ullah Z, de Renty C, DePamphilis ML. Checkpoint kinase 1 prevents cell cycle exit linked to terminal cell differentiation. Mol Cell Biol 2011; 31:4129 - 43; http://dx.doi.org/10.1128/MCB.05723-11; PMID: 21791608
- Puri PL, Bhakta K, Wood LD, Costanzo A, Zhu J, Wang JY. A myogenic differentiation checkpoint activated by genotoxic stress. Nat Genet 2002; 32:585 - 93; http://dx.doi.org/10.1038/ng1023; PMID: 12415271
- Simonatto M, Giordani L, Marullo F, Minetti GC, Puri PL, Latella L. Coordination of cell cycle, DNA repair and muscle gene expression in myoblasts exposed to genotoxic stress. Cell Cycle 2011; 10:2355 - 63; http://dx.doi.org/10.4161/cc.10.14.15948; PMID: 21685725
- Bar-Sagi D, Feramisco JR. Microinjection of the ras oncogene protein into PC12 cells induces morphological differentiation. Cell 1985; 42:841 - 8; http://dx.doi.org/10.1016/0092-8674(85)90280-6; PMID: 2996779
- Bagli DJ, D’Emilia JC, Summerhayes IC, Steele GD, Barlozzari T. c-Ha-ras-I oncogene-induced differentiation and natural killer cell resistance in a human colorectal carcinoma cell line. Cancer Res 1990; 50:2518 - 23; PMID: 2180571
- Celano P, Berchtold CM, Mabry M, Carroll M, Sidransky D, Casero RA Jr., et al. Induction of markers of normal differentiation in human colon carcinoma cells by the v-rasH oncogene. Cell Growth Differ 1993; 4:341 - 7; PMID: 8494794
- Feng Y, Bommer GT, Zhao J, Green M, Sands E, Zhai Y, et al. Mutant KRAS promotes hyperplasia and alters differentiation in the colon epithelium but does not expand the presumptive stem cell pool. Gastroenterology 2011; 141:1003 - 13, e1-10; http://dx.doi.org/10.1053/j.gastro.2011.05.007; PMID: 21699772
- Boukamp P. Non-melanoma skin cancer: what drives tumor development and progression?. Carcinogenesis 2005; 26:1657 - 67; http://dx.doi.org/10.1093/carcin/bgi123; PMID: 15905207