Abstract
Protein arginine methylation is catalyzed by protein arginine methyltransferases (PRMTs) and plays an important role in many cellular processes. Aberrant PRMT expression has been observed in several common cancer types; however, their precise contribution to the cell transformation process is not well understood. We previously reported that the PRMT1 gene generates several alternatively spliced isoforms, and our initial biochemical characterization of these isoforms revealed that they exhibit distinct substrate specificity and subcellular localization. We focus here on the PRMT1v2 isoform, which is the only predominantly cytoplasmic isoform, and we have found that its relative expression is increased in breast cancer cell lines and tumors. Specific depletion of PRMT1v2 using RNA interference caused a significant decrease in cancer cell survival due to an induction of apoptosis. Furthermore, depletion of PRMT1v2 in an aggressive cancer cell line significantly decreased cell invasion. We also demonstrate that PRMT1v2 overexpression in a non-aggressive cancer cell line was sufficient to render them more invasive. Importantly, this novel activity is specific to PRMT1v2, as overexpression of other isoforms did not enhance invasion. Moreover, this activity requires both proper subcellular localization and methylase activity. Lastly, PRMT1v2 overexpression altered cell morphology and reduced cell-cell adhesion, a phenomenon that we convincingly linked with reduced β-catenin protein expression. Overall, we demonstrate a specific role for PRMT1v2 in breast cancer cell survival and invasion, underscoring the importance of identifying and characterizing the distinct functional differences between PRMT1 isoforms.
Introduction
Arginine methylation, a common posttranslational modification, plays an important role in many cellular processes, including signal transduction, pre-mRNA splicing, DNA repair and intracellular localization of proteins.Citation1 The family of enzymes that catalyze this modification are called protein arginine methyltransferases (PRMTs). There are eight well-characterized PRMTs that are divided into groups based on the type of arginine methylation reaction they catalyze.Citation1,Citation2 Type I PRMTs (PRMT1, 2, 3, 4, 6, 8) catalyze asymmetric dimethylation, while type II PRMTs (PRMT5) catalyze symmetric dimethylation, and type III (PRMT7) generate monomethylarginines.Citation3 The list of arginine methylated protein substrates is constantly growing, and with it the discovery of new functions of PRMTs, as well as their potential involvement in human diseases.Citation4-Citation6
Aberrant expression of PRMTs has been observed in several cancers, including breast, lung, colorectal, bladder and leukemia.Citation7-Citation15 PRMT4/CARM1 expression is elevated in prostate and breast cancer, and it is thought to play a role in the regulation of hormone-dependent proliferation.Citation8,Citation16 Additionally, PRMT5 expression is upregulated in mantle cell lymphoma and enhances anchorage-independent cell growth.Citation14,Citation17 PRMT1 has been shown to be overexpressed in colorectal cancer and, along with PRMT6, is overexpressed in lung and bladder cancers.Citation11-Citation13 We and others have shown that PRMT1 mRNA and protein expression is elevated in breast cancer cells and tumors.Citation7,Citation9,Citation18 Functionally, PRMT1 has been shown to be an important component of a novel oncogenic transcriptional complex with mixed lineage leukemia (MLL) and can regulate MLL-mediated cell transformation.Citation10 PRMT1 can also methylate the estrogen receptor (ER) in breast cancer cells, which promotes ER extranuclear survival signaling and interaction with mediators of cell migration.Citation19 These studies show that PRMTs may have a role in cancer; however, their functional contributions to cancer development and progression are not well characterized.
Our previous work demonstrated that the PRMT1 gene can produce several isoforms through complex alternative splicing in the 5′end of the pre-mRNA.Citation7 Each has a unique N-terminal sequence and slightly different molecular weight. Prior studies have neglected to examine the specific functional contributions of these isoforms and have focused mainly on the most abundant isoform, PRMT1v1. Each isoform has distinct characteristics regarding enzymatic activity, substrate specificity and subcellular localization and therefore are predicted to be functionally different. PRMT1v2 is the only isoform with almost exclusively cytoplasmic expression. Sequence analysis showed that PRMT1v2 retains exon 2 in its coding sequence, a 54-base pair (bp) sequence that encodes an 18-amino acid insert in the protein. Importantly, within this sequence is a leucine-rich, CRM1-dependent, nuclear export sequence (NES; 15VATLANGMSLCitation24) that, when mutated, caused nuclear retention.Citation7 Furthermore, we investigated the expression of the isoforms identified, and found that while the overall expression of PRMT1 is elevated in breast cancer cells, PRMT1v2 had the greatest increase relative to the most abundant isoform, PRMT1v1.
Here we describe a novel role for the PRMT1v2 isoform in cancer cell growth, survival and invasion. This is the first study to evaluate the function of a specific PRMT1 isoform in cancer cells. We show that specific depletion of PRMT1v2 using RNA interference resulted in an induction of apoptosis and also decreased the invasiveness of an aggressive breast cancer cell line. We also show that overexpression of PRMT1v2 in a non-aggressive breast cancer cell line causes them to be more invasive. This occurs through the repression of β-catenin protein expression, since restoration of β-catenin expression levels inhibits PRMT1v2-induced invasion. This data uncovers isoform-specific functions of PRMT1 and shows a novel role for PRMT1v2 in promoting cancer cell survival and invasion with a potential implication for breast cancer.
Results
PRMT1v2 depletion affects breast cancer cell viability and growth
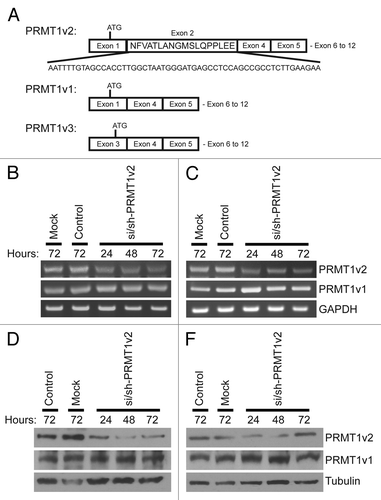
To assess the role of PRMT1v2 in breast cancer cells, we used RNA interference to reduce PRMT1v2 expression. Previously, we demonstrated that targeting an siRNA within the 54-base pair sequence of exon 2 effectively and specifically reduced PRMT1v2 expression in HeLa cells ().Citation7 However, efficient protein depletion required co-transfection of an siRNA duplex with an shRNA expression plasmid, both targeting the same sequence within exon 2 (Fig. S1A and B). Thus, we first wanted to test this approach in breast cancer cell lines. MCF7 cells were mock transfected, transfected with a non-targeting siRNA (control) or co-transfected with si/sh-PRMT1v2. Additionally, we generated a PRMT1v2-specific antibody raised against a peptide sequence of exon 2. Characterization of this antibody by western blot analysis of purified proteins confirmed that it exclusively detects PRMT1v2 (Fig. S1C). Compared with a pan-PRMT1 antibody raised against the C terminus of the protein,Citation20 it detects a single band corresponding to PRMT1v2 (~42.5 kDa) in MCF7 cells (Fig. S1D). We thus used this antibody for the detection of PRMT1v2 in our experiments. Efficient depletion of PRMT1v2 mRNA and protein expression was observed following 24 h of transfection using this approach (, respectively). Comparable knockdown efficiency was also observed in other distinct breast cancer cell lines, T47D (), Hs578T (Fig. S1E) and MDA-MB-231 (Fig. S1F). Reduced expression persisted for up to 72 h post-transfection (the longest time point examined); however, a gradual recovery of protein expression was often observed at 48 and 72 h. No reduction in PRMT1v1 mRNA or protein expression was seen in either MCF7 or T47D cells using this strategy.
Figure 1. Specific depletion of PRMT1v2. Schematic of PRMT1v2, PRMT1v1 and PRMT1v3 coding exon structure (A). Alternative splicing causes inclusion of exon 2 in the PRMT1v2 coding sequence. Total RNA was collected from MCF7 and T47D cells transfected with si/sh-PRMT1v2 at 24, 48 and 72 h post-transfection and mock and control siRNA (control) transfected cells at 72 h. PCR analysis of cDNA generated from total RNA using PRMT1 primers shows depletion of PRMT1v2 mRNA with no effect on PRMT1v1 in MCF7 (B) and T47D (C) cells. GAPDH serves as a loading control. Total protein lysates were collected from MCF7 and T47D cells transfected with si/sh-PRMT1v2 at 24, 48 and 72 h post-transfection and from mock and control transfected cells at 72 h. Western blot analysis using a PRMT1v2-specific antibody shows effective depletion of protein expression in MCF7 (D) and T47D (E) cells. No effect on PRMT1v1 protein expression was observed. Tubulin serves as a loading control.
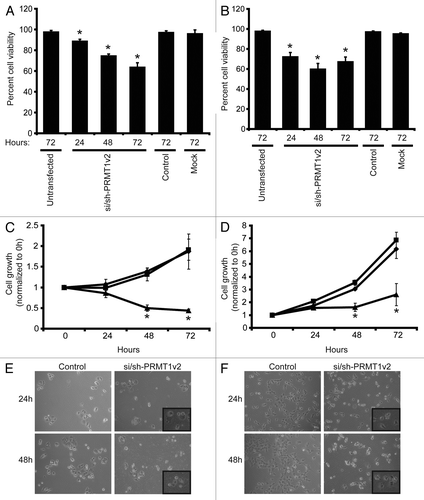
To study the effects of PRMT1v2 depletion on cancer cell growth and viability, MCF7, T47D, Hs578T and MDA-MB-231 breast cancer cells were transfected with si/sh-PRMT1v2 and assessed 24, 48 and 72 h post-transfection. PRMT1v2 depletion caused a significant decrease in live cell numbers in MCF7 (), T47D () and Hs578T cell lines (Fig. S2A), as determined by viable cell counts using Trypan blue exclusion. MTT assays were also used to follow cell growth over time. PRMT1v2 depletion caused a significant reduction in the cell growth of all four cell lines assessed (; Fig. S2B and C). No change in cell viability was observed whether control transfections were done using a non-targeting siRNA alone or this non-targeting siRNA co-transfected with a plasmid containing a non-targeting shRNA (Fig. S2D). PRMT1v2 depletion caused increased numbers of rounded cells and cells that exhibited membrane blebbing, morphologies, consistent with apoptotic cell death (; Fig. S2E).
Figure 2. Depletion of PRMT1v2 affects breast cancer cell viability and growth. Viable cell numbers were determined by viable cell counts using trypan blue exclusion 24, 48 and 72 h following mock, control or si/sh-PRMT1v2 transfection in MCF7 (A) and T47D (B) cells. Data are expressed as a percentage of viable cell counts to total cell counts and are the mean ± standard error of three independent experiments performed in triplicate (*p < 0.05). MCF7 (C) and T47D (D) cells were plated at equal numbers and then mock (♦), control (■) or si/sh-PRMT1v2 transfected (▲) and assessed by MTT assay at 0, 24, 48 and 72 h post-transfection. Data are the mean ± standard error of three independent experiments with six replicates per experiment (*p < 0.05). Morphology of control and si/sh-PRMT1v2 transfected MCF7 (E) and T47D (F) cells 24 and 48 h post-transfection. Images were taken at 20X magnification with insets focused on an area in the field of view.
Depletion of PRMT1v2 induces apoptosis
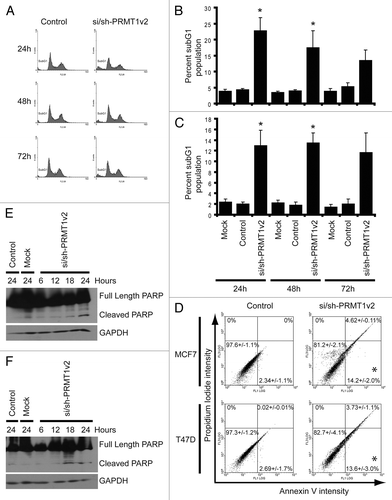
Flow cytometry was used to assess the effects of PRMT1v2 depletion on apoptosis. Analysis of DNA content showed that PRMT1v2 depletion caused a significant increase in DNA fragmentation as indicated by the presence of a subG1 peak, often indicative of apoptotic cell death, in both MCF7 () and T47D cells (; Fig. S3B) at 24 and 48 h. At 72 h, there is still a detectable subG1 peak; however, in both cell lines, the effect at this time point is not statistically significant. This reduced effect on cell death at 72 h is consistent with the gradual recovery of PRMT1v2 expression over time. Annexin V/propidium iodide (PI) co-staining was used to further validate the apoptotic phenotype following PRMT1v2 depletion. Cells were assessed after 24 h of PRMT1v2 depletion as this time point had the most efficient reduction in protein expression. MCF7 cells and T47D cells showed a significant increase in Annexin V-positive cells and a negligible PI-only-positive population (; Fig. S3D), consistent with apoptotic cell death.Citation21 Poly-ADP ribose polymerase (PARP) cleavage, an early apoptosis marker, was also observed in both MCF7 and T47D cells following PRMT1v2 depletion (, respectively). PARP cleavage likely occurs via caspase 7 or 9 in MCF7 cells, as they are caspase 3-deficient.Citation22,Citation23 These results demonstrate that inhibition of PRMT1v2 expression induces apoptosis, suggesting that its upregulation contributes to their inherent survival mechanism.
Figure 3. Depletion of PRMT1v2 induces apoptosis. Representative flow cytometric analyses of propidium iodide (PI)-stained MCF7 cells following control or si/sh-PRMT1v2 transfection at 24, 48 and 72 h (A). Percentage of subG1 population for MCF7 (B) and T47D (C) cells. Data are the mean ± standard error of five (for MCF7) and four (for T47D) independent experiments (*p < 0.05). Representative flow cytometric analyses of Annexin V and PI co-staining of MCF7 and T47D cells following control or si/sh-PRMT1v2 transfection for 24 h (D). Percentages in each quadrant represent the mean ± standard error of three independent experiments (*p < 0.05). Total protein lysates were collected from MCF7 and T47D cells that were mock and control transfected for 24 h or transfected with si/sh-PRMT1v2 for 6, 12, 18 and 24 h. Western blot analysis for the expression of PARP shows the appearance of its cleavage product in MCF7 (E) and T47D (F) cells transfected with si/sh-PRMT1v2. GAPDH was used as a loading control.
Depletion of PRMT1v2 inhibits breast cancer cell motility and invasion
The ability of cancer cells to metastasize begins with an enhancement of both their motility and their ability to invade through the extracellular matrix of surrounding tissues. To examine the role of PRMT1v2 in cancer cell motility and invasion we used a highly invasive breast cancer cell line, MDA-MB-231.Citation24 PRMT1v2 expression was specifically and effectively reduced in these cells using RNA interference as described above (). Cell motility was assessed using Transwell chambers. In these assays, following 24 h of PRMT1v2 depletion, control cells and cells depleted of PRMT1v2 are counted and replated at equal numbers into Transwell chambers. After an additional 24 h of incubation, cells that passed through the chambers are fixed, stained and counted. As expected, mock and control transfected MDA-MB-231 cells migrated very efficiently across the chamber membranes (). Depletion of PRMT1v2 in MDA-MB-231 cells resulted in a 36% decrease in the number of cells that crossed the chamber membrane, strongly suggesting a reduction in cell motility.
Figure 4. Depletion of PRMT1v2 inhibits breast cancer cell motility and invasion. MDA-MB-231 cells depleted of PRMT1v2 were assessed for effects on motility and invasion. Western analysis for PRMT1v2 and PRMT1v1 following 24 h depletion (A). GAPDH serves as a loading control. Following 24 h of PRMT1v2 depletion, cells replated at equal numbers into Transwell chambers and incubated for an additional 24 h. Motility was analyzed using Transwell chambers without a Matrigel (- Matrigel). Invasion was analyzed using Transwell chambers containing a Matrigel layer (+ Matrigel). Representative images of cells that have passed through the Transwell chamber -/+ Matrigel at 20X magnification (B). Cell numbers that passed through the Transwell chambers in the absence (C: motile cells/field) or presence (D: invasive cells/field) of Matrigel were determined. Data represents the mean ± standard error of three independent experiments (*p < 0.05 comparing to control).
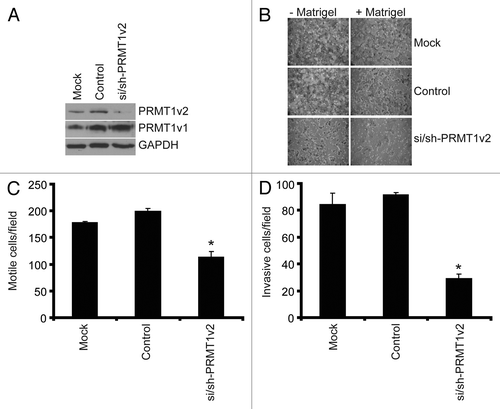
To determine the effects of PRMT1v2 on cell invasiveness (their motility combined with their ability to penetrate through an extracellular matrix-like physical barrier), the effects of PRMT1v2 depletion on the ability of cells to migrate through Transwell chamber membranes coated with Matrigel was also assayed. Strikingly, depletion of PRMT1v2 resulted in a 65% decrease in the ability of MDA-MB-231 cells to invade through the Matrigel-containing chambers (). Together, these results show that PRMT1v2 expression in this aggressive breast cancer cell line contributes significantly to its highly motile and invasive capacity, as determined using this in vitro assay.
Overexpression of PRMT1v2 promotes cancer cell motility and invasion
Since it is difficult to rule out and/or measure precisely the contribution of PRMT1v2 depletion-induced cell death in our Transwell assays, we decided to assess the effects of PRMT1v2 overexpression on cell motility and invasion to corroborate our observations. To investigate the effects of overexpression of PRMT1v2 on cell motility and invasion, we used MCF7 cells, which are a “luminal epithelial-like” breast cancer cell line, considered to be weakly invasive.Citation24 We generated cells that stably expressed GFP-tagged PRMT1v2. Stable cell lines expressing GFP and GFP-tagged PRMT1v1 were established as controls and for comparison. Exogenous expression levels were comparable for PRMT1v2-GFP and PRMT1v1-GFP (; Fig. S4A). Consistent with previous reports,Citation7,Citation25,Citation26 PRMT1v2-GFP expression was predominantly cytoplasmic, while PRMT1v1-GFP was mainly nuclear with lower cytoplasmic expression levels (Fig. S4B). As predicted from the literature, MCF7 cells displayed a low propensity to migrate through Transwell chamber membranes, even in the absence of Matrigel (). Interestingly, stable overexpression of PRMT1v2-GFP or PRMT1v1-GFP induced a significant increase in cell motility in Transwell chamber assays [3.2-fold increase with PRMT1v2 and 3.6-fold increase with PRMT1v1 ()]. Similarly, in “scratch wound” assays, cells stably expressing PRMT1v2-GFP or PRMT1v1-GFP had similar rates of wound closure, and both closed the wound significantly faster than control cells (Fig. S4C and D), reinforcing that our Transwell assays were indeed a representation of cell motility. While we do observe an increased growth rate in PRMT1v2-GFP-expressing cells after 6 d compared with control (Fig. S4E), the effect on cell growth after 72 h is negligible and thus should not significantly contribute to the effects on motility observed here.
Figure 5. Overexpression of PRMT1v2 in non-invasive breast cancer cells enhances invasion. Total RNA was collected from MCF7 cells stably expressing GFP, GFP-tagged PRMT1v1 (PRMT1v1-GFP) and GFP-tagged PRMT1v2 (PRMT1v2-GFP). PCR analysis of cDNA generated from total RNA using PRMT1 primers (A). GAPDH serves as a loading control. Total protein lysates from MCF7 cells stably expressing GFP, PRMT1v1-GFP or PRMT1v2-GFP were analyzed by western blotting using an anti-GFP antibody (B). Tubulin serves as a loading control. MCF7 stably expressing GFP, PRMT1v1-GFP or PRMT1v2-GFP were analyzed for motility and invasion using Transwell chambers without (- Matrigel) or with a Matrigel layer (+ Matrigel). Cells were plated into Transwell chambers at the same density and the numbers of cells that crossed the chamber membrane was counted after 72 h. Representative images of cells that have passed through the Transwell chamber -/+ Matrigel at 20X magnification (C). Cell numbers that passed through the chamber mebranes without a Matrigel layer (D: motile cells/field) or containing a Matrigel layer (E: invasive cells/field) were counted. Data represents the mean ± standard error of four independent experiments (*p < 0.05 comparing to GFP and #p < 0.05 comparing to PRMT1v1-GFP). MCF7 cells were transiently transfected with plasmid constructs containing a C-terminal EGFP-tagged PRMT1v1, PRMT1v2, PRMT1v3, PRMT1v2 NES mutant or a PRMT1v2 catalytically inactive methylase mutant for 24 h. Western analysis for GFP shows expression of each PRMT1 isoform and PRMT1v2 mutants in MCF7 cells 24 h post transfection (F). To examine invasion, following 24 h transfection, cells were seeded into Matrigel containing Transwell chambers and incubated for 72 h. Cell numbers that passed through the chamber membranes were counted (G). Data represents the mean ± standard error of three independent experiments (*p < 0.05 comparing to control, #p < 0.05 comparing to PRMT1v2).
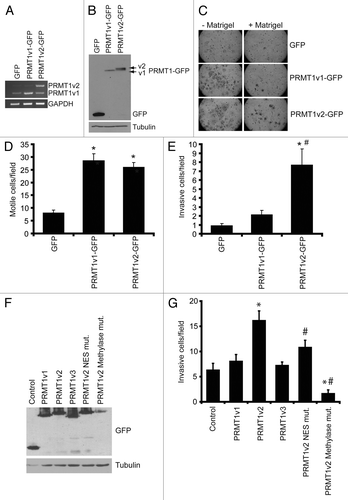
Strikingly, when we assessed invasion through Matrigel as a measure of in vitro metastatic potential, only stable PRMT1v2-GFP expression significantly increased the number of cells that invaded through Matrigel-coated membranes (6.2-fold compared with GFP alone; ). PRMT1v1-GFP expression did not cause a significant increase in the number of invading cells (; 1.9-fold, p = 0.06). Since these results were obtained using stable cell lines and may reflect differences arising during selection, we next assessed if transient overexpression would yield the same effects. Transient overexpression of PRMT1v2 in MCF7 cells also significantly enhanced their invasion, 2.5-fold compared with a GFP control transfection (). Importantly, no effect on invasion was observed with transient overexpression of either PRMT1v1 or an additional alternatively spliced isoform, PRMT1v3 (). To determine if cytoplasmic localization and methyltransferase activity was required for PRMT1v2 to promote invasion, we generated two mutant forms, an NES mutant and a methylase-inactive mutant. The NES mutant was generated by point mutation of two conserved hydrophobic residues in the nuclear export sequence to alanines (V15A/L18A). Mutation of these residues has previously been shown to cause an increased amount of PRMT1v2 to be retained in the nucleus.Citation7 The second PRMT1v2 mutant was a methylase inactive mutant and was generated by mutating three key amino acid residues to alanines within the binding region for the methyl donor, S-adenosyl methionine (68VLD70→68AAA70). This results in a complete loss of enzymatic activity and accumulation of the mutated PRMT1v2 in the nucleus.Citation26,Citation27 Transient overexpression of either of the mutant forms of PRMT1v2 did not enhance MCF7 cell invasiveness (). In fact, the methylase-inactive mutant caused a significant decrease in invasiveness as compared with control. However, part of this effect may be due to an increased presence of cell death that we and others have observed to be associated with overexpression of this mutant.Citation26 We also observed a consistently lower level of expression for the methylase-inactive mutant (), suggesting that high expression levels are not well tolerated. These results suggest that proper localization and methylase activity are important for PRMT1v2 to promote breast cancer cell invasion. Altogether, these results suggest that there are important functional differences between the PRMT1 isoforms relating to cell invasion, and that PRMT1v2 specifically enhances the ability of breast cancer cells to penetrate through an extracellular matrix-like barrier, which is a necessary characteristic for their invasive and metastatic potential.
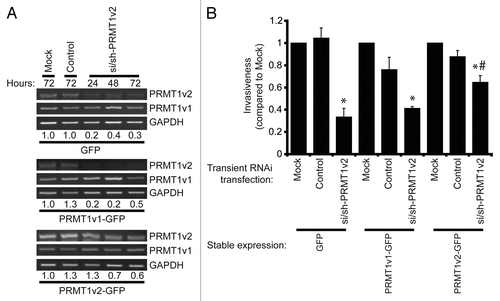
Lastly, to ensure that the observed effects were indeed due to the change in PRMT1v2 expression, we devised the following rescue experiment. We reasoned that stable overexpression of PRMT1v2 should confer resistance to the inhibition of invasion observed upon PRMT1v2 depletion using RNA interference. We first determined through PCR analysis of PRMT1v2 mRNA expression that MCF7 cells stably expressing PRMT1v2-GFP maintained higher levels of PRMT1v2 in the presence of si/sh-PRMT1v2 (). Following 24 h of PRMT1v2 depletion, cells were replated at equal numbers into Transwell chambers containing Matrigel and incubated for an additional 72 h. Although MCF7 cells are weakly invasive, and the numbers of cells that invade through Matrigel are very low, we were still able to observe a statistically significant inhibition of invasion in both GFP and PRMT1v1-GFP-expressing cells with PRMT1v2 depletion (). In contrast, stable overexpression of PRMT1v2 conferred partial resistance to the effect of the PRMT1v2 depletion (). Please note that although, as described above, stable overexpression of PRMT1v2-GFP does promote a significant increase in cell invasion relative to cells expressing GFP alone or PRMT1v1-GFP (6.2-fold and 3.3-fold, respectively). However, the values presented for this analysis were each independently normalized to their respective mock transfection. This allowed for a direct side-by-side comparison of the reduction in invasion upon PRMT1v2 depletion in each stable cell line. Altogether, these experiments demonstrate the specificity of our RNA interference approach, and show that changes in PRMT1v2 levels can influence the ability of breast cancer cells to invade through an extracellular matrix-like barrier.
Figure 6. Overexpression of PRMT1v2 partially rescues the inhibition of invasion resulting from PRMT1v2 depletion. MCF7 cells stably expressing GFP, PRMT1v1-GFP or PRMT1v2-GFP were transfected with si/sh-PRMT1v2. Total RNA was collected 24, 48 and 72 h post-si/sh-PRMT1v2 transfection and 72 h post-transfection for mock and control samples. PCR analysis of cDNA generated from total RNA using PRMT1 primers shows levels of PRMT1v2 and PRMT1v1 mRNA (A). Endogenous and exogenous PRMT1v2 levels are detected as a single band in this PCR analysis as the primers do not differentiate the two species. GAPDH serves as a loading control. The densitometry values (indicated below the respective lanes) for the samples in the representative PCRs were normalized to mock transfected cells. MCF7 cells stably expressing GFP, PRMT1v1-GFP or PRMT1v2-GFP were mock, control or si/sh-PRMT1v2 transfected for 24 h. Twenty-four hours post-transfection, cells were replated at equal numbers into Transwell chambers containing a Matrigel layer and incubated for 72 h. Cell numbers that passed through the chambers were counted (B). Data represents the mean ± standard error of three independent experiments (*p < 0.05 compared with matched control transfection, #p < 0.05 compared with MCF7 GFP transfected with si/sh-PRMT1v2).
Overexpression of PRMT1v2 alters breast cancer cell morphology
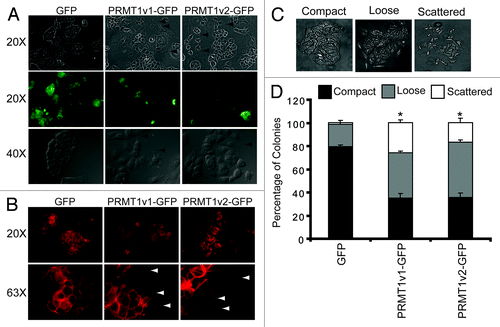
MCF7 cells are typically polygonal in shape and form organized compact colonies that have E-cadherin positive cell-cell junctions when grown in culture.Citation24,Citation28,Citation29 Upon close inspection of the colonies formed by cells stably expressing PRMT1v2-GFP, we noticed significant changes in cell morphology. Specifically, colonies formed by PRMT1v2-GFP-expressing cells appear disorganized, are less compact and have reduced E-cadherin membranous cell-cell junction staining compared with GFP-expressing controls (; Fig. S5A). Furthermore, many colonies appear to have dissociated completely and display filopodia, and small lamellipodia structures emanate from the membrane surfaces ( arrowheads), consistent with increased migratory properties.Citation30-Citation33 Lastly, F-actin, which is predominantly localized to cell-cell contacts in GFP-expressing control cells, is more dispersed through the cytoplasm and localized to the filopodia and lamellipodia structures at the edges of PRMT1v2-GFP-expressing cells (). These changes in colony structure were next quantified using a colony dispersion assay, comparing MCF7 cells stably expressing either GFP alone, PRMT1v1-GFP or PRMT1v2-GFP. This assay measures the ability of epithelial-like tumor cells to detach from colonies in culture mimicking a migratory phenotype.Citation29,Citation34,Citation35 Specifically, colonies were classified based on morphology as compact, loose or scattered (). Stable PRMT1v2-GFP expression caused a significant reduction (45%) in the number of compact colonies, and a concomitant increase in loose (28%) and scattered (15%) colony counts, compared with the GFP control (). PRMT1v1-GFP-expressing cells also showed a significant decrease in compact colonies (46%) and an increase in loose (19%) and scattered (25%) colony numbers. These changes in morphology are indicative of disrupted cell-cell adhesion,Citation35-Citation37 and are consistent with our observation that both the PRMT1v1 and PRMT1v2 isoforms can promote increased cell motility.
Figure 7. Overexpression of PRMT1v2 in MCF7 cells alters their morphology. Representative phase contrast and GFP fluorescence images of MCF7 cell stably expressing GFP, PRMT1v1-GFP or PRMT1v2-GFP proteins (A, arrowsheads indicate the presence of filopodia and lamellipodia, magnifications are indicated to the left). Fluorescence images of actin filament staining using FITC-conjugated phalloidin in MCF7 cells stably expressing GFP, PRMT1v1-GFP and PRMT1v2-GFP (B, arrowsheads indicate the presence of filopodia and lamellipodia, magnifications are indicated to the left). A colony dispersion assay was used to quantify the effects on colony formation. Representative images of compact, loose and scattered colonies (C). Distribution of compact, loose and scattered colonies in MCF7 cells stably expressing GFP, PRMT1v1-GFP or PRMT1v2-GFP (D). Ten thousand cells were plated and grown as described in ref. Citation29. Approximately 70 colonies were counted for each cell line per experiment. Colonies were scored by three independent investigators. Data are the mean ± standard error from four independent experiments (*p < 0.05, comparing compact colony numbers).
PRMT1v2 expression represses β-catenin expression causing increased cell invasion
The altered morphology observed with PRMT1v2-GFP expression indicated that disruption in cell-cell adhesion might be a mechanism through which PRMT1v2 promotes cell invasion. Adherens junctions (AJ) are critical protein complexes required for the maintenance of epithelial cell structure and polarity, and are involved in the retention of this morphology in MCF7 cells.Citation24,Citation29,Citation38 To assess the effects of stable PRMT1v2-GFP expression on cell-cell adhesion we examined the expression of two key AJ components, E-cadherin and β-catenin.Citation39-Citation41 No changes in E-cadherin mRNA or protein expression were observed with stable PRMT1v2-GFP overexpression (Fig. S5B). Nevertheless, reduced membranous cell-cell junction staining of both E-cadherin and β-catenin was observed in PRMT1v2-expressing cells compared with the GFP-expressing controls (Fig. S5A and C). Additionally, there also appears to be no obvious nuclear accumulation of β-catenin. Interestingly, stable PRMT1v2-GFP expression caused a significant reduction (58%) in β-catenin protein expression compared with cells expressing either GFP alone or PRMT1v1-GFP (). No difference in mRNA levels were observed between the three stable cell lines, suggesting that the effect of PRMT1v2 on β-catenin protein expression might be at the level of protein turnover. β-catenin protein stability is negatively regulated by phosphorylation,Citation2,Citation42,Citation43 therefore we assessed whether its phosphorylation status is altered in PRMT1v2-expressing cells and observed a lower level of phospho-β-catenin in PRMT1v2-GFP-expressing cells (). However, since these cells express a lower steady-state amount of total β-catenin, we determined the proportion of total β-catenin protein that is phosphorylated in each of the cell lines by calculating the phospho to total β-catenin ratio using densitometric analysis. This revealed that PRMT1v2-GFP-expressing cells have a significantly higher proportion of phosphorylated β-catenin compared with cells expressing GFP alone or PRMT1v1-GFP (). This strongly suggests that the lower levels of β-catenin protein expression observed in PRMT1v2-expressing cells are likely a consequence of phosphorylation-mediated degradation.
Figure 8. PRMT1v2 expression causes a decrease in β-catenin expression. Total RNA was collected from MCF7 cells stably expressing GFP, PRMT1v1-GFP or PRMT1v2-GFP. PCR analysis of cDNA generated from total RNA using β-catenin primers (A, mRNA). GAPDH serves as a loading control. Total protein lysates collected from MCF7 cells stably expressing GFP, PRMT1v1-GFP or PRMT1v2-GFP and were analyzed by western blotting for the expression of β-catenin protein (A, protein). Tubulin serves as a loading control. Densitometry of β-catenin protein expression normalized to loading control. Values are expressed relative to MCF7 GFP expressing cells (B). Data represents the mean ± standard error of seven independent experiments (*p < 0.01 comparing to GFP or PRMT1v1-GFP). Western blotting for levels of phosphorylated β-catenin using a phospho-specific antibody that recognizes phosphorylation at Ser33, Ser37 and Thr41 as well as total β-catenin and tubulin (C). Densitometry was used to determine the ratio of phosphorylated β-catenin to total β-catenin protein expression normalized to loading control (D). Values are expressed relative to MCF7 GFP-expressing cells. Data represents the mean ± standard error of three independent experiments (*p < 0.05 compared with GFP and PRMT1v1-GFP). MCF7 cells stably expressing GFP, PRMT1v1-GFP or PRMT1v2-GFP were transiently transfected with an expression plasmid containing flag-tagged β-catenin. Western blot analysis for flag 24 h post-transfection (E). Tubulin serves as a loading control. Following 24 h, transfection with flag-tagged β-catenin containing plasmid cells were replated at equal numbers in Transwell chambers containing a Matrigel layer and incubated for an additional 72 h. Cell numbers that passed through the chambers were counted (F). Data represents the mean ± standard error of three independent experiments (*p < 0.05 comparing to control, #p < 0.05 comparing to PRMT1v2 vector control).
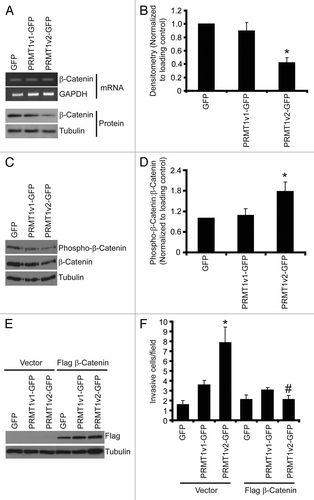
Since this reduction in β-catenin protein expression was specific to PRMT1v2-GFP-expressing cells, we reasoned that this might be linked to its unique property of promoting cell invasion. To determine if this repression of β-catenin protein expression contributes to the increase in cell invasion observed in cells stably expressing PRMT1v2-GFP, we overexpressed β-catenin by transient transfection. We first established that transient expression of a flag-tagged β-catenin protein could be observed at 24 h (). Therefore, following 24 h of transient expression of flag-tagged β-catenin protein, cells were replated into Transwell chambers coated with Matrigel at equal numbers and incubated for an additional 72 h. Remarkably, overexpression of β-catenin essentially abolished the effect of PRMT1v2-GFP on cell invasion, while it had no effect on cells expressing GFP alone or PRMT1v1-GFP (). Our results have uncovered a novel and unique pathway through which PRMT1v2 regulates β-catenin, thus promoting breast cancer cell invasion.
Discussion
In this study we describe a specific role for the alternatively spliced PRMT1 isoform, PRMT1v2, in promoting the survival and invasion of breast cancer cells. We demonstrate that inhibition of PRMT1v2 using RNA interference decreases cell survival through an induction of apoptosis. Moreover, we uncover a unique function of PRMT1v2 in promoting cell invasion through an extracellular matrix-like barrier. PRMT1v2 overexpression altered the morphology of non-aggressive cancer cells, causing a disruption in cell-cell adhesion and a reduction of β-catenin protein levels. Lastly, ectopic expression of β-catenin expression in cells expressing PRMT1v2 completely abrogated its effect on cell invasion, clearly establishing β-catenin as a component of this novel regulatory pathway present in breast cancer cells.
PRMT1v2 is generated as a result of alternative splicing in the 5′ end of the PRMT1 pre-mRNA, promoting inclusion of exon 2 in its translated sequence.Citation7,Citation44,Citation45 Inclusion of exon 2 in the PRMT1v2 sequence is a distinguishing feature that we have exploited to specifically downregulate its expression and to detect it in cells. It should be noted that exon 2 is also retained in the 5′-UTR of PRMT1v3 and PRMT1v5. However, a very low level of PRMT1v3 is present, and PRMT1v5 is virtually undetectable in the breast cancer cell lines used.Citation7 Dysregulated PRMT1 expression has been observed in several cancer types.Citation7-Citation15,Citation18 We previously reported that the mean expression of several PRMT1 isoforms is elevated in breast cancer cell lines; however, PRMT1v2 had the highest expression relative to the most abundant isoform, PRMT1v1. For this reason, we focused on PRMT1v2 in the current study. Recently, a clinical study evaluated the expression of PRMT1 isoforms, PRMT1v1, v2 and v3 in breast cancer tissues and established a correlation between PRMT1v1 mRNA expression and poor prognosis in breast cancer patients.Citation9 An immunohistochemical analysis of total PRMT1 protein (not differentiating specific isoforms) in breast tumor tissue sections showed increased expression mainly in the cytoplasm. However, we and others have shown that PRMT1v2 protein expression is predominantly localized to the cytoplasm,Citation7,Citation25,Citation26 so it is tempting to speculate that a significant fraction of PRMT1 protein detected in this study might in fact be PRMT1v2. Nevertheless, these results emphasize the importance of determining the specific functions of PRMT1 isoforms and their contribution to disease.
We found that the specific modulation of PRMT1v2 expression has a drastic impact on breast cancer cell growth and viability in vitro, even in the presence of unaffected expression levels of PRMT1v1, the most abundantly expressed isoform. More specifically, targeted depletion of PRMT1v2 by RNA interference in breast cancer cells caused a significant reduction in cell viability. This was observed in four different human breast cancer cell lines. Previous work has demonstrated that PRMT1 can affect growth and viability in several cell types, although the contribution of specific isoforms to these effects was never examined. Depletion of PRMT1 by RNA interference in osteosarcoma, lung and bladder cancer cells results in decreased cell growth.Citation13,Citation46 In breast cancer cells, depletion of PRMT1 decreased estrogen-induced PKB/Akt phosphorylation and activation and was accompanied by a decrease in cell proliferation.Citation19 In each of these studies, the decrease in cell growth as a result of PRMT1 depletion was attributed to growth arrest within the cell cycle, observed at G0/G1, G2/M or both.Citation13,Citation19,Citation46 We specifically show that the reduced cell growth and viability as a result of PRMT1v2 depletion is due to an induction of apoptosis as confirmed by annexin V staining and the presence of cleaved PARP. Of note, we see that as PRMT1v2 protein expression recovered over time, there was a diminished effect on cell viability, growth and apoptosis, supporting that these effects are likely due to the specific depletion of PRMT1v2.
Interestingly, PRMT1 has been linked to the DNA damage response and DNA repair mechanisms. PRMT1, along with CARM1, cooperates as a transcriptional coactivator of p53 and regulates target gene expression.Citation47 PRMT1 has also been shown to methylate 53BP1 and MRE11, which regulates their DNA-binding ability and function in DNA repair.Citation48-Citation50 This suggests that PRMT1 may be involved in promoting the resistance of cancer cells to DNA-damaging agents and, thus, could represent a potentially useful target in combination treatment strategies with these agents. Indeed, it was shown that PRMT1-deficient U2OS cells are hypersensitive to the topoisomerase II inhibitor, etoposide.Citation46 Furthermore, in MEFs, loss of PRMT1 expression leads to spontaneous DNA damage, interruption in cell cycle progression and chromosomal abnormalities, which ultimately results in cell death within ~1 wk.Citation46 Additionally, PRMT1 has been linked with the positive regulation of telomere length and stability at least in part through its methylation of TRF2, a component of the sheltering complex that normally functions to protect telomeres.Citation51 Specifically, stable knockdown of PRMT1 in cancer cells was shown to cause increased association of TRF2 with telomeres, thus resulting in telomere shortening. This suggests that PRMT1 overexpression in cancer cells may contribute to maintaining telomere length, a phenomenon associated with evading cellular senescence and survival in cancer cells. These studies have examined the long-term effects PRMT1 loss. We have clearly demonstrated that apoptosis is a short-term consequence of reduced PRMT1v2 expression in breast cancer cells. It would be interesting to determine whether PRMT1v2 also contributes to DNA damage and telomere maintenance regulatory pathways.
Intriguingly, conflicting roles for PRMT1 in apoptotic signaling have been postulated. In one study, methylation of apoptosis signal-regulating kinase 1 (ASK1) by PRMT1 inhibited activation of ASK1, thus protecting cells from stress-induced apoptosis.Citation52 This was observed in two breast cancer cell lines, MCF7 and MDA-MB-231. In contrast, PRMT1 was also shown to methylate the pro-apoptotic protein Bad, preventing its inactivation and, thus, promoting apoptosis in MCF7 cells.Citation53 Again, it remains unknown which PRMT1 isoforms are responsible for these effects, but it is possible that these opposing roles could in fact reflect functional differences and/or distinct substrate specificities between the isoforms, as we have previously documented.Citation7 Further studies will be required to determine the precise apoptotic pathways affected by PRMT1v2 in breast cancer cells.
A direct role for PRMT1 in cancer cell invasion has not been assessed. Here we have shown for the first time that PRMT1, and more importantly the PRMT1v2 isoform specifically, contributes to breast cancer cell invasiveness. Depletion of PRMT1v2 in a highly invasive breast cancer cell line, MDA-MB-231, significantly impaired both cell motility and invasiveness, while both transient and stable expression of PRMT1v2 in MCF7 cells, a weakly invasive cell line,Citation24 significantly enhanced invasion in vitro. Interesting, even though MCF7 cells express high levels of endogenous PRMT1v2,Citation7 additional exogenous PRMT1v2 expression is capable of enhancing their invasiveness. This suggests that there may be a threshold expression level of PRMT1v2 required for promoting invasion in certain cell types. It is conceivable that this threshold may vary with the expression of other mutant proteins that contribute to breast cancer cell invasion. Alternatively, or concurrently, it might reflect differential regulation of PRMT1 activity, which remains poorly understood. It has been shown that PRMTs can interact with non-methyl-accepting proteins, which can either enhance or inhibit their methyltransferase activity and also substrate specificity.Citation54,Citation55 The balance between enhancer protein and inhibitor protein expression could have a dramatic impact on the threshold expression level of PRMT1v2 required to exhibit effects. This also implies that the expression level of PRMT1v2 likely cannot be considered a clear cut marker of invasive potential. Furthermore, we have shown that proper localization and methylase activity of PRMT1v2 are critical to its role in promoting invasion, as transient expression of an NES mutant or methylase inactive mutant did not cause a significant enhancement. The methylase-inactive mutant actually caused a reduction in invasion; however, this was likely due, at least in part, to cell toxicity that we and others have observed with transfection of this mutant.Citation26 Nevertheless, it remains possible that methylase inactive PRMT1v2 might also act as a dominant negative, especially considering that PRMT1 is known to require dimerization for full catalytic activity.Citation26,Citation56
Overexpression of PRMT1v2 caused a significant impairment in cell-cell adhesion and disrupted colony formation in MCF7 cells. Colonies were more scattered, and these cells exhibited an increased presence of F-actin-containing filopodia and small lamellipodia structures at membrane surfaces reminiscent of active migration. E-cadherin and catenin proteins (α, β, γ) interact to form adherens junctions (AJ) that maintain epithelial cell polarity and tissue integrity.Citation57 E-cadherin, the transmembrane component, is responsible for the extracellular adhesion. Intracellularly, β-catenin links E-cadherin to the actin cytoskeleton, which is required for AJ stabilization.Citation40,Citation41 Loss of expression of any component of the E-cadherin/catenin complex through mutation or degradation disrupts the AJ stability. Disruption is associated with the invasiveness and metastasis of epithelial-derived tumors, such as breast cancer.Citation29,Citation33,Citation38,Citation40,Citation58,Citation59 Additionally, consistent with a disruption in cell-cell adhesion, we observed a reduction in F-actin, E-cadherin and β-catenin localization at the membrane.
An assessment of the expression of E-cadherin and β-catenin revealed that PRMT1v2 expression in MCF7 cells causes a decrease in the steady-state levels of β-catenin protein expression. No effect on E-cadherin mRNA or protein expression was observed. Reduction or loss of β-catenin protein expression has been observed in several cancer types and is associated with increased aggressiveness, metastasis and poor prognosis in patients.Citation60-Citation67 PRMT1v1 expression induced morphological changes and enhanced the motility of MCF7 cells similar to PRMT1v2. However, it did not significantly enhance invasion nor cause a reduction in β-catenin expression, suggesting it promotes these changes as well as its effects on cell motility through distinct mechanisms. There is precedent from previous studies that PRMT1 is capable of negatively regulating β-catenin expression through phosphorylation-triggered protein degradation.Citation2,Citation42,Citation43,Citation68 In PRMT1v2-expressing cells, we observe a higher proportion of phosphorylated β-catenin, suggesting PRMT1v2 might regulate β-catenin expression through such a mechanism. A recent study has shown that PRMT1 can methylate Axin, resulting in increased Axin protein stability.Citation68 Axin forms a complex with adenomatous polyposis coli (APC), casein kinase 1 (CK1) and glycogen synthase kinase 3β (GSK3β), which is responsible for phosphorylating β-catenin and thereby promoting its degradation. It was shown that both PRMT1v2 and PRMT1v1 were capable of methylating Axin in vitro, but this was not assessed in cells. Thus, it is conceivable that PRMT1v2 may regulate β-catenin via this pathway, since Axin, like PRMT1v2, is also a cytoplasmic protein. To determine the importance of the repression of β-catenin protein to PRMT1v2-enhanced cell invasion, we ectopically expressed β-catenin by transient transfection into PRMT1v2-expressing cells. These cells showed a significant reduction in invasion. Therefore, it appears that the effects of PRMT1v2 on cell invasion are, at least in part, a consequence of its ability to repress β-catenin protein expression. While the repression of β-catenin is one mechanism through which PRMT1v2 can promote cell invasion, it likely functions together with other invasion-promoting signals. These include the previous observation that PRMT1 triggers methylation-mediated association of ERα with PI3-kinase, Src and FAK complex,Citation19 all of which regulate cell migration and invasion.Citation69-Citation73 Furthermore, it has been shown that mutation of p53 is involved in the regulation of cell migration and invasion, increasing the metastatic potential of cancer cells.Citation74,Citation75 While PRMT1 was previously shown to participate in p53-dependent transcriptional activation,Citation47 it remains unknown whether this may be linked with the mechanisms through which PRMT1v2 promotes breast cancer cell invasion.
In summary, our results demonstrate functional differences between the PRMT1 isoforms, PRMT1v2, PRMT1v1 and PRMT1v3, and illustrate the importance of studying their individual functions and contributions to human disease. We show specifically that PRMT1v2 is capable of promoting cancer cell growth, survival and invasion. Of particular interest is the potential implication of PRMT1v2 in breast cancer, which will, of course, require further investigations using relevant in vivo analysis to identify its contributions to promoting tumor development and aggressiveness.
Materials and Methods
Cell lines
Cell lines were from the American Type Culture Collection. MDA-MB-231 cells were cultured Dulbecco’s modified Eagle’s medium (DMEM) with 2 mM glutamine, 1 mM sodium pyruvate and 10% fetal bovine serum (FBS). MCF7, T47D and Hs578T media was also supplemented with 2.75 μg/mL insulin.
Antibodies and reagents
Human polyclonal PRMT1 antibody was a gift from Dr. Stephane Richard (McGill University). Anti-His antibody was from Bioshops Canada Inc. (#TAG 001). GFP antibody was purchased from Roche Diagnostics (#11 814 460 001). PARP (#9542), and phospho-β-catenin (#9561) antibodies were from Cell Signaling Technologies. β-catenin antibody was from Millipore (#06–734). E-cadherin antibody was from BD Bioscience (#610181). Antibodies against α-tubulin (#T 6199) and GAPDH (#MMS-580S) were from Sigma and Covance, respectively.
PRMT1v2-specific antibody
Synthetic PRMT1v2 peptide, ANCIMENFVATLANGMSLQPPL-EE, was synthesized at W.M. Keck Foundation Biotechnology Resource Laboratory (Yale University). Polyclonal antibodies were generated by Cedarlane Laboratories using rabbits injected with the synthetic peptide coupled to KLH. Antibodies were affinity-purified over the antigenic peptide coupled to Affi-Gel 15 beads (Bio-Rad) following manufacturer's instructions, eluted in 100 mm Glycine pH 2.5, buffered with 1 M Tris–HCl pH 8.0, dialyzed against 1× phosphate-buffered saline (1× PBS: 137 mm NaCl, 2.7 mm KCl, 4.3 mm Na2HPO4, 1.4 mm KH2PO4, pH 7.4) and concentrated using a Centricon centrifugal device (Millipore).
RNA interference
RNA interference targeting PRMT1v2 was performed as described in reference Citation7. A non-targeting control siRNA was purchased from Life Technologies. Control vector (pRS) containing a scrambled shRNA cassette was purchases from Origene.
MTT assays
MTT assays were performed as described in reference Citation76. Absorbances were acquired using a Spectramax M2 microplate reader with Softmax Pro software.
Cell counts
Viable cell numbers were determined by counting 600 cells/condition and excluding the Trypan Blue-positive cells.
DNA constructs and stable cells
PRMT1v1-GFP, PRMT1v2-GFP, PRMT1v3-GFP, PRMT1v2 NES mutant and PRMT1v2 methylase inactive mutant constructs were generated as described in reference Citation7. MCF7 cells stably expressing GFP, PRMT1v1-GFP or PRMT1v2-GFP were created by selection with 1 mg/ml G418. After selection, cells were analyzed and sorted for GFP fluorescence. Flag β-catenin construct was a generous gift from Dr. David Lohnes (University of Ottawa).
Western blot analysis
Western blotting was performed similar to those described in reference Citation77.
Cell motility and invasion assays
Scratch wound assays were performed as described in reference Citation78. For Transwell chamber assays, 24-well BD control or Biocoat Matrigel invasion chambers containing 8-micron pores (BD Biosciences) were used to assess cell motility and invasion, respectively, according to the manufacturer’s specifications. Briefly, 50,000 cells in serum-free DMEM were seeded into each chamber and placed in a 24-well plate containing 500 μL complete DMEM and incubated for 72 h for MCF7 cells and 24 h for MDA-MB-231 cells. A minimum of four random fields at 20X magnification were counted to determine the number of cells that passed through the chamber membranes.
Scratch wound assay
MCF7 cells stably expressing GFP, PRMT1v2-GFP or PRMT1v1-GFP were grown to full confluence in 6-well plates containing complete DMEM. Once cells reached full confluence, plates were scratch wounded, washed with PBS and supplied with fresh complete media. Wounds were examined and measured at 0, 24, 48, 72 and 96 h.
Protein purification
PRMT1v1, -PRMT1v2, -PRMT1v3 protein were purified as described in reference Citation7.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde in PBS for 15 min, permeabilized with 0.5% Triton X-100 in PBS for 10 min and washed three times with PBS. Cells were then incubated with primary antibodies diluted in PBS for 1 h at room temperature (Anti-E-Cadherin (#13–1,700, Invitrogen), 1:500 or anti-β-Catenin, 1:350). Conjugated phalloidin was used to visualize actin fibers (Sigma). Cells were washed once with 0.1% Triton X-100 in PBS, twice with PBS and incubated for 1 h in the dark at room temperature with the appropriate fluorophore-labeled secondary antibody diluted in PBS (Alexa Fluor 594, Invitrogen, 1:300). Cells were washed again (once with 0.1% Triton X-100 in PBS and twice with PBS) and coverslips were mounted onto glass slides using the Vectashield Mounting medium with DAPI (Vector Laboratories).
Mircroscopy
Phase images for RNAi, scratch wound and invasion assays were acquired using a Zeiss Axiovert 40 CFL inverted microscope using a Canon digital camera. Fluorescence and accompanying phase imaging was performed on a Ziess Axio Imager.Z1 microscope and images acquired with an AxioCam HRm camera driven by Zeiss Axiovision 4.5 software.
Reverse transcriptase PCR analysis
Total RNA was isolated from cells using TRIzol (Life Technologies). First-strand cDNA synthesis was performed using AMV Reverse Transcriptase (Promega). PCRs were performed in a 20 μL reaction using Go Taq Green Master Mix (Promega). For PRMT1, cDNAs were amplified using the primers described in reference Citation44. β-catenin primer pair was forward (5′-AAAATGGCAGTGCGTT TAG-3′) and reverse (5′-TTTGAAGGCAGTCTGTCGTA-3′); E-cadherin primer pair was forward (5′-TGGGTTATTCCTCCCATCAG-3′) and reverse (5′-TTTGTCAGGGAGCTCAGG AT-3′). GAPDH was amplified with forward (5′-ACCACAGTCCATGCCATCAC-3′) and reverse (5′-TCCACCACCCTGTTGCTGTA-3′).
Colony scattering assay
Colony scattering assays were performed similar to those described in reference Citation29.
Flow cytometry
Adherent and non-adherent cells were prepared for Flow Cytometric analysis for DNA content as described in reference Citation76. Annexin V-FITC staining was performed using the Annexin V-FITC Apoptosis Kit (BioVision Inc.) according to the manufacturer’s protocol. Flow cytometry was performed using a Beckman Coulter Epics XL cytometer and data was analyzed using WinMDI 2.8. Annexin V/propidium iodide analysis quadrants were set up according to controls (Fig. S3C).
Statistical analysis
Statistical analysis was performed using the Student's t-test, and p < 0.05 was considered statistically significant.
| Abbreviations: | ||
| PRMT | = | protein arginine methyltransferease |
| mRNA | = | messenger ribo nucleic acid |
| DNA | = | deoxyribonucleic acid |
| CARM | = | coactivator-associated arginine methyltransferase 1 |
| MLL | = | mixed lineage leukemia |
| ER | = | estrogen receptor |
| bp | = | base pair |
| NES | = | nuclear export sequence |
| RNA | = | ribo nucleic acid |
| siRNA | = | small interfering ribo nucleic acid |
| shRNA | = | short hairpin ribo nucleic acid |
| kDa | = | kilo Daltons |
| h | = | hours |
| MTT | = | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PI | = | propidium iodide |
| PARP | = | Poly-ADP ribose polymerase |
| d | = | days |
| GFP | = | green fluorescence protein |
| V | = | valine |
| A | = | alanine |
| L | = | leucine |
| D | = | aspartic acid |
| UTR | = | untranslated region |
| MEF | = | mouse embryonic fibroblasts |
| PKB | = | protein kinase B |
| wk | = | weeks |
| ASK1 | = | apoptosis signal-regulating kinase 1 |
| Bad | = | Bcl-2-associated death promoter |
| 53BP1 | = | p53 binding protein 1 |
| AJ | = | adherens junctions |
| F-actin | = | filamentous actin |
| APC | = | adenomatous polyposis coli, CK1, casein kinase 1 |
| GSK3β | = | glycogen synthase kinase 3β |
| PI3-kinase | = | phosphatidyl inositol 3 kinase |
| FAK | = | focal adhesion kinase |
| DMEM | = | Dulbecco’s modified Eagle’s medium |
| FBS | = | fetal bovine serum |
| GAPDH | = | glyceraldehyde-3-phosphate dehydrogenase |
Additional material
Download Zip (1,017 KB)Acknowledgments
This work was supported by grants from the Cancer Research Society and a Canada Research Chair in RNA Metabolism Institutes of Health Research (to J.C.). R.M.B. is a postdoctoral fellow of the Canadian Institute of Health Research. I.G. was a research student of the Canadian Breast Cancer Foundation. Special thanks to Dr. Ian Lorimer for critically reading this manuscript.
References
- Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell 2009; 33:1 - 13; http://dx.doi.org/10.1016/j.molcel.2008.12.013; PMID: 19150423
- Hart M, Concordet JP, Lassot I, Albert I, del los Santos R, Durand H, et al. The F-box protein beta-TrCP associates with phosphorylated beta-catenin and regulates its activity in the cell. Curr Biol 1999; 9:207 - 10; http://dx.doi.org/10.1016/S0960-9822(99)80091-8; PMID: 10074433
- Zurita-Lopez CI, Sandberg T, Kelly R, Clarke SG. Human Protein Arginine Methyltransferase 7 (PRMT7) is a Type III Enzyme Forming omega-NG-Monomethylated Arginine Residues. J Biol Chem 2012; 287:29801 - 14; http://dx.doi.org/10.1074/jbc.M111.336271; PMID: 22761421
- Boisvert FM, Chénard CA, Richard S. Protein interfaces in signaling regulated by arginine methylation. Sci STKE 2005; 2005:re2; http://dx.doi.org/10.1126/stke.2712005re2; PMID: 15713950
- Ong SE, Mittler G, Mann M. Identifying and quantifying in vivo methylation sites by heavy methyl SILAC. Nat Methods 2004; 1:119 - 26; http://dx.doi.org/10.1038/nmeth715; PMID: 15782174
- Boisvert FM, Côté J, Boulanger MC, Richard S. A proteomic analysis of arginine-methylated protein complexes. Mol Cell Proteomics 2003; 2:1319 - 30; http://dx.doi.org/10.1074/mcp.M300088-MCP200; PMID: 14534352
- Goulet I, Gauvin G, Boisvenue S, Côté J. Alternative splicing yields protein arginine methyltransferase 1 isoforms with distinct activity, substrate specificity, and subcellular localization. J Biol Chem 2007; 282:33009 - 21; http://dx.doi.org/10.1074/jbc.M704349200; PMID: 17848568
- Hong H, Kao C, Jeng MH, Eble JN, Koch MO, Gardner TA, et al. Aberrant expression of CARM1, a transcriptional coactivator of androgen receptor, in the development of prostate carcinoma and androgen-independent status. Cancer 2004; 101:83 - 9; http://dx.doi.org/10.1002/cncr.20327; PMID: 15221992
- Mathioudaki K, Scorilas A, Ardavanis A, Lymberi P, Tsiambas E, Devetzi M, et al. Clinical evaluation of PRMT1 gene expression in breast cancer. Tumour Biol 2011; 32:575 - 82; http://dx.doi.org/10.1007/s13277-010-0153-2; PMID: 21229402
- Cheung N, Chan LC, Thompson A, Cleary ML, So CW. Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol 2007; 9:1208 - 15; http://dx.doi.org/10.1038/ncb1642; PMID: 17891136
- Kim YR, Lee BK, Park RY, Nguyen NT, Bae JA, Kwon DD, et al. Differential CARM1 expression in prostate and colorectal cancers. BMC Cancer 2010; 10:197; http://dx.doi.org/10.1186/1471-2407-10-197; PMID: 20462455
- Mathioudaki K, Papadokostopoulou A, Scorilas A, Xynopoulos D, Agnanti N, Talieri M. The PRMT1 gene expression pattern in colon cancer. Br J Cancer 2008; 99:2094 - 9; http://dx.doi.org/10.1038/sj.bjc.6604807; PMID: 19078953
- Yoshimatsu M, Toyokawa G, Hayami S, Unoki M, Tsunoda T, Field HI, et al. Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int J Cancer 2011; 128:562 - 73; http://dx.doi.org/10.1002/ijc.25366; PMID: 20473859
- Pal S, Baiocchi RA, Byrd JC, Grever MR, Jacob ST, Sif S. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J 2007; 26:3558 - 69; http://dx.doi.org/10.1038/sj.emboj.7601794; PMID: 17627275
- Papadokostopoulou A, Mathioudaki K, Scorilas A, Xynopoulos D, Ardavanis A, Kouroumalis E, et al. Colon cancer and protein arginine methyltransferase 1 gene expression. Anticancer Res 2009; 29:1361 - 6; PMID: 19414388
- Frietze S, Lupien M, Silver PA, Brown M. CARM1 regulates estrogen-stimulated breast cancer growth through up-regulation of E2F1. Cancer Res 2008; 68:301 - 6; http://dx.doi.org/10.1158/0008-5472.CAN-07-1983; PMID: 18172323
- Pal S, Vishwanath SN, Erdjument-Bromage H, Tempst P, Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol Cell Biol 2004; 24:9630 - 45; http://dx.doi.org/10.1128/MCB.24.21.9630-9645.2004; PMID: 15485929
- Vezzalini M, Aletta JM, Beghelli S, Moratti E, Della Peruta M, Mafficini A, et al. Immunohistochemical detection of arginine methylated proteins (MeRP) in archival tissues. Histopathology 2010; 57:725 - 33; http://dx.doi.org/10.1111/j.1365-2559.2010.03684.x; PMID: 21083602
- Le Romancer M, Treilleux I, Leconte N, Robin-Lespinasse Y, Sentis S, Bouchekioua-Bouzaghou K, et al. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol Cell 2008; 31:212 - 21; http://dx.doi.org/10.1016/j.molcel.2008.05.025; PMID: 18657504
- Côté J, Boisvert FM, Boulanger MC, Bedford MT, Richard S. Sam68 RNA binding protein is an in vivo substrate for protein arginine N-methyltransferase 1. Mol Biol Cell 2003; 14:274 - 87; http://dx.doi.org/10.1091/mbc.E02-08-0484; PMID: 12529443
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods 1995; 184:39 - 51; http://dx.doi.org/10.1016/0022-1759(95)00072-I; PMID: 7622868
- Mooney LM, Al-Sakkaf KA, Brown BL, Dobson PR. Apoptotic mechanisms in T47D and MCF-7 human breast cancer cells. Br J Cancer 2002; 87:909 - 17; http://dx.doi.org/10.1038/sj.bjc.6600541; PMID: 12373608
- Walsh JG, Cullen SP, Sheridan C, Lüthi AU, Gerner C, Martin SJ. Executioner caspase-3 and caspase-7 are functionally distinct proteases. Proc Natl Acad Sci USA 2008; 105:12815 - 9; http://dx.doi.org/10.1073/pnas.0707715105; PMID: 18723680
- Lacroix M, Leclercq G. Relevance of breast cancer cell lines as models for breast tumours: an update. Breast Cancer Res Treat 2004; 83:249 - 89; http://dx.doi.org/10.1023/B:BREA.0000014042.54925.cc; PMID: 14758095
- Herrmann F, Pably P, Eckerich C, Bedford MT, Fackelmayer FO. Human protein arginine methyltransferases in vivo--distinct properties of eight canonical members of the PRMT family. J Cell Sci 2009; 122:667 - 77; http://dx.doi.org/10.1242/jcs.039933; PMID: 19208762
- Herrmann F, Fackelmayer FO. Nucleo-cytoplasmic shuttling of protein arginine methyltransferase 1 (PRMT1) requires enzymatic activity. Genes Cells 2009; 14:309 - 17; http://dx.doi.org/10.1111/j.1365-2443.2008.01266.x; PMID: 19170758
- Wada K, Inoue K, Hagiwara M. Identification of methylated proteins by protein arginine N-methyltransferase 1, PRMT1, with a new expression cloning strategy. Biochim Biophys Acta 2002; 1591:1 - 10; http://dx.doi.org/10.1016/S0167-4889(02)00202-1; PMID: 12183049
- Liu X, Feng R. Inhibition of epithelial to mesenchymal transition in metastatic breast carcinoma cells by c-Src suppression. Acta Biochim Biophys Sin (Shanghai) 2010; 42:496 - 501; http://dx.doi.org/10.1093/abbs/gmq043; PMID: 20705589
- Shtutman M, Levina E, Ohouo P, Baig M, Roninson IB. Cell adhesion molecule L1 disrupts E-cadherin-containing adherens junctions and increases scattering and motility of MCF7 breast carcinoma cells. Cancer Res 2006; 66:11370 - 80; http://dx.doi.org/10.1158/0008-5472.CAN-06-2106; PMID: 17145883
- Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans 2005; 33:891 - 5; http://dx.doi.org/10.1042/BST20050891; PMID: 16246005
- Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 2005; 21:247 - 69; http://dx.doi.org/10.1146/annurev.cellbio.21.020604.150721; PMID: 16212495
- Schafer DA. Cell biology: barbed ends rule. Nature 2004; 430:734 - 5; http://dx.doi.org/10.1038/430734a; PMID: 15306794
- Tomaskovic-Crook E, Thompson EW, Thiery JP. Epithelial to mesenchymal transition and breast cancer. Breast Cancer Res 2009; 11:213; http://dx.doi.org/10.1186/bcr2416; PMID: 19909494
- Jourquin J, Yang N, Kam Y, Guess C, Quaranta V. Dispersal of epithelial cancer cell colonies by lysophosphatidic acid (LPA). J Cell Physiol 2006; 206:337 - 46; http://dx.doi.org/10.1002/jcp.20470; PMID: 16110477
- de Rooij J, Kerstens A, Danuser G, Schwartz MA, Waterman-Storer CM. Integrin-dependent actomyosin contraction regulates epithelial cell scattering. J Cell Biol 2005; 171:153 - 64; http://dx.doi.org/10.1083/jcb.200506152; PMID: 16216928
- Machesky LM. Lamellipodia and filopodia in metastasis and invasion. FEBS Lett 2008; 582:2102 - 11; http://dx.doi.org/10.1016/j.febslet.2008.03.039; PMID: 18396168
- Thiery JP, Chopin D. Epithelial cell plasticity in development and tumor progression. Cancer Metastasis Rev 1999; 18:31 - 42; http://dx.doi.org/10.1023/A:1006256219004; PMID: 10505544
- Hiscox S, Jiang WG, Obermeier K, Taylor K, Morgan L, Burmi R, et al. Tamoxifen resistance in MCF7 cells promotes EMT-like behaviour and involves modulation of beta-catenin phosphorylation. Int J Cancer 2006; 118:290 - 301; http://dx.doi.org/10.1002/ijc.21355; PMID: 16080193
- Yao H, Ashihara E, Maekawa T. Targeting the Wnt/β-catenin signaling pathway in human cancers. Expert Opin Ther Targets 2011; 15:873 - 87; http://dx.doi.org/10.1517/14728222.2011.577418; PMID: 21486121
- Van Aken E, De Wever O, Correia da Rocha AS, Mareel M. Defective E-cadherin/catenin complexes in human cancer. Virchows Arch 2001; 439:725 - 51; PMID: 11787845
- Kemler R. From cadherins to catenins: cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet 1993; 9:317 - 21; http://dx.doi.org/10.1016/0168-9525(93)90250-L; PMID: 8236461
- Latres E, Chiaur DS, Pagano M. The human F box protein beta-Trcp associates with the Cul1/Skp1 complex and regulates the stability of beta-catenin. Oncogene 1999; 18:849 - 54; http://dx.doi.org/10.1038/sj.onc.1202653; PMID: 10023660
- Winston JT, Strack P, Beer-Romero P, Chu CY, Elledge SJ, Harper JW. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev 1999; 13:270 - 83; http://dx.doi.org/10.1101/gad.13.3.270; PMID: 9990852
- Scorilas A, Black MH, Talieri M, Diamandis EP. Genomic organization, physical mapping, and expression analysis of the human protein arginine methyltransferase 1 gene. Biochem Biophys Res Commun 2000; 278:349 - 59; http://dx.doi.org/10.1006/bbrc.2000.3807; PMID: 11097842
- Scott HS, Antonarakis SE, Lalioti MD, Rossier C, Silver PA, Henry MF. Identification and characterization of two putative human arginine methyltransferases (HRMT1L1 and HRMT1L2). Genomics 1998; 48:330 - 40; http://dx.doi.org/10.1006/geno.1997.5190; PMID: 9545638
- Yu Z, Chen T, Hébert J, Li E, Richard S. A mouse PRMT1 null allele defines an essential role for arginine methylation in genome maintenance and cell proliferation. Mol Cell Biol 2009; 29:2982 - 96; http://dx.doi.org/10.1128/MCB.00042-09; PMID: 19289494
- An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell 2004; 117:735 - 48; http://dx.doi.org/10.1016/j.cell.2004.05.009; PMID: 15186775
- Boisvert FM, Déry U, Masson JY, Richard S. Arginine methylation of MRE11 by PRMT1 is required for DNA damage checkpoint control. Genes Dev 2005; 19:671 - 6; http://dx.doi.org/10.1101/gad.1279805; PMID: 15741314
- Boisvert FM, Rhie A, Richard S, Doherty AJ. The GAR motif of 53BP1 is arginine methylated by PRMT1 and is necessary for 53BP1 DNA binding activity. Cell Cycle 2005; 4:1834 - 41; http://dx.doi.org/10.4161/cc.4.12.2250; PMID: 16294045
- Yu Z, Vogel G, Coulombe Y, Dubeau D, Spehalski E, Hébert J, et al. The MRE11 GAR motif regulates DNA double-strand break processing and ATR activation. Cell Res 2012; 22:305 - 20; http://dx.doi.org/10.1038/cr.2011.128; PMID: 21826105
- Mitchell TR, Glenfield K, Jeyanthan K, Zhu XD. Arginine methylation regulates telomere length and stability. Mol Cell Biol 2009; 29:4918 - 34; http://dx.doi.org/10.1128/MCB.00009-09; PMID: 19596784
- Cho JH, Lee MK, Yoon KW, Lee J, Cho SG, Choi EJ. Arginine methylation-dependent regulation of ASK1 signaling by PRMT1. Cell Death Differ 2012; 19:859 - 70; http://dx.doi.org/10.1038/cdd.2011.168; PMID: 22095282
- Sakamaki J, Daitoku H, Ueno K, Hagiwara A, Yamagata K, Fukamizu A. Arginine methylation of BCL-2 antagonist of cell death (BAD) counteracts its phosphorylation and inactivation by Akt. Proc Natl Acad Sci USA 2011; 108:6085 - 90; http://dx.doi.org/10.1073/pnas.1015328108; PMID: 21444773
- Robin-Lespinasse Y, Sentis S, Kolytcheff C, Rostan MC, Corbo L, Le Romancer M. hCAF1, a new regulator of PRMT1-dependent arginine methylation. J Cell Sci 2007; 120:638 - 47; http://dx.doi.org/10.1242/jcs.03357; PMID: 17264152
- Berthet C, Guéhenneux F, Revol V, Samarut C, Lukaszewicz A, Dehay C, et al. Interaction of PRMT1 with BTG/TOB proteins in cell signalling: molecular analysis and functional aspects. Genes Cells 2002; 7:29 - 39; http://dx.doi.org/10.1046/j.1356-9597.2001.00497.x; PMID: 11856371
- Zhang X, Cheng X. Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure 2003; 11:509 - 20; http://dx.doi.org/10.1016/S0969-2126(03)00071-6; PMID: 12737817
- Yonemura S. Cadherin-actin interactions at adherens junctions. Curr Opin Cell Biol 2011; 23:515 - 22; http://dx.doi.org/10.1016/j.ceb.2011.07.001; PMID: 21807490
- Morrogh M, Andrade VP, Giri D, Sakr RA, Paik W, Qin LX, et al. Cadherin-catenin complex dissociation in lobular neoplasia of the breast. Breast Cancer Res Treat 2012; 132:641 - 52; http://dx.doi.org/10.1007/s10549-011-1860-0; PMID: 22080244
- Birchmeier W, Behrens J. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim Biophys Acta 1994; 1198:11 - 26; PMID: 8199193
- Faleiro-Rodrigues C, Macedo-Pinto IM, Maia SS, Vieira RH, Lopes CS. Biological relevance of E-cadherin-catenin complex proteins in primary epithelial ovarian tumours. Gynecol Obstet Invest 2005; 60:75 - 83; http://dx.doi.org/10.1159/000084614; PMID: 15785075
- Faleiro-Rodrigues C, Macedo-Pinto I, Pereira D, Lopes CS. Loss of beta-catenin is associated with poor survival in ovarian carcinomas. Int J Gynecol Pathol 2004; 23:337 - 46; http://dx.doi.org/10.1097/01.pgp.0000139711.22158.14; PMID: 15381903
- Dolled-Filhart M, McCabe A, Giltnane J, Cregger M, Camp RL, Rimm DL. Quantitative in situ analysis of beta-catenin expression in breast cancer shows decreased expression is associated with poor outcome. Cancer Res 2006; 66:5487 - 94; http://dx.doi.org/10.1158/0008-5472.CAN-06-0100; PMID: 16707478
- De Leeuw WJ, Berx G, Vos CB, Peterse JL, Van de Vijver MJ, Litvinov S, et al. Simultaneous loss of E-cadherin and catenins in invasive lobular breast cancer and lobular carcinoma in situ. J Pathol 1997; 183:404 - 11; http://dx.doi.org/10.1002/(SICI)1096-9896(199712)183:4<404::AID-PATH1148>3.0.CO;2-9; PMID: 9496256
- Yoshida R, Kimura N, Harada Y, Ohuchi N. The loss of E-cadherin, alpha- and beta-catenin expression is associated with metastasis and poor prognosis in invasive breast cancer. Int J Oncol 2001; 18:513 - 20; PMID: 11179480
- Nakopoulou L, Mylona E, Papadaki I, Kavantzas N, Giannopoulou I, Markaki S, et al. Study of phospho-beta-catenin subcellular distribution in invasive breast carcinomas in relation to their phenotype and the clinical outcome. Mod Pathol 2006; 19:556 - 63; http://dx.doi.org/10.1038/modpathol.3800562; PMID: 16474376
- Pontes J Jr., Srougi M, Borra PM, Dall’ Oglio MF, Ribeiro-Filho LA, Leite KR. E-cadherin and beta-catenin loss of expression related to bone metastasis in prostate cancer. Appl Immunohistochem Mol Morphol 2010; 18:179 - 84; http://dx.doi.org/10.1097/PAI.0b013e3181640bca; PMID: 18685493
- Ebert MP, Yu J, Hoffmann J, Rocco A, Röcken C, Kahmann S, et al. Loss of beta-catenin expression in metastatic gastric cancer. J Clin Oncol 2003; 21:1708 - 14; http://dx.doi.org/10.1200/JCO.2003.10.017; PMID: 12721245
- Cha B, Kim W, Kim YK, Hwang BN, Park SY, Yoon JW, et al. Methylation by protein arginine methyltransferase 1 increases stability of Axin, a negative regulator of Wnt signaling. Oncogene 2011; 30:2379 - 89; http://dx.doi.org/10.1038/onc.2010.610; PMID: 21242974
- Hernandez-Aya LF, Gonzalez-Angulo AM. Targeting the phosphatidylinositol 3-kinase signaling pathway in breast cancer. Oncologist 2011; 16:404 - 14; http://dx.doi.org/10.1634/theoncologist.2010-0402; PMID: 21406469
- Navarro-Tito N, Robledo T, Salazar EP. Arachidonic acid promotes FAK activation and migration in MDA-MB-231 breast cancer cells. Exp Cell Res 2008; 314:3340 - 55; http://dx.doi.org/10.1016/j.yexcr.2008.08.018; PMID: 18804105
- Zheng S, Huang J, Zhou K, Zhang C, Xiang Q, Tan Z, et al. 17β-Estradiol enhances breast cancer cell motility and invasion via extra-nuclear activation of actin-binding protein ezrin. PLoS One 2011; 6:e22439; http://dx.doi.org/10.1371/journal.pone.0022439; PMID: 21818323
- Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol 2007; 170:1445 - 53; http://dx.doi.org/10.2353/ajpath.2007.070011; PMID: 17456751
- Weber GL, Parat MO, Binder ZA, Gallia GL, Riggins GJ. Abrogation of PIK3CA or PIK3R1 reduces proliferation, migration, and invasion in glioblastoma multiforme cells. Oncotarget 2011; 2:833 - 49; PMID: 22064833
- Hwang CI, Choi J, Zhou Z, Flesken-Nikitin A, Tarakhovsky A, Nikitin AY. MET-dependent cancer invasion may be preprogrammed by early alterations of p53-regulated feedforward loop and triggered by stromal cell-derived HGF. Cell Cycle 2011; 10:3834 - 40; http://dx.doi.org/10.4161/cc.10.22.18294; PMID: 22071625
- Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 2009; 137:87 - 98; http://dx.doi.org/10.1016/j.cell.2009.01.039; PMID: 19345189
- Baldwin RM, Garratt-Lalonde M, Parolin DA, Krzyzanowski PM, Andrade MA, Lorimer IA. Protection of glioblastoma cells from cisplatin cytotoxicity via protein kinase Ciota-mediated attenuation of p38 MAP kinase signaling. Oncogene 2006; 25:2909 - 19; http://dx.doi.org/10.1038/sj.onc.1209312; PMID: 16331246
- Hubers L, Valderrama-Carvajal H, Laframboise J, Timbers J, Sanchez G, Côté J. HuD interacts with survival motor neuron protein and can rescue spinal muscular atrophy-like neuronal defects. Hum Mol Genet 2011; 20:553 - 79; http://dx.doi.org/10.1093/hmg/ddq500; PMID: 21088113
- Sang RL, Johnson JF, Taves J, Nguyen C, Wallert MA, Provost JJ. alpha(1)-Adrenergic receptor stimulation of cell motility requires phospholipase D-mediated extracellular signal-regulated kinase activation. Chem Biol Drug Des 2007; 69:240 - 50; http://dx.doi.org/10.1111/j.1747-0285.2007.00502.x; PMID: 17461971