Abstract
A balance must be struck between cell growth and stress responses to ensure that cells proliferate without accumulating damaged DNA. This balance means that optimal cell proliferation requires the integration of pro-growth and stress-response pathways. mTOR (mechanistic target of rapamycin) is a pleiotropic kinase found in complex 1 (mTORC1). The mTORC1 pathway governs a response to mitogenic signals with high energy levels to promote protein synthesis and cell growth. In contrast, the p53 DNA damage response pathway is the arbiter of cell proliferation, restraining mTORC1 under conditions of genotoxic stress. Recent studies suggest a complicated integration of these pathways to ensure successful cell growth and proliferation without compromising genome maintenance. Deciphering this integration could be key to understanding the potential clinical usefulness of mTORC1 inhibitors like rapamycin. Here we discuss how these p53-mTORC1 interactions might play a role in the suppression of cancer and perhaps the development of cellular senescence and organismal aging.
Keywords: :
The mTORC1 Growth Response: Dionysus and Silenus at the Banquet
mTOR (mechanistic target of rapamycin) is a serine/threonine kinase in the phosphatidylinositol 3-kinase (PI3K)-related family. It is found in the mTOR complex 1 (mTORC1) and is conserved across species from yeast to mammals. mTORC1 regulates cell growth (mass) and proliferation (cell division) in response to the environment, thereby ensuring that proliferation occurs only when sufficient nutrients are available without destructive stresses such as DNA damage. We describe a simplified version of this pathway, but much greater detail can be found in several excellent reviews.Citation1-Citation4
There are both upstream and downstream components to the mTORC1 pathway. Upstream of mTORC1 are growth factors that bind to receptor tyrosine kinases in the cell membrane. Receptor occupancy induces activation of PI3K, leading to phosphorylation of PIP3 (phosphatidylinositol-3,4,5-triphosphate), a second messenger in the cell membrane. The tumor suppressor PTEN (phosphatase and tensin homolog deleted on chromosome 10) attenuates this pathway through PIP3 dephosphorylation. Otherwise, PIP3 activates AKT, which, in turn, inhibits the TSC1 and TSC2 (tuberous sclerosis complex), a negative regulator of mTORC1. TSC1/2 are GTPase-activating proteins that inhibit the small GTPase RHEB (RAS homolog enriched in brain), thereby prohibiting mTORC1 activation.Citation5,Citation6
Downstream of mTORC1 are components of the mRNA translation machinery. Activated mTORC1 regulates mRNA translation to enhance protein synthesis. mTORC1, in part, by phosphorylating S6K1 (S6 kinase 1), induces ribosome biogenesis and translation.Citation2,Citation3 In addition, mTORC1 phosphorylates 4E-BPs (elF4E-binding proteins) to terminate binding to elF4E and relieve the block on translation. Thus, mTORC1 enables cell growth in response to mitogenic signaling in the presence of high nutrient/energy levels and low stress.
Rapamycin and rapamycin derivatives (rapalogs) specifically inhibit mTOR and have been instrumental in studying the mTORC1 pathway. Rapamycin is a macrocyclic lactone discovered on the island of Rapa Nui (Easter Island).Citation7 The bacterium Streptomyces hygroscopicus naturally produces rapamycin as an antifungal agent. Mechanistically, rapamycin binds to FKBP12, a protein folding chaperone required for mTORC1 activity, and, thus, inhibits mTORC1.Citation8 There is a history of using rapamycin in the clinic as an adjuvant to the immunosuppressant cyclosporine for organ transplant patients, with few toxic side effects (even with chronic use). mTOR inhibitors are being examined for their ability to prevent post-transplant cancers.Citation9 In addition to use in transplant patients, rapamycin also has potential value in ameliorating any disease whose etiology includes cell growth, the most obvious being cancer.Citation10 Yet, exuberant cell growth may contribute to other maladies, especially those related to aging. Thus, rapamycin and other agents that target the mTORC1 pathway are candidates for a host of therapies.
The p53 DNA Damage Response: Nomos, Themis and Dike Set Divine Order
There must be an antagonist to balance these high energy and growth processes. For mTORC1, one such antagonist is p53. The p53 pathway is best known for suppressing tumors and is mutated in more than half of all human cancers.Citation11 p53, a transcription factor,Citation12 suppresses cellular proliferation in response to a variety of stimuli, including genotoxic stress and low energy levels.Citation13-Citation15 In response to genotoxic stress, ATM (ataxia telangiectasia mutated) phosphorylates p53 on serine 15/18 (pS15 in human, pS18 in mice) to stimulate p53 transcriptional activity.Citation16
p53 pS15/18 induces transcription. The majority of p53’s anticancer function derives from transactivation of a large number of genes, most of which suppress cell proliferation in response to various stresses, including damaged DNA.Citation15 These p53 targets are generally classified into four groups: (1) proliferation arrest (p21, 14–3-3α), (2) apoptosis (BAX, NOXA, PUMA, APAF1), (3) death receptor (KILLER/DR5, FAS and PIDD) and (4) mTORC1 inhibition (sestrins).Citation17 In addition, p53 induces a negative autoregulatory loop by transactivating MDM2, which targets p53 for ubiquitination and subsequent degradation. ATM also phosphorylates MDM2 on serines 395, 386 and 429 to induce p53 ubiquitination. This negative regulation is important because high p53 levels can be deleterious. In mice, for example, MDM2 deletion is early embryonic lethal, and lethality is rescued by p53 deletion.Citation18,Citation19 In addition to ubiquitination, other post-translational p53 modifications include phosphorylation and acetylation, neddylation, sumoylation and methylation, all of which are essential for modulating p53 activity. Thus, p53 is a highly regulated protein that inhibits cell proliferation and induces apoptosis in response to a variety of cellular stresses, including DNA damage. The p53 pathway is critical for regulating the pro-growth mTORC1 pathway.
The p53 DNA Damage Response Confronts mTORC1: Zeus Battles Cronus for His Day of Reckoning
p53 inhibits the mTORC1 pathway in response to cellular stresses, including DNA damageCitation4 (). Activated p53 inhibits mTORC1 through AMPK (5′ adenosine monophosphate-activated kinase) and REDD1 (regulated in development and DNA damage responses 1) via the TSC1/2 complexes.Citation20 This DNA damage response occurs in a low-energy environment, since AMPK responds to a high AMP/ATP ratio in the cell (, red box). REDD1 is a p53 transcriptional target under several kinds of stress conditions, including DNA damage.Citation21 p53 also decreases S6K1 activity, 4E-BP1 dephosphorylation and protein synthesis, independent of its role in transcription regulation.Citation22 Cytoplasmic p53 is covalently linked to 5.8S rRNA and is associated with ribosomes.Citation23,Citation24 Furthermore, p53 induces PTEN transcription that ultimately inhibits mTORC1, since PTEN inhibits PIP3/AKT.Citation25 Thus, p53 inhibits mTORC1 and protein synthesis through multiple mechanisms.
Figure 1. The integration of DNA damages response with cell growth. (1) The ATM response (red box) activates p53 to inhibit mTORC1 through AMPK/TSC1/2. (2) The p38α/AKT/mTORC1/S6K1 response (green box) inhibits MDM2 suppression of p53 which inhibits mTORC1. (3) The combination of p53/p21 cell cycle arrest and mTORC1-induced cell growth may result in cellular senescence, which, in turn, may impact cancer and longevity.
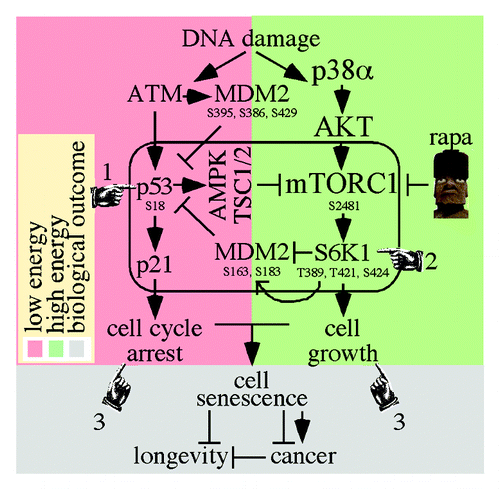
Conversely, mTORC1 activity increases p53 function in what appears to be a compensatory response to DNA damage with high energy levels (, green box). p38α/AKT/mTORC1/S6K1 upregulated p53 function in tissue culture cells exposed to genotoxic agents, and rapamycin negated this effect (i.e., suppressed p53) through S6K1.Citation26 More specifically, p38α/AKT/mTORC1/S6K1 negatively regulated MDM2, which increased p53 levels in response to genotoxic stress, including DNA double-strand breaks.Citation26 In this case, activated mTORC1 phosphorylates S6K1 on T389, T421 and S424. Subsequently, activated S6K1 phosphorylates MDM2 on S163/183 (S166/186 in human). In addition, activated S6K1 binds to MDM2 pS163/183 to inhibit nuclear localization and p53 ubiquitination, thereby stabilizing p53. Rapamycin reverses this process.
Though it appears counterintuitive for a pro-growth pathway to enhance an anti-proliferation pathway, this makes biological sense if one considers it as a negative feedback loop that integrates metabolic and energy signals with the DNA damage response. To support this possibility, high serum levels enhance S6K1 activation and p53 induction for cells exposed to strong mitogenic signaling and genotoxic stress. In addition to this feedback loop, p53 activity is stimulated in response to activated mTORC1 caused by loss of TSC1/2.Citation27,Citation28 TSC1/2 loss causes tuberous sclerosis and is associated with multiple benign tumors that are maintained in a benign state, in part by the increase in p53 activity.Citation29 Therefore, p53 and mTORC1 are linked together to regulate protein translation with cell fate determination based on the combined levels of energy and cellular stress.
Given that both ATM and mTORC1/S6K1 induce a p53 response to DNA damage, our model shows how these two independent pathways could be integrated in an interdependent manner. First, in response to DNA damage, p53 inhibits mTORC1 through AMPK/TSC1/2 (, pointer #1). Second, in response to DNA damage, p38α/AKT/mTORC1/S6K1 phosphorylates MDM2 and activated S6K1 binds to MDM2 thereby elevating p53 levels (, pointer #2) which, in turn, inhibits mTORC1. Thus, a circle of regulation (, rounded rectangle) would inhibit the growth of cells experiencing DNA damage. It is possible that high energy levels would favor the second pathway (, green box), perhaps in addition to the first (, red box) as an extra layer of regulation. This circle of regulation would integrate energy levels with genotoxic stress to regulate cell cycle arrest and cell growth in a negative feedback loop and provide an extra measure of growth regulation and tumor suppression.
Arrested Cells Become Quiescent or Senescent: Nyx Bears Geras and Old Age Shrouds All
The integration of growth pathways like mTORC1 with cell cycle arrest pathways like p53/p21 can push cell fate toward either quiescence or senescence ( pointer #3).Citation30 Quiescence and senescence are both characterized by the absence of proliferation, but the former is reversible with low metabolism, while the latter is permanent with high metabolism.Citation31,Citation32 As mentioned above, p53 arrests the cell cycle in response to DNA damage. Most p53-arrested cells re-enter the cell cycle after the damage is repaired. However, if the damage is severe, then the arrested state could become permanent, or the cell could become senescent.
The precise mechanism that drives arrested cells into senescence is not understood. It is presumed that p53 is at least partly culpable, perhaps by inducing prolonged p21 expression. However, p53 might not induce cellular senescence beyond stimulating p21 transcription, so the role of p53 in inducing senescence is unclear. It is also possible that p53 actually inhibits cellular senescence (a process coined gerosuppression).Citation30 For example, prolonged p21 overexpression induces senescence, but simultaneous p53 overexpression prohibited senescence in favor of quiescence.Citation33 Furthermore, mTORC1 could induce senescence in these p21-expressing cells (a process coined geroconversion).Citation30 mTOR activation converts quiescent cells into senescent cells when they were growth arrested by nutlin-3a (nutlin-3a activates p53-induced quiescence but inhibits p53-induced senescence).Citation34 Rapamycin reverses this process. Furthermore, TSC1-dependent mTORC1 suppression was crucial for maintaining naive T cells in a quiescent state.Citation35 Finally, p53-mediated DNA damage responses could be inconsequential for tumor suppression. Mice that express a mutant p53 defective for acetylation of three lysines (3KR: K117R, K161R and K162R) are unable to mount a DNA damage response but are protected from cancer.Citation36 Even though the DNA damage response is negated, p533KR/3KR mutant mice can still regulate glycolysis and reactive oxidation species production, activities that also influence growth pathways like mTORC1.Citation37,Citation38 Thus, p53 and mTORC1 (and their integration) can influence the choice between cellular quiescence and senescence (or apoptosis).
p53 Inhibits mTORC1 to Suppress Cancer: Nemesis Enacts Divine Retribution against Hubris
mTORC1 inhibitors like rapamycin show promise as anticancer therapies.Citation10 Based on p53/mTORC1 interactions that ultimately elevate p53, it is of interest to know whether rapamycin can suppress tumors that lack p53. This knowledge is important because p53 is mutated in about half of all human tumors.Citation11 Some reports suggest p53 is important for rapamycin-mediated tumor suppression because p53 reduces cellular proliferation after mTORC1 inhibition. For example, RAD001 (a rapalog) suppressed hepatocyte proliferation during chronic liver injury that was dependent on p53.Citation39 Further, RAD001 combined with cisplatin suppressed the growth of hepatocellular carcinoma cells, which was enhanced by p53 expression.Citation40 Another genotoxin, doxorubicin, caused acute cardiac dysfunction and reduced cardiac mass through p53-dependent mTOR inhibition.Citation40 Furthermore, a p53 activator (chloroquine) in combination with RAD001 reduced mammary tumors in a mouse model.Citation41 Finally, consistent with rapamycin’s ability to block bladder cancer development, deletion of p53 and PTEN led to invasive bladder cancer with upregulated mTOR.Citation42 These data support the possibility that p53 is important for rapamycin-mediated tumor activity.
There is also evidence that does not support the notion that rapamycin is dependent on p53 to fight cancer. For example, in a cancer treatment model, RAD001 synergized with ionizing radiation to increase cell death in pre-B acute lymphocytic leukemia cells independent of p53.Citation43 In addition, rapamycin prevented early onset of oral mucosal papillomas deleted for p53 and expressing mutant K-Ras,Citation44 and rapamycin delayed the onset or progression of tumors in p53−/− and p53+/− mice to extend their lifespan.Citation45,Citation46 Thus, rapamycin’s dependence on p53 for suppressing tumor growth could be influenced by context and might not be absolute.
DNA Damage Responses Vs. Cell Growth and Aging: Achilles’ Death Spawns Immortality
Data suggests that DNA damage responses influence longevity. The cyclin-dependent kinase inhibitors p21 and p16 are considered aging biomarkers, and their overexpression induces cellular senescence.Citation47 In addition, removal of p16-expressing cells improves survival and delays aging for the BubR1-deficeint progeroid mouse model.Citation48 Furthermore, at the cellular level, p53 overexpression induces replicative senescence,Citation49 while p53-deletion induces immortality.Citation49 Naturally, cellular immortality does not equate to organismal longevity, but instead to cancer predisposition (the arrow that found Achilles’ heel).
At the organismal level, the impact that p53 has on longevity is complex. Mice with extra copies of intact p53 have an enhanced DNA damage response and less cancer, but a normal lifespan.Citation50,Citation51 Likewise, mice with extra copies of the INK4a/Arf locus, which encodes both p16INK4a, a positive regulator of retinoblastoma tumor suppressor protein, and Arf, a positive regulator of p53, are cancer-resistant but have a normal lifespan.Citation52 However, mice with extra copies of both the p53 locus and the INK4a/Arf locus are both cancer-resistant and long-lived. These studies suggest that enhanced—but regulated—p53 activity might promote longevity in conjunction with other cell cycle regulators such as p16INK4a, independent of its ability to suppress cancer. On the other hand, mice that carry one wild-type p53 gene and one p53 gene that produces an N-terminally truncated protein show early aging and a shortened lifespan, despite a low cancer incidence.Citation53,Citation54 Because p53 normally acts as a tetramer, the truncated p53 interacts with full-length p53 to increase the stability, nuclear localization and activity of the tetramer.Citation55 Thus, enhanced but unregulated (or differently regulated) p53 promotes aging. N-terminally truncated p53 occurs naturally, so these products may influence lifespan.Citation56 Taken together, the current evidence suggests that balanced p53 activity is essential for optimal longevity. This idea is consistent with the data discussed above showing a dynamic tension between the pro-growth activities of mTORC1 and the growth-suppressive activities of p53.
Likewise, several lines of evidence suggest that the mTORC1 pathway shortens lifespan—that is, mTORC1 activity enhances organismal senescence, not just cellular senescence. In support of this possibility, S6K1 deletion increases lifespan in mice.Citation57 Furthermore, the NIH Intervention Testing Program showed that microencapsulated and enterically released rapamycin (eRapa) increased lifespan when fed to genetically heterogeneous and inbred mice.Citation58-Citation60 It is likely that tumor suppression contributes to rapamycin-mediated lifespan extension, since several forms of tumors presented at a lower rate or a delayed onset. In addition, rapamycin reduced cancer by 46% for human kidney transplant patients.Citation9 However, rapamycin likely ameliorates multiple aspects of aging beyond tumors because it increases lifespan for a variety of evolutionarily diverse species that do not suffer from cancer, including yeast, worms and flies.Citation61 In addition, eRapa enhances learning and memory in young mice and maintains memory in old mice, and it acts as an anti-depressant.Citation62 Rapamycin also attenuated many age-related changes in mice, independent of cancer.Citation63 Furthermore, rapamycin improved survival and cardiac/skeletal muscle function for lamin A/C-deficient mice.Citation64 Lamin A deficiencies lead to multiple diseases in humans, one being Hutchinson-Gilford progeria syndrome that is due to a truncated form of prelamin A called progerin. Of interest, small amounts of progerin also occur naturally during aging and may contribute to the normal aging process. Progerin is aberrantly farnesylated lamin and farnesyltransferase inhibitors ameliorate Hutchinson-Gilford progeria syndrome in childrenCitation65 and in mouse models.Citation66 Therefore, it is noteworthy that rapamycin also ameliorates a lamin A defect, which must occur by indirect mechanisms because there is no evidence that rapamycin directly alters lamin A expression or processing, so rapamycin does not have any means for direct intervention. Thus, rapamycin can ameliorate aging for both wild-type and lamin A-deficient mice, suggesting that mTORC1 contributes to aging in both cases.
Conclusion: Orpheus Takes a Look Back
Cell growth is essential for development and survival. Likewise, the mTORC1 pathway is essential across species yet can curtail longevity if left unrestrained. Like Dionysus and Silenus at a banquet, intemperate mTORC1 causes problems. Without some restraint, excessive growth can result in a variety of afflictions, including cancer and age-related pathologies (and perhaps a waistline that would confound Atlas). Cell cycle responses answer the call to mete out justice or at least maintain the feast at an acceptable level of ecstasy. The relationship between the pro-growth pathways and the stress response pathways are likely to be extremely complicated and heavily influenced by environment and cell type. Nonetheless, this integration is key to understand growth and aging. With an understanding of these pathways and their integration, therapeutics could be developed and used to prevent, delay or treat a good number of pathologies like cancer and cardiovascular disease, sarcopenia and possibly other age-related afflictions.
Related Research Data
References
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274 - 93; http://dx.doi.org/10.1016/j.cell.2012.03.017; PMID: 22500797
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 2009; 10:307 - 18; http://dx.doi.org/10.1038/nrm2672; PMID: 19339977
- Tsang CK, Qi H, Liu LF, Zheng XF. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discov Today 2007; 12:112 - 24; http://dx.doi.org/10.1016/j.drudis.2006.12.008; PMID: 17275731
- Reiling JH, Sabatini DM. Stress and mTORture signaling. Oncogene 2006; 25:6373 - 83; http://dx.doi.org/10.1038/sj.onc.1209889; PMID: 17041623
- Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol 2003; 5:578 - 81; http://dx.doi.org/10.1038/ncb999; PMID: 12771962
- Sato T, Nakashima A, Guo L, Tamanoi F. Specific activation of mTORC1 by Rheb G-protein in vitro involves enhanced recruitment of its substrate protein. J Biol Chem 2009; 284:12783 - 91; http://dx.doi.org/10.1074/jbc.M809207200; PMID: 19299511
- Vézina C, Kudelski A, Sehgal SN. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo) 1975; 28:721 - 6; http://dx.doi.org/10.7164/antibiotics.28.721; PMID: 1102508
- Koser PL, Eng WK, Bossard MJ, McLaughlin MM, Cafferkey R, Sathe GM, et al. The tyrosine89 residue of yeast FKBP12 is required for rapamycin binding. Gene 1993; 129:159 - 65; http://dx.doi.org/10.1016/0378-1119(93)90264-4; PMID: 8325502
- Piselli P, Serraino D, Segoloni GP, Sandrini S, Piredda GB, Scolari MP, et al, The Immunosuppression and Cancer Study Group. Risk of de novo cancers after transplantation: Results from a cohort of 7217 kidney transplant recipients, Italy 1997-2009. Eur J Cancer 2012; 8049:00727 - 7; PMID: 23062667
- Hasty P. Rapamycin: the cure for all that ails. J Mol Cell Biol 2010; 2:17 - 9; http://dx.doi.org/10.1093/jmcb/mjp033; PMID: 19805415
- Shangary S, Wang S. Targeting the MDM2-p53 interaction for cancer therapy. Clin Cancer Res 2008; 14:5318 - 24; http://dx.doi.org/10.1158/1078-0432.CCR-07-5136; PMID: 18765522
- Beckerman R, Prives C. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol 2010; 2:a000935; http://dx.doi.org/10.1101/cshperspect.a000935; PMID: 20679336
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell 2009; 137:413 - 31; http://dx.doi.org/10.1016/j.cell.2009.04.037; PMID: 19410540
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9:402 - 12; http://dx.doi.org/10.1038/nrm2395; PMID: 18431400
- Meek DW. The p53 response to DNA damage. DNA Repair (Amst) 2004; 3:1049 - 56; http://dx.doi.org/10.1016/j.dnarep.2004.03.027; PMID: 15279792
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 2003; 3:155 - 68; http://dx.doi.org/10.1038/nrc1011; PMID: 12612651
- Budanov AV, Lee JH, Karin M. Stressin’ Sestrins take an aging fight. EMBO Mol Med 2010; 2:388 - 400; http://dx.doi.org/10.1002/emmm.201000097; PMID: 20878915
- Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995; 378:206 - 8; http://dx.doi.org/10.1038/378206a0; PMID: 7477327
- Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995; 378:203 - 6; http://dx.doi.org/10.1038/378203a0; PMID: 7477326
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA 2005; 102:8204 - 9; http://dx.doi.org/10.1073/pnas.0502857102; PMID: 15928081
- Ellisen LW, Ramsayer KD, Johannessen CM, Yang A, Beppu H, Minda K, et al. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol Cell 2002; 10:995 - 1005; http://dx.doi.org/10.1016/S1097-2765(02)00706-2; PMID: 12453409
- Horton LE, Bushell M, Barth-Baus D, Tilleray VJ, Clemens MJ, Hensold JO. p53 activation results in rapid dephosphorylation of the eIF4E-binding protein 4E-BP1, inhibition of ribosomal protein S6 kinase and inhibition of translation initiation. Oncogene 2002; 21:5325 - 34; http://dx.doi.org/10.1038/sj.onc.1205662; PMID: 12149653
- Fontoura BM, Atienza CA, Sorokina EA, Morimoto T, Carroll RB. Cytoplasmic p53 polypeptide is associated with ribosomes. Mol Cell Biol 1997; 17:3146 - 54; PMID: 9154813
- Fontoura BM, Sorokina EA, David E, Carroll RB. p53 is covalently linked to 5.8S rRNA. Mol Cell Biol 1992; 12:5145 - 51; PMID: 1406686
- Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, et al. Regulation of PTEN transcription by p53. Mol Cell 2001; 8:317 - 25; http://dx.doi.org/10.1016/S1097-2765(01)00323-9; PMID: 11545734
- Lai KP, Leong WF, Chau JF, Jia D, Zeng L, Liu H, et al. S6K1 is a multifaceted regulator of Mdm2 that connects nutrient status and DNA damage response. EMBO J 2010; 29:2994 - 3006; http://dx.doi.org/10.1038/emboj.2010.166; PMID: 20657550
- Lee CH, Inoki K, Karbowniczek M, Petroulakis E, Sonenberg N, Henske EP, et al. Constitutive mTOR activation in TSC mutants sensitizes cells to energy starvation and genomic damage via p53. EMBO J 2007; 26:4812 - 23; http://dx.doi.org/10.1038/sj.emboj.7601900; PMID: 17962806
- Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, et al. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest 2003; 112:1223 - 33; PMID: 14561707
- Habib SL, Yadav A, Mahimainathan L, Valente AJ. Regulation of PI 3-K, PTEN, p53, and mTOR in Malignant and Benign Tumors Deficient in Tuberin. Genes Cancer 2011; 2:1051 - 60; http://dx.doi.org/10.1177/1947601912445376; PMID: 22737271
- Blagosklonny MV. Tumor suppression by p53 without apoptosis and senescence: conundrum or rapalog-like gerosuppression?. Aging (Albany NY) 2012; 4:450 - 5; PMID: 22869016
- Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY) 2012; 4:159 - 65; PMID: 22394614
- Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol 2011; 192:547 - 56; http://dx.doi.org/10.1083/jcb.201009094; PMID: 21321098
- Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci USA 2010; 107:9660 - 4; http://dx.doi.org/10.1073/pnas.1002298107; PMID: 20457898
- Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY) 2010; 2:344 - 52; PMID: 20606252
- Yang K, Neale G, Green DR, He W, Chi H. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat Immunol 2011; 12:888 - 97; http://dx.doi.org/10.1038/ni.2068; PMID: 21765414
- Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012; 149:1269 - 83; http://dx.doi.org/10.1016/j.cell.2012.04.026; PMID: 22682249
- Blagosklonny MV. Aging: ROS or TOR. Cell Cycle 2008; 7:3344 - 54; http://dx.doi.org/10.4161/cc.7.21.6965; PMID: 18971624
- Leontieva OV, Blagosklonny MV. Yeast-like chronological senescence in mammalian cells: phenomenon, mechanism and pharmacological suppression. Aging (Albany NY) 2011; 3:1078 - 91; PMID: 22156391
- Buitrago-Molina LE, Pothiraju D, Lamlé J, Marhenke S, Kossatz U, Breuhahn K, et al. Rapamycin delays tumor development in murine livers by inhibiting proliferation of hepatocytes with DNA damage. Hepatology 2009; 50:500 - 9; http://dx.doi.org/10.1002/hep.23014; PMID: 19642171
- Zhu W, Soonpaa MH, Chen H, Shen W, Payne RM, Liechty EA, et al. Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation 2009; 119:99 - 106; http://dx.doi.org/10.1161/CIRCULATIONAHA.108.799700; PMID: 19103993
- Loehberg CR, Strissel PL, Dittrich R, Strick R, Dittmer J, Dittmer A, et al. Akt and p53 are potential mediators of reduced mammary tumor growth by cloroquine and the mTOR inhibitor RAD001. Biochem Pharmacol 2012; 83:480 - 8; http://dx.doi.org/10.1016/j.bcp.2011.11.022; PMID: 22142888
- Puzio-Kuter AM, Castillo-Martin M, Kinkade CW, Wang X, Shen TH, Matos T, et al. Inactivation of p53 and Pten promotes invasive bladder cancer. Genes Dev 2009; 23:675 - 80; http://dx.doi.org/10.1101/gad.1772909; PMID: 19261747
- Saunders P, Cisterne A, Weiss J, Bradstock KF, Bendall LJ. The mammalian target of rapamycin inhibitor RAD001 (everolimus) synergizes with chemotherapeutic agents, ionizing radiation and proteasome inhibitors in pre-B acute lymphocytic leukemia. Haematologica 2011; 96:69 - 77; http://dx.doi.org/10.3324/haematol.2010.026997; PMID: 20952516
- Raimondi AR, Molinolo A, Gutkind JS. Rapamycin prevents early onset of tumorigenesis in an oral-specific K-ras and p53 two-hit carcinogenesis model. Cancer Res 2009; 69:4159 - 66; http://dx.doi.org/10.1158/0008-5472.CAN-08-4645; PMID: 19435901
- Comas M, Toshkov I, Kuropatwinski KK, Chernova OB, Polinsky A, Blagosklonny MV, et al. New nanoformulation of rapamycin Rapatar extends lifespan in homozygous p53−/− mice by delaying carcinogenesis. Aging (Albany, NY Online) 2012; 4:728 - 35
- Komarova EA, Antoch MP, Novototskaya LR, Chernova OB, Paszkiewicz G, Leontieva OV, et al. Rapamycin extends lifespan and delays tumorigenesis in heterozygous p53+/− mice. Aging (Albany, NY Online) 2012; 4:719 - 27
- Coppé JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem 2011; 286:36396 - 403; http://dx.doi.org/10.1074/jbc.M111.257071; PMID: 21880712
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011; 479:232 - 6; http://dx.doi.org/10.1038/nature10600; PMID: 22048312
- Harvey M, Sands AT, Weiss RS, Hegi ME, Wiseman RW, Pantazis P, et al. In vitro growth characteristics of embryo fibroblasts isolated from p53-deficient mice. Oncogene 1993; 8:2457 - 67; PMID: 8103211
- García-Cao I, García-Cao M, Martín-Caballero J, Criado LM, Klatt P, Flores JM, et al. “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J 2002; 21:6225 - 35; http://dx.doi.org/10.1093/emboj/cdf595; PMID: 12426394
- Mendrysa SM, O’Leary KA, McElwee MK, Michalowski J, Eisenman RN, Powell DA, et al. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev 2006; 20:16 - 21; http://dx.doi.org/10.1101/gad.1378506; PMID: 16391230
- Matheu A, Pantoja C, Efeyan A, Criado LM, Martín-Caballero J, Flores JM, et al. Increased gene dosage of Ink4a/Arf results in cancer resistance and normal aging. Genes Dev 2004; 18:2736 - 46; http://dx.doi.org/10.1101/gad.310304; PMID: 15520276
- Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 2002; 415:45 - 53; http://dx.doi.org/10.1038/415045a; PMID: 11780111
- Maier B, Gluba W, Bernier B, Turner T, Mohammad K, Guise T, et al. Modulation of mammalian life span by the short isoform of p53. Genes Dev 2004; 18:306 - 19; http://dx.doi.org/10.1101/gad.1162404; PMID: 14871929
- Moore L, Lu X, Ghebranious N, Tyner S, Donehower LA. Aging-associated truncated form of p53 interacts with wild-type p53 and alters p53 stability, localization, and activity. Mech Ageing Dev 2007; 128:717 - 30; http://dx.doi.org/10.1016/j.mad.2007.10.011; PMID: 18061646
- Bourdon JC, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, et al. p53 isoforms can regulate p53 transcriptional activity. Genes Dev 2005; 19:2122 - 37; http://dx.doi.org/10.1101/gad.1339905; PMID: 16131611
- Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009; 326:140 - 4; http://dx.doi.org/10.1126/science.1177221; PMID: 19797661
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009; 460:392 - 5; PMID: 19587680
- Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci 2011; 66:191 - 201; http://dx.doi.org/10.1093/gerona/glq178; PMID: 20974732
- Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, et al. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle 2011; 10:4230 - 6; http://dx.doi.org/10.4161/cc.10.24.18486; PMID: 22107964
- Kapahi P, Chen D, Rogers AN, Katewa SD, Li PW, Thomas EL, et al. With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab 2010; 11:453 - 65; http://dx.doi.org/10.1016/j.cmet.2010.05.001; PMID: 20519118
- Halloran J, Hussong SA, Burbank R, Podlutskaya N, Fischer KE, Sloane LB, et al. Chronic inhibition of mammalian target of rapamycin by rapamycin modulates cognitive and non-cognitive components of behavior throughout lifespan in mice. Neuroscience 2012; 223:102 - 13; http://dx.doi.org/10.1016/j.neuroscience.2012.06.054; PMID: 22750207
- Wilkinson JE, Burmeister L, Brooks SV, Chan CC, Friedline S, Harrison DE, et al. Rapamycin slows aging in mice. Aging Cell 2012; 11:675 - 82; http://dx.doi.org/10.1111/j.1474-9726.2012.00832.x; PMID: 22587563
- Ramos FJ, Chen SC, Garelick MG, Dai DF, Liao CY, Schreiber KH, et al. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Transl Med 2012; 4:ra103; http://dx.doi.org/10.1126/scitranslmed.3003802; PMID: 22837538
- Gordon LB, Kleinman ME, Miller DT, Neuberg DS, Giobbie-Hurder A, Gerhard-Herman M, et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA 2012; 109:16666 - 71; http://dx.doi.org/10.1073/pnas.1202529109; PMID: 23012407
- Fong LG, Frost D, Meta M, Qiao X, Yang SH, Coffinier C, et al. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science 2006; 311:1621 - 3; http://dx.doi.org/10.1126/science.1124875; PMID: 16484451