Abstract
Aurora A kinase has drawn considerable attention as a therapeutic target for cancer therapy. However, the underlying molecular and cellular mechanisms of the anticancer effects of Aurora A kinase inhibition are still not fully understood. Herein, we show that depletion of Aurora A kinase by RNA interference (RNAi) in hepatocellular carcinoma (HCC) cells upregulated FoxO1 in a p53-dependent manner, which induces cell cycle arrest. Introduction of an RNAi-resistant Aurora A kinase into Aurora A-knockdown cells resulted in downregulation of FoxO1 expression and rescued proliferation. In addition, silencing of FoxO1 in Aurora A-knockdown cells allowed the cells to exit cytostatic arrest, which, in turn, led to massive cell death. Our results suggest that FoxO1 is responsible for growth arrest at the G2/M phase that is induced by Aurora A kinase inhibition.
Introduction
Aurora A is a serine/threonine protein kinase and is required for the centrosome duplication and bipolar spindle assembly needed for mitotic progression.Citation1 In cycling cells, the mRNA and protein levels of Aurora A kinase are low at the G1/S transition and gradually increase as the cells progress through the S phase and the G2/M transition then return to a low level as cells resume the G1 phase.Citation2 Aberrant expression or regulation of Aurora A is frequently observed in several malignancies, including colorectal, breast, colon, pancreatic, ovarian, bladder, gastric and hepatocellular carcinomas. Dysregulation is associated with advanced tumor stage and poor prognosis.Citation2-Citation5 Therefore, recent research has focused on developing drugs that inhibit aberrant activation of Aurora A kinase, and several selective inhibitors of Aurora A, including MLN 8237 and MLN 8054, have been developed and tested in clinical trials.Citation6-Citation10
Aurora A interacts with a number of proteins that are required for commitment to mitosis and cell cycle checkpoints including TPX2, TACC, FBXL7 and PLK1.Citation1,Citation11 Recent reports indicate that Aurora A regulates Akt, GSK-3β, mTOR, BRCA1, BRCA2 and IκBα.Citation12-Citation16 Several studies have demonstrated the interaction between Aurora A kinase and p53. Aurora A directly phosphorylates p53, enhancing MDM2-mediated ubiquitination of p53, and inhibition of Aurora A leads to an increase in p53 stability and results in cell cycle arrest in G2/M phase.Citation17 In addition, p53 is an important determinant for polyploidy or aberrant mitosis-induced apoptosis.Citation18,Citation19 Many studies using different cancer cell lines have shown that inhibition of Aurora A, either by RNAi or small-molecule inhibitors, induces G2 arrest or mitotic arrest, which, in turn, triggers apoptosis.Citation20-Citation23 On the other hand, a recent study demonstrated that treatment with MLN 8054 induces cellular senescence.Citation24 Although many studies have shown that inhibition of Aurora A inhibits growth and confers pro-apoptotic effects, little is known about the underlying mechanism.
Forkhead transcription factor FoxO1 is the most abundant isoform in insulin-responsive tissue, such as liver, and regulates the expression of genes that are involved in apoptosis, cell cycle, metabolism, stress response and differentiation.Citation25 A recent study showed that FoxO3a is an important regulator of chemosensitivity to combined treatment of alisertib and ara-C in human AML cells.Citation26 FoxO3a transcription factors induce cell cycle arrest in G1 by transcriptionally activating cyclin-dependent kinase inhibition (CDKI), p27 and the Rb (Retinoblastoma) family member, p130.Citation27,Citation28 FoxO3a proteins are also required for cell cycle progression by promoting the expression of cyclin B1 and polo-like kinase (Plk).Citation29 In response to oxidative stress, FoxO4 proteins participate in G2/M checkpoint through upregulation of GADD45 expression.Citation30 In addition, several studies reported that FoxM1 regulates expression of G2-specific genes and is required for proper mitotic progression.Citation31 Given the important roles of FoxO transcription factors in chromosome stability and in the mitotic process,Citation29,Citation31 we examined whether Aurora A is functionally engaged with FoxO in mitotic progression.
Here, we show that depletion of Aurora A upregulates FoxO1 via transcriptional activation, resulting in cell cycle arrest at the G2/M phase in a hepatocellular carcinoma (HCC) cell line. Reintroduction of functional Aurora A kinase into Aurora A-knockdown cells led to downregulation of FoxO1 and its downstream target, p21. Moreover, depletion of FoxO1 in the absence of Aurora A kinase allowed cells to exit mitotic arrest, resulting in cell death. Our results suggest that FoxO1 is a potential target of Aurora A, and propose a new regulatory mechanism of mitosis that is activated by Aurora A inhibition.
Results
FoxO1 expression is upregulated in the absence of Aurora A kinase
To gain insight into the possible involvement of FoxO1 and Aurora A in cell cycle progression, we measured their RNA and protein levels at different stages of the cell cycle in HepG2 cells. FoxO1 was expressed at a lower level in M phase than in G1, S or G2 phase, whereas Aurora A kinase was expressed at the highest level during M phase (). Cell cycle phases were confirmed by FACS analysis (). These data imply that Aurora A and FoxO1 proteins are inversely related in M phase. Thus, we hypothesized that FoxO1 and Aurora A kinase are functionally engaged in the mitotic progression.
Figure 1. Expression of FoxO1 and Aurora A kinase is inversely related. (A) Western blot analysis of total FoxO1, phospho-FoxO1 (S256), p21 and Aurora A expression at different cell cycle stages in HepG2 cells. Equal loading of protein was confirmed by GAPDH. (B) Quantitative real-time PCR of FoxO1 and Aurora A expression at different cell cycle stages of HepG2 cells. RNA levels were normalized to GAPDH. (C) Cell cycle phases of HepG2 cells were confirmed by FACA analysis. (D) Knockdown of Aurora A was confirmed by quantitative real-time RT-PCR. The graph represents means ± SD for the change in the mRNA level relative to GAPDH from three independent experiments. (E) Expression of FoxO1 and FoxO3a in Aurora A-knockdown cells was measured by quantitative real-time RT-PCR. GAPDH expression was examined as an internal control. Data represent the means ± SD of three independent experiments.
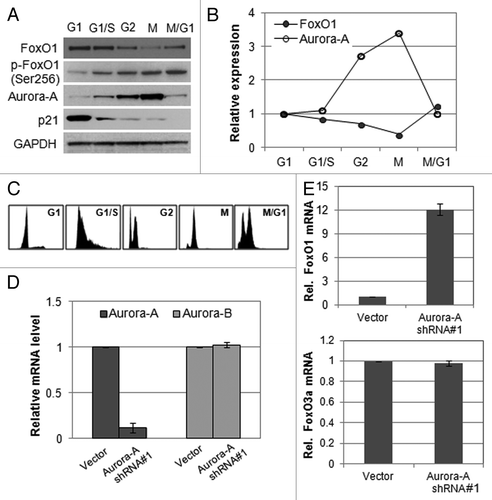
To further investigate whether Aurora A is linked with FoxO1, we depleted Aurora A kinase in HepG2 cells by transducing Aurora A kinase-specific shRNA (short hairpin RNA) or vector (pLKO.1) lentiviral particles and measured the expression of FoxO1. Aurora A shRNA-expressing HepG2 cells showed an 80% reduction in Aurora A mRNA levels, while the expression of Aurora B kinase was unaffected (). We observed that, after Aurora A depletion, mRNA levels of FoxO1 was upregulated, whereas FoxO3a expression was not changed ().
FoxO1 expression is upregulated in p53-proficient cells but not in p53-mutant or -null cells
It has been reported that Aurora A kinase interacts with p53, and both proteins are functionally associated at multiple levels.Citation17,Citation32-Citation34 To evaluate the contribution of p53 to the upregulation of FoxO1 in response to Aurora A inhibition, we depleted Aurora A kinase in additional HCC cell lines, including Huh-7 (p53 mutant), Hep3B (p53 deleted) and SK-HEP-1 (p53 wild-type) cells, and determined FoxO1 expression by western blot analysis. While FoxO1 expression was significantly increased in HepG2 and SK-HEP-1 cells by Aurora A knockdown (), its expression was not changed in Hep3B and Huh-7 cells despite the significant reductions in Aurora A kinase (). In addition, the level of phospho-FoxO1 was significantly reduced in HepG2 and SK-HEP-1 cells (). These data showed that silencing of Aurora A kinase resulted in upregulation of FoxO1 along with reduction of phosphorylation. These data suggest that FoxO1 expression is upregulated in p53-proficient cells but not in p53-deficient cells.
Figure 2. Depletion of Aurora A induces upregulation of FoxO1 expression in HepG2 and SK-HEP-1 cells, and RNAi-resistant Aurora A downregulates FoxO1 expression. (A) Western blot analysis of total FoxO1, phospho-FoxO1 (S256), p21 and Aurora A expression in vector or Aurora A shRNA-expressing HepG2 and SK-HEP-1 cells. (B) Western blot analysis of total FoxO1, phospho-FoxO1 (S256), p21 and Aurora A expression in vector or Aurora A shRNA-expressing Huh-7 and Hep3B cells. Two different Aurora A shRNA were used. Equal loading of protein was confirmed by GAPDH. (C) Nucleotides substituted (base changes are underlined) in the vector expressing the RNAi-resistant Aurora A. (D) HepG2 cells were transduced with pLKO.1 or Aurora A shRNA lentiviral particles. At 48 h after the transduction, cells were transfected with RNAi-resistant Aurora A containing or empty vector. Protein lysates were extracted 48 h later and followed by western blot analysis with the indicated antibodies. (E) Cell proliferation in control and Aurora A-knockdown cells that expressed either empty vector or RNAi resistant Aurora A were measured on the indicated days. Data represent the means ± SD of three independent experiments.
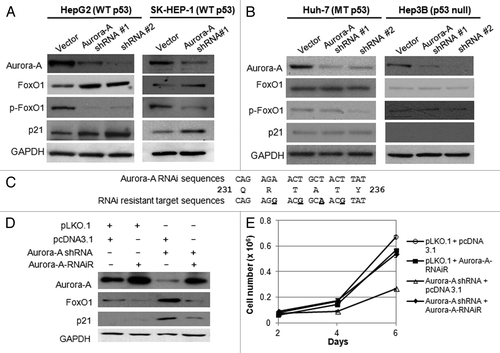
To validate that the upregulation of FoxO1 was solely due to the absence of functional Aurora A kinase, a plasmid expressing the RNAi-resistant wild-type Aurora A kinase was generated by introducing site-directed mutations within the RNAi target sequences (). Reintroduction of functional Aurora A kinase resulted in downregulation of FoxO1 and p21 () and rescue of cell proliferation (). This suggests that Aurora A regulates expression of FoxO1 either directly or indirectly, and that FoxO1 mediates the inhibition of growth in Aurora A-depleted cells.
Recent reports indicate that inhibition of Aurora A induces senescence.Citation24 Because we observed upregulation of p21, which is known to induce senescenceCitation35 in the absence of Aurora A kinase (), we examined the SA-β-gal (senescence-associated-β-galactosidase) activity in Aurora A shRNA HepG2 cells. There were no detectable SA-β-gal-positive cells among the Aurora A-knockdown cells (data not shown).
Depletion of Aurora A results in G2/M arrest in HepG2 cells, accompanied by upregulation of FoxO1
We monitored the cell growth of Aurora A-knockdown HepG2 cells for 8 d and found their proliferation was inhibited compared with control cells (). There have been many reports of delays in the G2/M transition or of mitotic arrest after the inhibition of Aurora A, using RNAi or small-molecule inhibitors.Citation20,Citation22 To determine the nature of the growth inhibition shown in , HepG2, Huh-7 and Hep3B cells were synchronized at the G1/S boundary by a double thymidine block. They were then released into fresh medium and examined the DNA ploidy using flow cytometry analysis. The result showed that knockdown of Aurora A in HepG2 cells leads to G2/M arrest with an accumulation of 4N DNA content as compared with control. However, Aurora A-knockdown Huh-7 and Hep3B cells progressed through G2/M and accumulated with greater 4N or 8N DNA content (). These data suggest that depletion of Aurora A in p53-proficient HepG2 cells, which upregulates Foxo1 expression, lead to G2/M arrest, while p53-deficient Hep3B or Huh-7 cells were allowed to exit mitosis and proceed to an additional round of cell cycle.
Figure 3. Silencing of Aurora A induces G2/M arrest. (A) The growth of vector and Aurora A shRNA HepG2 cells were measured on the indicated days. Data shown are the mean ± SD of three independent experiments. (B) Cell cycle profile of synchronized vector control and Aurora A shRNA HepG1, Huh-7 and Hep3B cells was analyzed by flow cytometry analysis. Percent of G2/M cells is shown. (C) The level of FoxO1, cyclin B1 and Aurora A during M phase was measured by western blot analysis. Nocodazole (100 ng/ml) arrested control and Aurora A shRNA HepG2 and Hep3B cells were released from nocodazole block, and protein lysates were prepared at the indicated time thereafter. Equal loading of protein was confirmed by GAPDH.
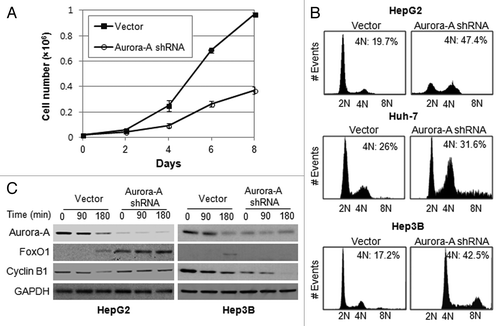
To examine the expression of FoxO1 in mitotic progression, vector or Aurora A shRNA-expressing HepG2 and Hep3B cells were arrested with nocodazole and released to allow the progress to mitosis by removing the arresting agent, and the level of FoxO1 during M phase was measured by western blot analysis. While FoxO1 levels were not increased in the Aurora A-knockdown Hep3B cells (), there was significantly more FoxO1 in the Aurora A shRNA-expressing HepG2 cells, and the elevated levels were sustained throughout M phase (). To assure that cells undergo mitotic progression, we examined expression of mitotic-specific cyclin B1 by western blotting. The data showed that cyclin B1 was degraded in a time-dependent manner in Hep3B cells (), suggesting that the cells proceeded through G2 /M progression. However, degradation of cyclin B1 appeared to be delayed in Aurora A-knockdown HepG2 cells compared with control cells, reflecting that silencing of Aurora A leads to G2/M arrest ().
FoxO1 is responsible for G2/M arrest in the Aurora A kinase-depleted HepG2 cells
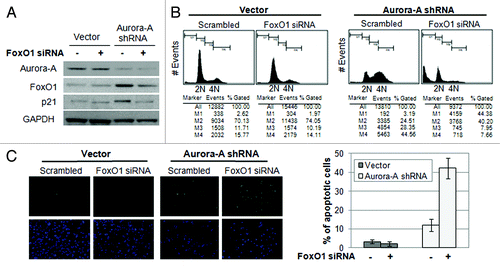
As we observed, an increase of FoxO1 expression and a delay in mitotic completion in Aurora A-knockdown cells, we hypothesized that the increase in FoxO1 is responsible for G2/M arrest in Aurora A-knockdown cells. To examine the function of FoxO1 on cell cycle progression, we depleted FoxO1 using FoxO1-specific siRNA in control and Aurora A-knockdown HepG2 cells (). The cell cycle profile of the cells was analyzed after 72 h by using flow cytometry analysis. Aurora A-knockdown cells transfected with FoxO1 siRNA exited from G2/M growth arrest () and induced massive cell death (). These results suggest that the FoxO1 induced by Aurora A knockdown was responsible for cytostatic growth arrest in the G2/M phase.
Figure 4. Depletion of FoxO1 in Aurora A-knockdown cells lead to exit G2/M arrest and induce apoptosis. (A) FoxO1-specific siRNA was transfected into vector control and Aurora A shRNA HepG2 cells, and knockdown of FoxO1 was confirmed by western blot analysis. (B) Cell cycle profiles in scrambled or FoxO1-specific siRNA-transfected control and Aurora A-knockdown HepG2 cells were monitored at 72 h after transfection by flow cytometry analysis. Representative profiles and percent of subG1, G0/G1, S, G2/M cells from one of two independent experiments are shown. (C) The percentages of TUNEL-positive nuclei in scrambled or FoxO1-specific siRNA transfected control and Aurora A-knockdown cells 96 h after transfection. Data represent the means ± SD of two independent experiments.
Aurora A kinase regulates FoxO1 expression at the transcriptional level in a p53-dependent manner
As we observed upregulation of FoxO1 in p53-proficient cells but not in p53-deficient cells, we further asked whether functional p53 is required for upregulation of FoxO1 in the absence of Aurora A kinase. To this end, we transfected Hep3B cells with either wild-type p53 or mutant p53 (G135A), a transcriptional-inactive mutant, and detected the induction of FoxO1 expression in the presence or absence of Aurora A kinase. Notably, FoxO1 and p21 levels were increased in wild-type p53-expressing cells in the absence of Aurora A kinase (), suggesting that depletion of Aurora A kinase leads to upregulation of FoxO1 in a p53-dependent manner.
Figure 5. Aurora A regulates FoxO1 expression at the transcriptional level in a p53-dependent manner. (A) Hep3B cells were co-transfected with a combination of indicated plasmids, incubated for 48 h, and western blot analysis was performed. (B) FoxO1 mRNA half-life was determined by actinomycin D treatment. Vector control and Aurora A shRNA cells were treated with actinomycin D (10 μg/ml) for the indicated time points. FoxO1 mRNA levels were measured by real-time qRT-PCR. (C) For luciferase reporter assay, vector control and Aurora A-knockdown cells were transfected with luciferase reporter plasmid containing FoxO1 promoter. pGL3 vector was used as a negative control. Data shown are mean ± SD of three independent experiments. *p < 0.01. (D) HepG2 cells were transfected with a combination of the Aurora A shRNA and RNAi-resistant Aurora A variant in the presence of luciferase reporter plasmid containing FoxO1 promoter. pGL3 vector was used as a negative control. Data shown are mean ± SD of three independent experiments. *p < 0.05. (E) Hep3B cells were co-transfected with a combination of the indicated plasmids in the presence of luciferase reporter plasmid containing FoxO1 promoter. pGL3 vector was used as a negative control. Data shown are mean ± SD of three independent experiments. *p < 0.05.
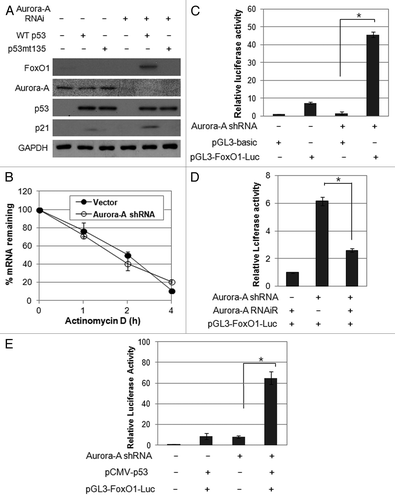
We explored the underlying molecular mechanism of FoxO1 induction by Aurora A knockdown. De novo mRNA synthesis was blocked using actinomycin D (10 μg/ml), and the half-life of FoxO1 mRNA was monitored using real-time qPCR to determine whether the increase in FoxO1 levels resulted from changes in the stability of FoxO1. No significant difference in FoxO1 mRNA stability of was observed between vector and Aurora A shRNA cells (), suggesting that the increase of FoxO1 mRNA by Aurora A inhibition was not due to increased stability.
We next examined whether Aurora A kinase controls transcription of FoxO1. To this end, the promoter of the FoxO1 gene, which spans a 1.8-kb sequence upstream of the first ATG, was linked to a luciferase reporter gene. Co-transfection of this plasmid and the Aurora A shRNA resulted in an approximately 5-fold increase in luciferase activity (). In addition, introduction of functional Aurora A reduced on luciferase activity under control of FoxO1 promoter (), indicating that Aurora A specifically regulates FoxO1 expression via transcriptional activation, either directly or indirectly. This is the first time that regulation of FoxO1 was shown to be affected by Aurora A kinase. To determine whether p53 is required for transcription of FoxO, Hep3B cells were co-transfected with promoter of the FoxO1 gene, p53 and Aurora A shRNA and analyzed the promoter activity. The results showed that the promoter activity of FoxO1 was significantly induced by overexpression of p53 (), further confirming that Aurora A may regulate FoxO1 expression in a p53-dependent manner.
Since we observed the effect of p53 expression on FoxO1 promoter-luciferase activity in the absence of Aurora A kinase, we analyzed potential transcription binding sites to identify the putative p53-responsive cis-acting elements in FoxO1 promoter using a computer search. This analysis showed that there is no putative p53-binding consensus sequence, indicating that the effect of p53 on FoxO1 promoter is indirect and requires other trans-acting factors.
Discussion
Numerous reports of preclinical and clinical trials have shown that the inhibition of Aurora A kinase has consistent anticancer effects. Silencing or inhibiting Aurora A kinase is effective for inhibition of growth and apoptosis in various tumor types.Citation36,Citation37 However, the detailed underlying mechanism of the growth inhibition that is induced by Aurora A-targeted suppression is still not fully understood. In this study, we showed that depletion of Aurora A kinase by RNAi in a human HCC cell line led to upregulation of FoxO1 via transcriptional activation in a p53-dependnet manner, and that the elevated FoxO1 coincides with a G2/M arrest. Conversely, the silencing of FoxO1 in Aurora A-knockdown cells drove the cells out of mitotic arrest and led to cell death. Our results suggest that FoxO1 transcription factor induces growth arrest in G2/M phase in the absence of Aurora A kinase and propose that combined inhibition of FoxO1 and Aurora A kinase has potential as a therapeutic strategy for promoting apoptosis.
FoxO1 is the most abundant member of the FoxO family of mammalian Forkhead transcription factors in insulin-responsive tissue, including the liver, and plays important roles in the regulation of various biological processes, including cell cycle progression, apoptosis and metabolism.Citation38 Increasing evidence shows that FoxO transcription factor plays a major role in cell cycle regulation.Citation29,Citation31,Citation39 FoxO3a transactivates cyclin B1 and Plk genes in G2 to complete mitosis and to progress to G1.Citation29 FoxO4 proteins function in the G2/M checkpoint when cells are under oxidative stress.Citation30 The data presented in our study show that depletion of Aurora A upregulates FoxO1, resulting in cell cycle arrest at the G2/M phase. Proteolytic degradation of cyclin B1 and a decrease in its transcription are a prerequisite for completion of mitosis and progression through G1 phase.Citation40-Citation42 Because degradation of cyclin B1 is slower in Aurora A-knockdown HepG2 cells than in controls during M phase, we cannot preclude the possibility that elevated FoxO1 replenishes cyclin B1 by upregulating its expression directly or indirectly.
Inhibition of FoxO1 using RNAi in the Aurora A-knockdown HepG2 cells promoted exit from G2/M arrest and resulted in significant apoptosis. We postulate that elevated FoxO1 protein might be responsible for growth arrest at G2/M phase in Aurora A-knockdown cells, and these arrested cells are allowed to exit through an additional round of the cell cycle when FoxO1 expression is inhibited by RNAi. In p53-proficient cells, the mitotic checkpoint in the cells might remain active due to functional p53, which eventually results in significant apoptotic cell death. Several selective inhibitors of Aurora kinases,including MK0457 and AZD1152, have been developed and tested in clinical trials. However, their use has been linked to prolonged stable disease states rather than cytocidal in several patients with different tumor types.Citation43 Our results suggest that combined inhibition of FoxO1 and Aurora A kinase might be a potential strategy for treatment of p53-positive HCC patients.
Aurora A kinase plays several important roles in mitotic progression in such processes as spindle assembly, centrosome maturation, chromosome segregation and checkpoint regulation. Citation44,Citation45 Inhibition of Aurora A, either by RNAi or by small molecule inhibitors in different cancer cell lines induces G2 or mitotic arrest, which, in turn, trigger apoptosis.Citation22,Citation23 Small molecule inhibitors of Aurora A also likely block Aurora B kinase and possibly other, unrelated, kinases. This makes it difficult to attribute any effects to inhibition of Aurora A kinase activity. We therefore took a genetic approach that used RNA interference to unravel the regulatory mechanism involved in Aurora A inhibition. Our data show that depletion of Aurora A kinase using RNAi results in G2/M arrest, accompanied by a 4N accumulation and an elevated level of FoxO1. These results suggest that Aurora A is functionally engaged with FoxO1 in G2/M progression. In HepG2 cells, expression of Aurora A kinase is highest during M phase, whereas the transcriptional activity of FoxO1 is lowest in M phase as compared with that in G1, S and G2 phases. This inverse relationship suggests that Aurora A might negatively regulate the FoxO transcription factor that regulates the transition from proliferative growth to quiescence, to maintain cell cycle progression. It has been demonstrated that p53 controls the upstream regulators of FoxO1 function.Citation46 In this study, we confirmed that FoxO1 is upregulated by transiently overexpressing p53 in p53-null Hep3B cells. Moreover, we showed that p53-proficient HepG2 cells upregulate FoxO1 expression in the absence of Aurora A kinase and are prevented from exiting mitotic arrest. Therefore, we speculate that FoxO1 might play a role in the p53-dependent mitotic checkpoint, in which Aurora A kinase may negatively regulate activities of p53.
Introduction of functional p53 to Aurora A-knockdown cells significantly increased luciferase activity under the control of FoxO1 promoter. However, sequence analysis revealed no putative p53-consensus sequences within the FoxO1 promoter. We speculate that p53 might be recruited indirectly to the FoxO1 promoter through binding to other trans-acting elements that are induced by silencing of Aurora A kinase. This may explain why expression of functional p53 itself did not induce luciferase activity of FoxO1 promoter.
In summary, our data showed that depletion of Aurora A leads to upregulation of FoxO1 transcription factor via transcriptional activation, which, in turn, leads to G2/M arrest in a p53-dependent manner. Moreover, combined inhibition of FoxO1 and Aurora A kinase induces massive cell death. We provide evidence that FoxO1 is a potential target of Aurora A and propose a new regulatory mechanism for growth arrest that is induced by Aurora A inhibition.
Materials and Methods
Cell culture and treatments
HepG2 and Hep3B cell lines were purchased from American Type Culture Collection (ATCC®). SK-HEP-1and Huh-7 cell line was obtained from the Korean Cell Line Bank (KCKB). Cells were grown in culture medium in Dulbecco’s minimal essential medium (DMEM) supplemented with 10% FBS (GIBCO) and antibiotics (50 U/ml penicillin and 50 μg/ml streptomycin). For G1 arrest, cells were synchronized by serum starvation (0.2% FBS) for 24 h. G1-arrested cells were released by the addition of normal growth medium (10% FBS) and cultured for 4 h for G1/S progression. Cells were synchronized at the G1/S border by inhibition of DNA synthesis using 2 mM thymidine (Sigma-Aldrich), and were released for 3 h for G2 phase. For M phase, cells were incubated with 100 ng/ml nocodazole (Sigma-Aldrich) for 16 h. Nocodazole-treated cells were washed with PBS and changed to fresh culture medium for 4 h for M to G1 transition. Cell cycle synchronization and progression was confirmed by FACS analysis. For determination of mRNA stability, cells were treated with 10 μg/ml actinomycin D (Sigma-Aldrich). Then RNA was extracted at different time points, and real-time qPCR was performed as described below.
Antibodies
Aurora A antibody was purchased from BD Transduction Laboratories. p21, cyclin B1, FoxO1, phospho-FoxO1 (S256) antibodies were purchased from Cell Signaling Technology, Inc. p53 (clone E26) antibody was from Epitomics INC. GAPDH antibody was purchased from Sigma-Aldrich.
RNA interference
Predesigned Mission® shRNA for human Aurora A kinase and control vector (pLKO.1) were purchased from Sigma-Aldrich. Two independent shRNAs for targeting Aurora A kinase (Aurora A shRNA#1 and #2) were used. Pre-designed FoxO1 siRNAs were purchased from Bioneer Corporation and the following sequences were used: 5′-CUGCAUAGCAUCAAGUCUU-3′ and 5′-AAGACUUGAUGCUAUGCAG-3′; 5′-GCUGCUGUAGAUAAGGACU-3′ and 5′-AGUCCUUAUCUACAGCAGC-3′.
Plasmid construction and site-directed mutagenesis
Aurora A cDNA was amplified and sub-cloned into the pcDNA3.1/Neo (+) vector. The primers used for PCR amplification were as follows: 5′-CTCGAGATGGACCGATCTAAAGAAAACTGC-3′ and 5′-GAATTCCTAAGACTGTTTGCTAGCTGATTC-3′. pCMV-p53 expression vector, which carries wild-type p53 or mutant p53 (p53mt135) was purchased from Clontech Laboratories, Inc. The p53mt135 gene differs from the p53 gene by a G to A conversion at nucleotide 1017, which causes a conformational change that prevents interaction with p53 DNA-binding sites. The RNAi-resistant Aurora A cDNA was generated by double-stranded DNA mutagenesis using the QuickChange Site-Directed Mutagenesis Kit (Agilent Technologies). The sense mutagenic primer was 5′-GTTTGATGAGCAGAGGACGGCAACGTATATAACAGAATTG-3′.
Luciferase reporter assay
For the luciferase reporter assay, the promoter of the FoxO1 gene was amplified and cloned into the pGL3-basic (Promega). The following primers were used: 5′-GGTACCCCTAATTTTTCCTTTTTTCCCCTC-3′ and 5′-AAGCTTGAGTGGAAGCGCGAGCCCAGAAC-3′. Control and Aurora A-knockdown cells were transfected with luciferase reporter plasmid containing 1.8 kb of FoxO1 promoter. Transfection was performed with Turbofect™ according to the manufacturer’s instruction (Fermentas INC.). Forty-eight hours after transfection, cells were harvested, and luciferase assays were performed. pGL3-basic vector was used as a negative control. β-galactosidase expression vector was co-transfected and measured its activity for normalization of transfection efficiency.
Quantitative RT-PCR
Real-time qRT-PCR was performed using a StepOne real-time PCR system with a 2 × POWER SYBR Green PCR Master mix (Applied Biosystems). The following primers were used for RT-PCR: Aurora A, 5′-AATGATTGAAGGTCGGATGC-3′ and 5′-TTCTCTGAGCATTGGCCTCT-3′; Aurora B, 5′-GCTCAAGGGAGAGCTGAAGA-3′ and 5′-GACAGATTGAAGGGCAGAGG-3′; FoxO1, 5′-AAGAGCGTGCCCTACTTCAA-3′ and 5′-AGGCCATTTGGAAAACTGTG-3′; FoxO3a, 5′-GACCTGCTCACTTCGGACTC-3′ and 5′-GGACTCACTCAAGCCCATGT-3′; Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-GTGAAGGTCGGAGTCAACG-3′ and 5′-GGTGAAGACGCCAGTGGACTC-3′.
TUNEL assay
Apoptosis was assessed with a commercially available kit (In Situ Cell Death detection kit, Fluorescein, Roche Applied Science) detecting terminal deoxynucleotide transferase (TdT)-mediated nick end labeling (TUNEL).
Acknowledgments
This research was Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2010–0023847 to S.-Y.L.). S.-Y.L. was supported by the Priority Research Center Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (2009–0094050).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol 2003; 4:842 - 54; http://dx.doi.org/10.1038/nrm1245; PMID: 14625535
- Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J 1998; 17:3052 - 65; http://dx.doi.org/10.1093/emboj/17.11.3052; PMID: 9606188
- Jeng YM, Peng SY, Lin CY, Hsu HC. Overexpression and amplification of Aurora-A in hepatocellular carcinoma. Clin Cancer Res 2004; 10:2065 - 71; http://dx.doi.org/10.1158/1078-0432.CCR-1057-03; PMID: 15041727
- Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet 1998; 20:189 - 93; http://dx.doi.org/10.1038/2496; PMID: 9771714
- Sakakura C, Hagiwara A, Yasuoka R, Fujita Y, Nakanishi M, Masuda K, et al. Tumour-amplified kinase BTAK is amplified and overexpressed in gastric cancers with possible involvement in aneuploid formation. Br J Cancer 2001; 84:824 - 31; http://dx.doi.org/10.1054/bjoc.2000.1684; PMID: 11259099
- Kumano M, Miyake H, Terakawa T, Furukawa J, Fujisawa M. Suppressed tumour growth and enhanced chemosensitivity by RNA interference targeting Aurora-A in the PC3 human prostate cancer model. BJU Int 2010; 106:121 - 7; http://dx.doi.org/10.1111/j.1464-410X.2009.09047.x; PMID: 19912186
- Warner SL, Bearss DJ, Han H, Von Hoff DD. Targeting Aurora-2 kinase in cancer. Mol Cancer Ther 2003; 2:589 - 95; PMID: 12813139
- Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PN Jr., Gandara DR. Aurora kinases as anticancer drug targets. Clin Cancer Res 2008; 14:1639 - 48; http://dx.doi.org/10.1158/1078-0432.CCR-07-2179; PMID: 18347165
- Wang XX, Liu R, Jin SQ, Fan FY, Zhan QM. Overexpression of Aurora-A kinase promotes tumor cell proliferation and inhibits apoptosis in esophageal squamous cell carcinoma cell line. Cell Res 2006; 16:356 - 66; http://dx.doi.org/10.1038/sj.cr.7310046; PMID: 16617331
- Li D, Zhu J, Firozi PF, Abbruzzese JL, Evans DB, Cleary K, et al. Overexpression of oncogenic STK15/BTAK/Aurora A kinase in human pancreatic cancer. Clin Cancer Res 2003; 9:991 - 7; PMID: 12631597
- Coon TA, Glasser JR, Mallampalli RK, Chen BB. Novel E3 ligase component FBXL7 ubiquitinates and degrades Aurora A, causing mitotic arrest. Cell Cycle 2012; 11:721 - 9; http://dx.doi.org/10.4161/cc.11.4.19171; PMID: 22306998
- Dar AA, Belkhiri A, El-Rifai W. The aurora kinase A regulates GSK-3beta in gastric cancer cells. Oncogene 2009; 28:866 - 75; http://dx.doi.org/10.1038/onc.2008.434; PMID: 19060929
- Taga M, Hirooka E, Ouchi T. Essential roles of mTOR/Akt pathway in Aurora-A cell transformation. Int J Biol Sci 2009; 5:444 - 50; http://dx.doi.org/10.7150/ijbs.5.444; PMID: 19564927
- Briassouli P, Chan F, Savage K, Reis-Filho JS, Linardopoulos S. Aurora-A regulation of nuclear factor-kappaB signaling by phosphorylation of IkappaBalpha. Cancer Res 2007; 67:1689 - 95; http://dx.doi.org/10.1158/0008-5472.CAN-06-2272; PMID: 17308110
- Yang G, Chang B, Yang F, Guo X, Cai KQ, Xiao XS, et al. Aurora kinase A promotes ovarian tumorigenesis through dysregulation of the cell cycle and suppression of BRCA2. Clin Cancer Res 2010; 16:3171 - 81; http://dx.doi.org/10.1158/1078-0432.CCR-09-3171; PMID: 20423983
- Chefetz I, Holmberg JC, Alvero AB, Visintin I, Mor G. Inhibition of Aurora-A kinase induces cell cycle arrest in epithelial ovarian cancer stem cells by affecting NFĸB pathway. Cell Cycle 2011; 10:2206 - 14; http://dx.doi.org/10.4161/cc.10.13.16348; PMID: 21623171
- Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet 2004; 36:55 - 62; http://dx.doi.org/10.1038/ng1279; PMID: 14702041
- Kaestner P, Stolz A, Bastians H. Determinants for the efficiency of anticancer drugs targeting either Aurora-A or Aurora-B kinases in human colon carcinoma cells. Mol Cancer Ther 2009; 8:2046 - 56; http://dx.doi.org/10.1158/1535-7163.MCT-09-0323; PMID: 19584233
- Gizatullin F, Yao Y, Kung V, Harding MW, Loda M, Shapiro GI. The Aurora kinase inhibitor VX-680 induces endoreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint function. Cancer Res 2006; 66:7668 - 77; http://dx.doi.org/10.1158/0008-5472.CAN-05-3353; PMID: 16885368
- Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med 2004; 10:262 - 7; http://dx.doi.org/10.1038/nm1003; PMID: 14981513
- Hata T, Furukawa T, Sunamura M, Egawa S, Motoi F, Ohmura N, et al. RNA interference targeting aurora kinase a suppresses tumor growth and enhances the taxane chemosensitivity in human pancreatic cancer cells. Cancer Res 2005; 65:2899 - 905; http://dx.doi.org/10.1158/0008-5472.CAN-04-3981; PMID: 15805292
- Du J, Hannon GJ. Suppression of p160ROCK bypasses cell cycle arrest after Aurora-A/STK15 depletion. Proc Natl Acad Sci USA 2004; 101:8975 - 80; http://dx.doi.org/10.1073/pnas.0308484101; PMID: 15178765
- Manfredi MG, Ecsedy JA, Meetze KA, Balani SK, Burenkova O, Chen W, et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci USA 2007; 104:4106 - 11; http://dx.doi.org/10.1073/pnas.0608798104; PMID: 17360485
- Huck JJ, Zhang M, McDonald A, Bowman D, Hoar KM, Stringer B, et al. MLN8054, an inhibitor of Aurora A kinase, induces senescence in human tumor cells both in vitro and in vivo. Mol Cancer Res 2010; 8:373 - 84; http://dx.doi.org/10.1158/1541-7786.MCR-09-0300; PMID: 20197380
- Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci 2007; 120:2479 - 87; http://dx.doi.org/10.1242/jcs.001222; PMID: 17646672
- Kelly KR, Nawrocki ST, Espitia CM, Zhang M, Yang JJ, Padmanabhan S, et al. Targeting Aurora A kinase activity with the investigational agent alisertib increases the efficacy of cytarabine through a FOXO-dependent mechanism. Int J Cancer 2012; 131:2693 - 703; http://dx.doi.org/10.1002/ijc.27579; PMID: 22488249
- Kops GJ, Medema RH, Glassford J, Essers MA, Dijkers PF, Coffer PJ, et al. Control of cell cycle exit and entry by protein kinase B-regulated forkhead transcription factors. Mol Cell Biol 2002; 22:2025 - 36; http://dx.doi.org/10.1128/MCB.22.7.2025-2036.2002; PMID: 11884591
- Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 2000; 404:782 - 7; http://dx.doi.org/10.1038/35008115; PMID: 10783894
- Alvarez B, Martínez-A C, Burgering BM, Carrera AC. Forkhead transcription factors contribute to execution of the mitotic programme in mammals. Nature 2001; 413:744 - 7; http://dx.doi.org/10.1038/35099574; PMID: 11607034
- Furukawa-Hibi Y, Yoshida-Araki K, Ohta T, Ikeda K, Motoyama N. FOXO forkhead transcription factors induce G(2)-M checkpoint in response to oxidative stress. J Biol Chem 2002; 277:26729 - 32; http://dx.doi.org/10.1074/jbc.C200256200; PMID: 12048180
- Laoukili J, Kooistra MR, Brás A, Kauw J, Kerkhoven RM, Morrison A, et al. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol 2005; 7:126 - 36; http://dx.doi.org/10.1038/ncb1217; PMID: 15654331
- Liu Q, Kaneko S, Yang L, Feldman RI, Nicosia SV, Chen J, et al. Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J Biol Chem 2004; 279:52175 - 82; http://dx.doi.org/10.1074/jbc.M406802200; PMID: 15469940
- Mao JH, Wu D, Perez-Losada J, Jiang T, Li Q, Neve RM, et al. Crosstalk between Aurora-A and p53: frequent deletion or downregulation of Aurora-A in tumors from p53 null mice. Cancer Cell 2007; 11:161 - 73; http://dx.doi.org/10.1016/j.ccr.2006.11.025; PMID: 17292827
- Nair JS, Ho AL, Schwartz GK. The induction of polyploidy or apoptosis by the Aurora A kinase inhibitor MK8745 is p53-dependent. Cell Cycle 2012; 11:807 - 17; http://dx.doi.org/10.4161/cc.11.4.19323; PMID: 22293494
- Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8:729 - 40; http://dx.doi.org/10.1038/nrm2233; PMID: 17667954
- Dar AA, Goff LW, Majid S, Berlin J, El-Rifai W. Aurora kinase inhibitors--rising stars in cancer therapeutics?. Mol Cancer Ther 2010; 9:268 - 78; http://dx.doi.org/10.1158/1535-7163.MCT-09-0765; PMID: 20124450
- Hoellein A, Pickhard A, von Keitz F, Schoeffmann S, Piontek G, Rudelius M, et al. Aurora kinase inhibition overcomes cetuximab resistance in squamous cell cancer of the head and neck. Oncotarget 2011; 2:599 - 609; PMID: 21865609
- Nakae J, Park BC, Accili D. Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J Biol Chem 1999; 274:15982 - 5; http://dx.doi.org/10.1074/jbc.274.23.15982; PMID: 10347145
- Zhu G, Spellman PT, Volpe T, Brown PO, Botstein D, Davis TN, et al. Two yeast forkhead genes regulate the cell cycle and pseudohyphal growth. Nature 2000; 406:90 - 4; http://dx.doi.org/10.1038/35021046; PMID: 10894548
- Hwang A, Maity A, McKenna WG, Muschel RJ. Cell cycle-dependent regulation of the cyclin B1 promoter. J Biol Chem 1995; 270:28419 - 24; http://dx.doi.org/10.1074/jbc.270.47.28419; PMID: 7499347
- Pines J. Mitosis: a matter of getting rid of the right protein at the right time. Trends Cell Biol 2006; 16:55 - 63; http://dx.doi.org/10.1016/j.tcb.2005.11.006; PMID: 16337124
- Lindon C, Pines J. Ordered proteolysis in anaphase inactivates Plk1 to contribute to proper mitotic exit in human cells. J Cell Biol 2004; 164:233 - 41; http://dx.doi.org/10.1083/jcb.200309035; PMID: 14734534
- Jackson JR, Patrick DR, Dar MM, Huang PS. Targeted anti-mitotic therapies: can we improve on tubulin agents?. Nat Rev Cancer 2007; 7:107 - 17; http://dx.doi.org/10.1038/nrc2049; PMID: 17251917
- Hirota T, Kunitoku N, Sasayama T, Marumoto T, Zhang D, Nitta M, et al. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell 2003; 114:585 - 98; http://dx.doi.org/10.1016/S0092-8674(03)00642-1; PMID: 13678582
- Marumoto T, Honda S, Hara T, Nitta M, Hirota T, Kohmura E, et al. Aurora-A kinase maintains the fidelity of early and late mitotic events in HeLa cells. J Biol Chem 2003; 278:51786 - 95; http://dx.doi.org/10.1074/jbc.M306275200; PMID: 14523000
- van der Horst A, Burgering BM. Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol 2007; 8:440 - 50; http://dx.doi.org/10.1038/nrm2190; PMID: 17522590