Abstract
The RB pathway plays a critical role in proliferation control that is commonly subverted in tumor development. However, restoration of RB pathway function can be elicited in many tumor cells by the inhibition of CDK4/6 activity that leads to dephosphorylation of RB and subsequent repression of E2F-mediated transcription. In this context, active RB/E2F complexes inhibit the expression of a critical program of coding genes that promote cell cycle progression. However, the non-coding RNA target genes downstream from RB that could be relevant for tumor biology remain obscure. Here, miRNA gene expression profiling identified the miR106b cluster as being efficiently repressed with CDK4/6 inhibition in an E2F and RB-dependent manner. Importantly, the miR106B-cluster is intragenic of MCM7, and through a series of functional studies, the basis of MCM7 regulation and concordant expression of the miRNA species within the 106b cluster was determined. Importantly, RB-mediated repression of the 106b cluster enhances the transcript levels of p21Cip1 and PTEN. These data provide a mechanistic basis for cross-talk between the RB pathway and p21 and PTEN through the regulation of the MCM7/miR106b locus.
Introduction
Aberrations in the Retinoblastoma tumor suppressor (RB) pathway contribute to human malignancies.Citation1-Citation3 While defined by the prototypical loss of the RB tumor suppressor, disruption of the pathway occurs through multiple mechanisms in different cancers. Particularly, deregulation of CDK4/6 activity through loss of p16INK4A or amplification/overexpression of CDK4 or cyclin D1 are prevalent in a host of tumor types.Citation4,Citation5 The RB pathway has been extensively studied, and it is well-established that activation of RB function leads to cell cycle inhibition that is associated with the downregulation of genes that are controlled through the E2F family of transcription factors.Citation6,Citation7 Physiologically, RB activation occurs as a result of the blockade of phosphorylation during cell cycle exit as induced by lack of mitogen or specific anti-proliferative stresses.Citation1,Citation3 The subsequent hypophosphorylated form of RB efficiently prevents cell cycle progression and also invokes other cellular effects, such as senescence, non-invasive properties and differentiation.Citation8 These phenotypes are highly relevant for the tumor-suppressive function of RB and are believed to be controlled by transcriptional programs, coordinated downstream from RB. In breast cancer, it has been shown that deregulation of this transcriptional program is associated with aggressive subtypes of breast cancer (e.g., basal and luminal B) and, respectively, is associated with a relatively poor prognosis.Citation9,Citation10
While much of the focus for RB has been on the regulation of coding genes, it has become clear that non-coding RNA species are particularly important as key effectors of disease pathogenesis, biological markers and therapeutic targets in cancer. Non-coding RNA encompass a variety of discrete entities, including micro RNA (miRNA), ultra conserved long non-coding RNA and pyknons. While several miRNA species are regulated via E2F and RB, the mechanisms coordinating such altered expression of miRNA,Citation11-Citation14 the targets and overall mode of regulation has remained obscure relative to coding genes.
Importantly, just as with coding genes, specific miRNA species are associated with different aspects of breast cancer.Citation14-Citation16 For example, deregulated miRNA expression has been implicated in aberrant proliferation, genome stability and epithelial-mesenchymal transition.Citation17-Citation19 Analyses of miRNA expression in breast cancer tumor specimens have demonstrated association with specific breast cancer subtypes. Additionally, specific miRNA species are correlated with specific facets of breast cancer biology and prognosis. These combined studies supported an effort to understand miRNA species regulated by the RB pathway, the mechanism of their regulation and the corresponding impact related to breast cancer biology.
Results
Modulation of the miRNA species through the RB pathway
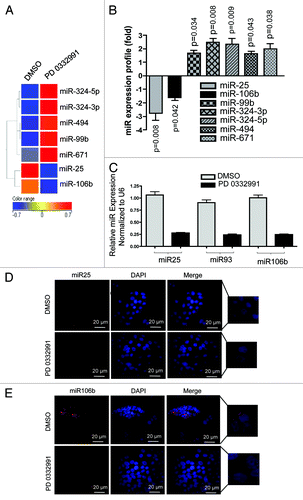
To understand how the RB pathway impacts the expression of miRNA species, expression profiling was performed. For these studies, we used the CDK4/6 specific inhibitor PD-0332991 to activate the RB pathway in ER-positive MCF7 breast cancer cell line. This agent is a potent modulator of RB pathway function that is clinically significant, as it is being evaluated as a therapeutic agent in multiple diseases, including breast cancer.Citation20,Citation21 As shown, treatment of MCF7 cells with PD-0332991 resulted in significantly altered expression of seven miRNA species (). In particular, we were intrigued by miR-25 and miR-106b, as they were strongly suppressed by CDK4/6 inhibition (). These miRNA species are part of the co-regulated miR106b-cluster that also includes miR-93.Citation18 The three miRNAs were evaluated by qRT-PCR and demonstrated consistent repression with CDK4/6 inhibition () and also in another RB-proficient cell line MDA-MB 231 (Fig. S1D). These results were further confirmed through the analyses of RNA abundance using fluorescence in situ hybridization (). Therefore, CDK4/6 inhibition is associated with the suppression of this specific miRNA cluster.
Figure 1. MicroRNA profiling and validation in MCF7 cells in response to CDK4/6 inhibitor. (A) Heat map of differentially regulated microRNAs. (B) Graphic representation of differentially expressed microRNAs in response to DMSO or PD 0332991. (C) qRT-PCR validation of miR 25, 93 and 106b. (D) In situ hybridization detection of miR-25. (E) In situ hybridization detection of miR-106b. Each data point is a mean ± SD from three or more independent experiments. p < 0.05 were considered as significant.
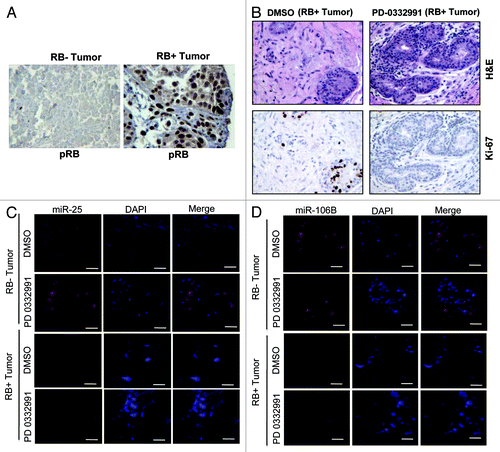
To determine whether this event could be relevant in the context of human breast cancer, we employed breast cancer explants. These explants are an ex vivo culture of primary breast tumors that were treated with PD-0332991.Citation22 In these tumors, RB status was determined by immunohistochemistry (). The explants are responsive to CDK4/6 inhibition, as evidenced by the reduction in Ki67 with treatment with PD-0332991, which is manifest in an RB-dependent fashion (). Fluorescence in situ hybridization for the miRNA revealed downregulation of miRNA 25 and 106b in the human tumor tissue (), and this effect was associated with the presence of RB in the tissue ().
Figure 2. MicroRNA 106b-cluster analysis in human breast tumor explants treated with PD 0332991. (A) Immunohistochemical localization of pRB in RB- and RB+ human breast tumors. (B) H&E and ki67 staining in RB+ human solid breast tumors. (C) In situ hybridization detection of miR25 in RB- and RB+ human breast tumors. (D) In situ hybridization detection of miR106b. (n = 11 ER+RB+ tumors and 2 ER-RB- tumors.)
Regulation of the miR106b cluster locus via the RB pathway
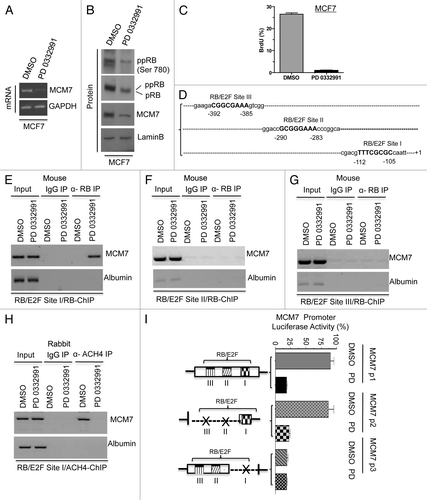
The miR106b-cluster is located in an intragenic region of the MCM7 gene.Citation18 MCM7 is a known RB/E2F target geneCitation7; however, the promoter elements that control expression remain loosely defined. With CDK4/6 inhibition, the MCM7 transcript and resultant protein were significantly downregulated (). MCM7 is known to form a DNA replication complex with MCM2, MCM4, MCM6 and CDC6, which are also repressed with PD-0332991 treatment (Fig. S1A). Correspondingly, upon CDK4/6 inhibition, BrdU incorporation was significantly inhibited, consistent with the inhibition of DNA replication (). Based on these findings, we explored, in detail, the regulation of MCM7 via the RB pathway. The MCM7 proximal promoter contains three consensus E2F-binding sites (). Using chromatin immunoprecipitation analyses, we found that RB preferentially bound to the site closest to the transcriptional start site (). Chromatin immunoprecipitation assays revealed that RB failed to bind significantly at the other two RB/E2F consensus binding sites (RB/E2F sites II and III). As a control, no binding was observed to this site in cells rendered RB deficient by knockdown (Fig. S1B). RB binding was associated with reduced acetylated histone occupancy at the MCM7 promoter (). To determine the functional significance of the RB-binding site, the promoter was cloned, and a series of mutants were produced. The full-promoter was clearly repressed by CDK4/6 inhibition (). While deletion of the proximal E2F binding element reduced overall transcriptional activity, this reduced activity was no longer responsive to the CDK4/6 inhibition (). Thus, the E2F-site proximal to the transcriptional start site is critical for the regulation of MCM7.
Figure 3. Analysis of transcriptional control in MCM7 gene in MCF7 cells in response to CDK4/6 inhibitor. (A) Semi-quantitative RT-PCR analysis of MCM7 and GAPDH mRNA expression. (B) Western blotting analysis of pRB, phospho RB (Ser 780), MCM7 and loading control lamin b. (C) Analysis of BrdU incorporation by flow cytometric analysis. (D) Schematic representation of RB/E2F-biding nucleotide sequences on MCM7 promoter. (E) Identification pRB association on RB/E2F consensus binding site I on MCM7 promoter (ChIP assay). (F) RB ChIP on RB/E2F consensus binding site II of MCM7 promoter in response to DMSO or PD 0332991 in MCF7 cells. (G) RB ChIP on RB/E2F consensus binding site III of MCM7 promoter in response to DMSO or PD 0332991 in MCF7 cells. (H) Identification acetylated histone 4 association on RB/E2F binding site I on MCM7 promoter (ChIP assay). (I) MCM7 promoter analysis (luciferase assay). Each data point is a mean ± SD from three or more independent experiments. p < 0.05 were considered as significant.
MCM7 expression correlates with miR106b expression
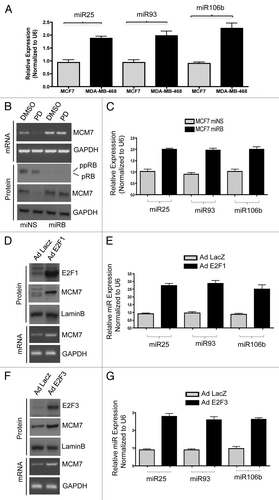
To determine if MCM7 regulation via the RB pathway directly correlated with the expression of miR25, miR93 and miR106b, a series of functional permutations in the pathway were performed. Initially, breast cancer models harboring mutation of RB gene (MDA-MB468) were compared against MCF7 cells. As shown in , MDA-MB468 cells expressed higher levels of miR25, miR93 and miR106b. Since there are numerous differences between MCF7 and MDA-MB468 cells, we employed knockdown of RB in MCF7 cells. RB knockdown resulted in a modest increase in MCM7 expression, but obviated the repression of MCM7 that is observed with PD-0332991 (). This resulted in an increase in miR25, miR93 and miR106b levels (). To directly interrogate the functionality of E2F in controlling the expression of these miRNAs, we used adenoviral-mediated ectopic expression of E2F1 and E2F3. Both E2F proteins resulted in a substantive increase in MCM7 mRNA and protein levels, and a concomitant increase in miR25, miR93 and miR106b levels (). These data indicate that RB functioning through E2F transcription factors controls the expression of these miRNAs.
Figure 4. Comparative analysis of miR106b cluster expression in different breast cancer cell lines in response to PD 0332991 and RB status. (A) qRT-PCR analysis of miR106b cluster in RB-proficient MCF7, RB mutant MDA-MB 468 cells and the signals are normalized to U6 internal control. (B) Semi-quantitative RT-PCR analysis of MCM7 mRNA and loading control GAPDH in miNS and miRB MCF7 cells (top panel) and immunoblot analysis of pRB, MCM7 and loading control lamin b (bottom panel). (C) qRT-PCR analysis of miR106b cluster in miNS and miRB MCF7 cells, and the signals are normalized to U6 internal control. (D) Semi-quantitative RT-PCR analysis of MCM7 and GAPDH mRNA in E2F1-overexpressing MCM7 cells (bottom panel) and immunoblot analysis of MCM7, E2F1 and lamin b in E2F1-overexpressing MCM7 cells (top panel). (E) qRT-PCR analysis of miR106b- cluster in E2F1-overexpressing MCF7 cells and the signals are normalized to U6 internal control. (F) Semi-quantitative RT-PCR analysis of MCM7 and GAPDH mRNA (bottom panel) and immunoblot analysis of MCM7, E2F3 and lamin b in E2F3-overexpressing MCM7 cells (top panel). (G) qRT-PCR analysis of miR106b cluster in E2F3-overexpressing MCF7 cells and the signals are normalized to U6 internal control. Each data point is a mean ± SD from three or more independent experiments. p < 0.05 were considered as significant.
Identification of targets of the miR106b cluster
To define the functional consequence of miRNA deregulation, it is critical to define functional targets. We used the microRNA.org/Target Scan program to define likely targets for the miRNA species that were regulated through the RB pathway (Fig. S2). For miR25, miR93 and miR106b, the CDK inhibitor p21Cip1 and the tumor suppressor PTEN are highly conserved targets. We initially focused on p21Cip1 as a functional target for mir106b-cluster. With the suppression of CDK4/6, we observed the upregulation of p21 Cip1 mRNA and the p21 Cip1 protein (). These findings correlated with the p21Cip1 transcript levels in human explants treated with the CDK4/6 inhibitor (). To demonstrate that this finding was due to the miRNA 106b-cluster species, we expressed precursor miRNA 25, 93 and 106b individually in cells and observed downregulation of p21 mRNA and protein (; Fig. S3A); conversely, anti-miR transfection resulted in an increased level of p21 protein (Fig. S3B). To directly interrogate if this was mediated through effect on the p21Cip1 miR-binding sites, a reporter with the p21 3′ UTR was constructed (Fig. S3C) to monitor RNA stability. As shown, CDK4/6 inhibition induced the accumulation of luciferase from this reporter (). Expression of precursor miRNAs in cells reduced the accumulation of p21Cip1 luciferase, while anti-miRs induced p21Cip1 luciferase activity (). These alterations in the level of the p21 3′UTR luciferase were dependent on RB, as there were no effects of CDK4/6 inhibition in RB-deficient MCF7 cells and RB-mutated MD MB468 cells (Fig. S3D and E). Together, these data show that CDK4/6 activity impinges on p21Cip1, at least in part through the regulation of miRNA species.
Figure 5. Analysis of p21 regulation in in vitro and ex vivo breast cancer models in response to miR106b-cluster and PD 0332991. (A) qRT-PCR analysis of p21 mRNA and the signals were normalized to GAPDH internal control. (B) Western blotting analysis of p21Cip1/Waf1 and lamin b loading control in MCF7 cells. (C) qRT-PCR analysis of p21 mRNA in RB- and RB+ human solid tumors in response to CDK4/6 inhibitor. (D) qRT-PCR analysis of p21Cip1/Waf1 in response to ectopic expression of precursor mirR 25, 93 and 106b. (E) p21 3′ luciferase activity in MCF7 cells in response to PD 0332991, and signals were normalized to renilla luciferase. (F) p21 3′ luciferase activity in MCF7 cells in response to ectopic expression of precursor mirR 25, 93 and 106b, and signals were normalized to renilla luciferase. (G) p21 3′ luciferase activity in MCF7 cells in response to ectopic expression of anti-mirR 25, 93 and 106b, and signals were normalized to renilla luciferase. Each data point is a mean ± SD from three or more independent experiments. (Tumors, n = 11 ER+RB+ tumors and 2 ER-RB- tumors.) p < 0.05 were considered as significant.
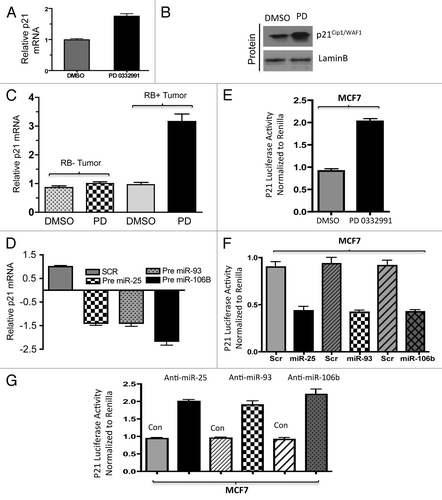
Another target of the miR106B-cluster is the tumor suppressor PTEN. Similar analyses were performed as shown in and demonstrated that PTEN levels are regulated by CDK4/6 inhibition, resulting in increased expression of PTEN mRNA and protein (). These findings correlated with PTEN mRNA levels in human breast tumors treated with PD-0332991 (). To demonstrate that altered regulation was due to miRNA 106b-cluster species, we expressed the precursor miRNA in cells and observed downregulation of PTEN mRNA and PTEN protein (; Fig. S4A). The anti-miR 25, 93 and 106b transfection moderately increased the PTEN protein (Fig. S4B). To directly interrogate if this was mediated through effects on the PTEN miR-binding sites, a reporter with the PTEN 3′ UTR was constructed (Fig. S4C) to monitor RNA stability. As shown, CDK4/6 inhibition induced the accumulation of luciferase from this reporter (). Conversely, expression of pre-miRNAs in cells reduced the accumulation of PTEN luciferase, while anti-miRs induced PTEN luciferase ().
Figure 6. Analysis of PTEN regulation in in vitro and ex vivo breast cancer models in response to miR106b-cluster and PD 0332991. (A) qRT-PCR analysis of PTEN mRNA (left panel) and immunoblot (right panel). (B) Immunolocalization of PTEN protein in MCF7 cells. (C) qRT-PCR analysis of PTEN mRNA in RB+ and RB- human solid tumors in response to CDK4/6 inhibitors. (D) qRT-PCR analysis of PTEN mRNA in response to ectopic expression of precursor mirR 25, 93 and 106b in MCF7 cells. (E) PTEN 3′ luciferase activity in MCF7 cells in response to PD 0332991 and signals were normalized to renilla luciferase. (F) PTEN 3′ luciferase activity in MCF7 cells in response to ectopic expression of precursor mirR 25, 93 and 106b, and signals were normalized to renilla luciferase. (G) PTEN 3′ luciferase activity in MCF7 cells in response to ectopic expression of anti-mirR 25, 93 and 106b, and signals were normalized to renilla luciferase. Each data point is a mean ± SD from three or more independent experiments. (tumors n = 11 ER+RB+ tumors and 2 ER-RB- tumors.) p < 0.05 were considered as significant.
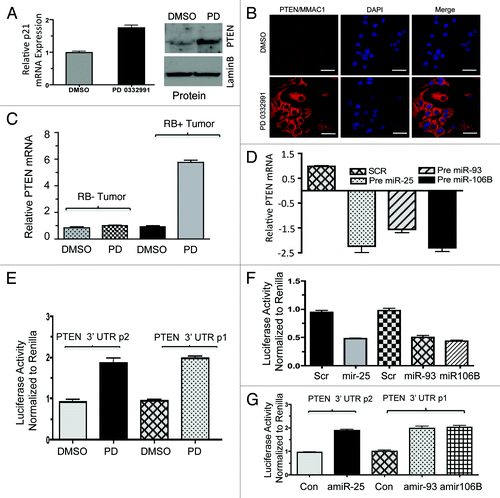
Direct analyses of p21 and PTEN mRNA targeting by the miRNA 106B-cluser was determined using an RNA-immunoprecipitation approach. Argonaute-2 is one of the microRNA/RISC complex (RNA-induced silencing complex) proteins. We used anti-Argonaute-2 antibodies to immunoprecipitate RNA/microRNA complexes (schematic approach, Fig. S5A). Success of immunoprecipitation was verified by immunoblotting with anti-Argonate-2 antibody (). The purified RNA was subjected to semi-quantitative RT-PCR and qRT-PCR. The results showed that p21Cip1 and PTEN mRNAs and protein were upregulated with CDK4/6 inhibition (; Fig. S5B). Parallel real-time RT-PCR was performed for microRNA 25, 93 and 106b. Internal control GAPDH mRNA was unaltered upon CDK4/6 inhibition. As shown, the amount of p21Cip1 and PTEN RNA associated with the miRNA machinery was significantly reduced by CDK4/6 inhibition ().
Figure 7. RNA immunoprecipitation, p21, PTEN mRNA and miR106b-cluster analysis in response to CDK4/6 inhibitor in MCF7 cells. (A) Immunoprecipitation of Ago2, western blot of Ago2 and loading control lamin b. (B) qRT-PCR signals of Ago2 bound p21Cip1/Waf1 and PTEN/MMAC1, GAPDH mRNA in response to DMSO or PD 0332991. (C) qRT-PCR signals of Ago2-bound miR25. (D) qRT-PCR signals of Ago2-bound miR93. (E) qRT-PCR signals of Ago2-bound mi106b, and the signals were normalized to U6 internal control. Each data point is a mean ± SD from three or more independent experiments. p < 0.05 were considered as significant.
Functional significance of RB-regulated miRNA species
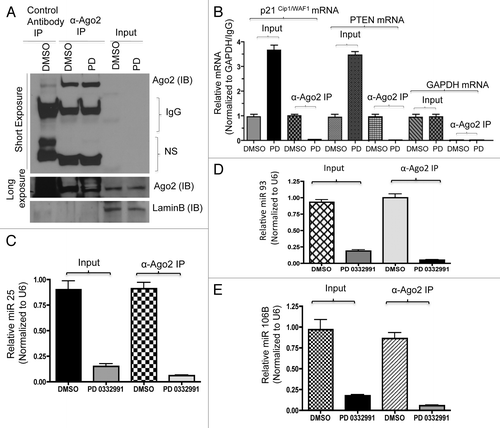
Lastly, in order to investigate the functional role of miR106b in cellular phenotypes, we cloned the mir106b-cluster and used lentiviral transduction to develop stable overexpression models. To investigate the miR cluster role in proliferation, we performed the BrdU incorporation assays. Cells were either untreated or treated with PD-0332991; under this condition, the expression of the cluster had no significant effect on BrdU incorporation (). Such a finding is consistent with the continued repression of E2F-target genes required for cell cycle progression (e.g., MCM7), under which condition proliferation will remain inhibited. Since RB also modulates invasive properties, we determined the effect of mir106b on invasive properties. Wound healing (cell migration) and cell invasion assays (boyden chamber) were employed in control MCF7 cells and cells expressing mir106B cluster. The cells expressing mir106B cluster showed enhanced cell migration and invasion ().
Figure 8. Functional analysis of miR106b-cluster in MCF7 cells in response to CDK4/6 inhibitor. (A) Flow cytometric analysis of BrdU uptake in MCF7 cells expressing miR106b-cluster (left panel), and QRT-PCR analysis of p21 and PTEN mRNA levels in MCF7 control and cells expressing mir106B-cluster in response PD 0332991 right panel. (B) Wound-healing assay. (C) Cell migration assay/invasion. (D) Quantitation of cell migration/invasion assay. Each data point is a mean ± SD from three or more independent experiments. p < 0.05 were considered as significant.
Discussion
Non-coding RNA plays an important role in tumor biology.Citation23-Citation25 Here, we demonstrate that in breast cancer models, the mir106b cluster is regulated through the RB pathway. Specifically, the miRNA species within the cluster are repressed when CDK4/6 activity is inhibited and stimulated when RB function is compromised. The mir106b cluster is embedded within the MCM7 gene, which is a direct RB/E2F target gene.Citation16,Citation26-Citation30 The mir106b cluster targets the expression of several genes that are directly disease-relevant and that provide a mechanism for cross-talk from the canonical RB pathway to PTEN and p21Cip1. Together, these data demonstrate a new pathway through which RB function can modulate additional pathways.
RB is a well-known tumor suppressor that is functionally compromised in many tumor types via multiple mechanisms. In general, the loss of RB function is associated with the deregulation of the transcription of coding genes that contribute to diverse biological processes.Citation9,Citation31 In particular, RB modulates the expression of genes involved in DNA replication and can correspondingly deregulate multiple facets of replication control. While MCM7 has a key role in replication control,Citation32,Citation33 it also harbors the mir106b cluster that is disease-relevant and encodes three miRNA species that target multiple disease-relevant coding genes (e.g., PTEN and p21Cip1).Citation18,Citation34-Citation37 As shown here, RB is a key regulator of the MCM7 gene, and this occurs through direct binding to a proximal E2F element in the promoter. The repression of MCM7 transcription is associated with the repression of miRNA expression and resultant induction of p21Cip1 and PTENMMAC1.
In breast cancer models, RB loss is associated with aggressive forms of disease that harbor an intrinsically poor prognosis. MCM7 is part of the RB loss signature of genes and has been shown by our group and others to be associated with luminal b, her2-positive and basal breast cancer subtypes.Citation9,Citation10 These forms of breast cancer have a higher proliferative index and are also associated with more aggressive forms of disease prone to distant metastasis. In keeping with the association between MCM7 expression and mir106b cluster, the miRNA species within this cluster are concordantly elevated in basal, her2-postive and luminal b breast cancers.Citation38 Thus, it is reasonable to speculate that, in human cancer, they are under the same control mechanisms that are coordinated by the RB pathways as determined mechanistically here. Consistent with this idea, in primary human tumor explants, repression of the mir106b cluster is coordinated through the RB pathway.
While RB is known to regulate cell cycle progression, emerging data point to an important role for RB in the invasive properties of breast cancer.Citation8,Citation39 The mechanism underlying this process remains ill-defined. Recent studies have suggested a role for the RB pathway in regulating the epithelial-to-mesenchymal transition through miR200.Citation8 Particularly, it has been shown that RB loss contributes to invasive phenotypes, and RB activation can inhibit invasion. In our studies, we did not observe a clear effect of RB pathway activation on miR200; however, the mir106b pathway has been functionally demonstrated to contribute to invasive properties through PTEN and the TGF-β pathway.Citation11,Citation26,Citation34 Consistent with this work, we observed that expression of the mir106b cluster significantly stimulated invasion, but had minimal effect on cell cycle progression. This is not surprising, as RB represses multiple key coding genes required for cell cycle progression, which presumably cannot be rescued via a specific miRNA locus. However, the findings herein reinforce the concept that miRNAs controlled by RB contribute to invasive phenotypes that are of high relevance to breast cancer.
Materials and Methods
Cell culture and drug treatments
Breast cancer cells lines including, MCF7 Wt, MCF7 miNS, MCF7 miRB, HEK293FT, MDA-MB 231 and MDA MB 468, were maintained in DMEM containing 10% fetal bovine serum, 100 U/ml penicillin/streptomycin and 2 mM l-glutamine. All cells were cultured at 37°C and 5% CO2. All cells were counted for experimental seeding using trypan blue exclusion. The cells were treated with 500 nM PD 0332991 or DMSO for 24 h and processed for further analysis.
MicroRNA profiling and validation
Total DNA-free RNA was extracted from MCF7 cells treated with DMSO or PD 0332991 (500 nM) for 24 h. The RNA quality was assessed using Agilent 2100 bioanalyzer (Agilent). MicroRNA profiling was performed in Cancer Genomics, (Kimmel Cancer Center, Thomas Jefferson University) using Affymetrix GeneChIP miRNA Arrays (Affimetrix). The data was analyzed by Affimetrix MiRNA QC tool and Genespring V 11.5 software with a 0.05 p value (Agilent). Total RNA was used to validate miR 25, 93 and 106b by qRT-PCR (Applied Biosystems), and U6 was used as an internal control.
qRT-PCR and mRNA analysis
Total DNA-free RNA was isolated by a standard protocol from cells and FFPE human breast tumors treated with CDK4/6 or DMSO, and the RNA was reverse transcribed. Real-time PCR was performed using 50 ng of cDNA and SYBR Green PCR master mix to amplify MCM7, p21Cip1/Waf1 and PTEN/MMAC1 mRNAs as described.Citation40-Citation43 The mRNA signals were normalized to GAPDH mRNA internal control and the primers used are presented in the Table S1. Mir 25, miR93 and mir106b were analyzed by QRT-PCR by using specific RT-PCR primers from Applied Biosystem, Inc. The microRNA signals were normalized to U6 mRNA internal control.
Immunoprecipitation and immunoblot analysis
Total cell extract was isolated from MCF7 Wt, miNS and miRB cells exposed to DMSO or PD 0332991, or transfected with miR106B cluster or anti-miR106B cluster. The total cell extract was subjected to immunoprecipitation with anti-Ago2 antibody or SDS-PAGE analysis and transferred on to Immobilon-P membranes. Membranes were probed with antibodies against RB (G3–245; BD PharMingen), ppRB(Ser 780), PTEN/MMAC1and Ago2 (Cell Signaling Technology), CDC6, MCM2, MCM4, MCM6, MCM7, p21Cip1/Waf1 and Lamin B (M-20) (Santa Cruz Biotechnology, Inc.), and lamin b served as a loading control.
MCM7-promoter analysis
MCM7 promoter containing three E2F/RB binding sites (I, II and III) were cloned in pGL4 basic reporter,Citation44 and the primers used in the study are presented in the Table S1. The constructs were transfected individually in MCF7 cells using Lipofectamine 2000 (Invitrogen), and renilla pRL-TK luciferase contruct was co-transfected as an internal control. After 24 h of post-transfection, the cells were treated with DMSO or PD 0332991 (500 μM) for 24 h. After 48 h of post-transfection, the cells were harvested and lysed and luciferase activity was measured by using dual-luciferase assay kit (Promega), and the luciferase was normalized with Renilla luciferase.
Chromatin immunoprecipitation assay (ChIP)
ChIP was performed as previously described.Citation45 Briefly, cells were cross-linked with formaldehyde. Cells were harvested, lysed and sheared. Sheared chromatin was measured and equal amounts of chromatin from all growth conditions were pre-cleared with protein A/G beads (GE Healthcare). Ten percent of total chromatin was used as an input control. The remaining chromatin was used for immunoprecipitation with antibodies against RB (1F8, Neo Markers), ACH4 (Cell signaling). Immunoprecipitation was performed in a 500 µl volume with RIPA buffer containing sheared chromatin and protease inhibitors. The immunoprecipitaed chromatin incubated at 65°C overnight in order to de-crosslink. Immunoprecipitated DNA was purified using a PCR purification kit (Qiagen) and subjected to semi-qPCR. Total chromatin DNA (Input) or from ChIP DNA was amplified by PCR using the primer sequences targeting E2F/RB binding sites on MCM7 (primers presented in Table S1). Rabbit, Mouse IgG and albumin promoter amplifications were served as a negative control. PCR-amplified products were subjected to 2% agarose gel electrophoresis and were visualized with ethidium bromide.
Measurement of p21 and PTEN 3′ UTR luciferase activity
p21 and PTEN 3′UTRs were cloned in pMIR-vector as described,Citation26,Citation37,Citation46 and the primers are presented in Table S1. The 3′UTRs (25 nM) were transiently transfected in MCF7 cells using Lipofectamine 2000 (Invitrogen), and renilla luciferase was co-transfected as an internal control. After 24 h of post-transfection, the cells were treated with DMSO or CDK4/6 inhibitor 9 500 uM). In some experiments, p21 and PTEN 3′ UTRs were co-transfected with precursor (Ambion/Life Technologies) miR 25 or 93 or 106b or anti-miR25 or anti-mir93 or miR106B. Renilla pRL-TK luciferase was co-transfected in each transfection. Also, precursor microRNAs alone were transfected in MCF7 cells with Oligofectamine. After 24–48 h of post-transfection, cells were harvested, lysed, and the luciferase was measured by using dual-luciferase assay kit (Promega), and the luciferase was normalized with renilla luciferase as described.Citation26,Citation47 MicroRNAs were analyzed by QRT-PCR using specific microRNA probes as described in manufacturer's protocol (Applied Biosystem). MicroRNA signals were normalized to U6 mRNA internal control PTEN and p21 mRNA were analyzed by QRT-PCR, and the signals were normalized to GAPDH mRNA internal control. The p21 and PTEN mRNA primers are provided in Table S1.
RNA-immunoprecipitation and qRT–PCR
MCF7 cells were treated with DMSO or PD 0332991 for 24 h, and the RNA immunoprecipitation was performed using Ago2 antibody as described.Citation48,Citation49 RNA was extracted from immunoprecipitates using Trizol-Reagent (Invitrogen). cDNA was synthesized, and qRT-PCRs were performed for p21, PTEN mRNA and miR106B cluster using Applied Biosystem. The immunoprecipitation efficiency of the Ago2 antibody was confirmed by the immunobloting of the immunoprecipitates from Ago2 IPs with anti-Ago2. GAPDH and U6 were served as internal controls for mRNA and microRNA analysis.
Lentiviral construction, transduction and generation of stable miR106b-cluster in MCF7 cells
MiR-106B cluster was cloned from MCF7 cells via PCR amplification using the primers as presented in Table S1. Once amplified, the miR106b- cluster was cloned into pENTR/D-TOPO (Invitrogen) and LR recombined into plenti/TO/V5-DEST. Desired sequence was verified via DNA sequencing. Once recombined into the destination vector, this expression plasmid was co-transfected alongside the packaging plasmids pLP1, pLP2 and pLP/VSVG in HEK293FT cells to generate viral particles. Cells were infected with viral particles for a period of 72 h and selected using standard methodologies. plenti/TO/v-5-GW/LacZ (Invitrogen) was used as an infection control.
Flow cytometery
Flow cytometry was performed on MCF7 cells treated with DMSO or PD 0332991 or cells expressing miR106B cluster as previously described.Citation45 Prior to harvesting, cells were pulse-labeled with BrdU for 1 h (GE, Healthcare).
Wound-healing and cell migration/invasion assay
The wound-healing assay was performed as described previously.Citation50,Citation51 MCF7 cells expressing miR106B-cluster and control cells were plated on 6-cm culture plates and allowed to be confluent. Straight scratches were made on the plates, and the cells were washed and replaced with fresh medium. Wounded monolayers were photographed under the inverted microscope at 0, 24 and 48 h after post-scratch. The extent of wound repair and cell migration was evaluated by comparing the area of the wound in control as well as cells expressing miR106b cluster. Cell migration/invasion assay was performed as described,Citation51 MCF7 cells harboring miR106B-cluster and control cells were seeded (5 × 104 cells) on Boyden Chambers (BioCoat 354578) under low serum conditions. Complete growth medium was added to the wells as a chemoattractant. Boyden chambers were placed in wells containing growth medium, and for the negative control, Boyden chambers containing cells were placed on wells containing no growth factors. The cells were incubated overnight in the incubator at 37°C. The Boyden chambers were removed and washed with PBS and cells on the upper surface of the boyden chamber membranes were removed, not disturbing the cells on the lower surface of the membrane. The cells at the lower surface of the membrane were stained by placing with DAPI. Cells were scored by fluorescent microscopy.
Immunofluoresence, in situ hybridization and immunohistochemistry
MCF7 cells were grown on coverslips, treated with DMSO or PD 0332991 and fixed with 0.2% paraformaldehyde or methanol. Some of the coverslips were stained for PTEN antibody and imaged using confocal microscopy as described.Citation16,Citation26 Some of the coverslips were processed for in situ hybridization of miR 25 and 106B using biotin labeled LNA probes (Exiqon) following the manufacturer's protocol. Modifications include 20 μg/ml Proteinase K for 10 min and incubated overnight at 55°C hybridization with 10 nM of LNA probe in a formamide containing hybridization buffer (50% Formamide, 5 X SSC, 0.1% Tween, 50 μg/ml Heparin, 500 μg/ml yeast tRNA). Detection was achieved with Streptavidin Cy3 (Invitrogen) solution, and the cells were counterstained with DAPI (Poly Scientific). Similarly, human primary breast tumors were collected from Jefferson Hospital and treated with PD 0332991. Parafin-embedded sections were stained for H&E, RB and BrdU were performed as describedCitation51 on the BenchMark XT Slide Preparation System (Ventana Medical Systems). Tissues were scored using ASCO/CAP guidelines. RB were stained and scored as previously described.Citation51 Some of the ER (+), RB (-) and ER (+), RB (+) tumor sections were processed for in situ hybridization detection of miR 25 and 106B using biotin-labeled LNA probes (Exiqon) and imaged using confocal microscopy.
Statistical analysis
Statistical analyses were performed using GraphPadPrism (version 4.0c) software (GraphPad PrismSoftware, Inc.). All the data were analyzed for statistical significance using Student’s t-test/ one-way ANOVA. For all experiments, p < 0.05 were considered as significant.
Additional material
Download Zip (434.1 KB)Acknowledgments
The study was supported by a grant from NCI CA12934.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer 2008; 8:714 - 24; http://dx.doi.org/10.1038/nrc2401; PMID: 19143056
- Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer 2008; 8:671 - 82; http://dx.doi.org/10.1038/nrc2399; PMID: 18650841
- Goodrich DW. The retinoblastoma tumor-suppressor gene, the exception that proves the rule. Oncogene 2006; 25:5233 - 43; http://dx.doi.org/10.1038/sj.onc.1209616; PMID: 16936742
- Mittnacht S. Control of pRB phosphorylation. Curr Opin Genet Dev 1998; 8:21 - 7; http://dx.doi.org/10.1016/S0959-437X(98)80057-9; PMID: 9529601
- Wang JY, Knudsen ES, Welch PJ. The retinoblastoma tumor suppressor protein. Adv Cancer Res 1994; 64:25 - 85; http://dx.doi.org/10.1016/S0065-230X(08)60834-9; PMID: 7879661
- Ishida S, Huang E, Zuzan H, Spang R, Leone G, West M, et al. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol Cell Biol 2001; 21:4684 - 99; http://dx.doi.org/10.1128/MCB.21.14.4684-4699.2001; PMID: 11416145
- Markey MP, Angus SP, Strobeck MW, Williams SL, Gunawardena RW, Aronow BJ, et al. Unbiased analysis of RB-mediated transcriptional repression identifies novel targets and distinctions from E2F action. Cancer Res 2002; 62:6587 - 97; PMID: 12438254
- Arima Y, Hayashi H, Sasaki M, Hosonaga M, Goto TM, Chiyoda T, et al. Induction of ZEB proteins by inactivation of RB protein is key determinant of mesenchymal phenotype of breast cancer. J Biol Chem 2012; 287:7896 - 906; http://dx.doi.org/10.1074/jbc.M111.313759; PMID: 22262832
- Ertel A, Dean JL, Rui H, Liu C, Witkiewicz AK, Knudsen KE, et al. RB pathway disruption in breast cancer: differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle 2010; 9:4153 - 63; http://dx.doi.org/10.4161/cc.9.20.13454; PMID: 20948315
- Herschkowitz JI, He X, Fan C, Perou CM. The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Res 2008; 10:R75; http://dx.doi.org/10.1186/bcr2142; PMID: 18782450
- Petrocca F, Visone R, Onelli MR, Shah MH, Nicoloso MS, de Martino I, et al. E2F1-regulated microRNAs impair TGFbeta-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell 2008; 13:272 - 86; http://dx.doi.org/10.1016/j.ccr.2008.02.013; PMID: 18328430
- Bao J, Li D, Wang L, Wu J, Hu Y, Wang Z, et al. MicroRNA-449 and microRNA-34b/c function redundantly in murine testes by targeting E2F transcription factor-retinoblastoma protein (E2F-pRb) pathway. J Biol Chem 2012; 287:21686 - 98; http://dx.doi.org/10.1074/jbc.M111.328054; PMID: 22570483
- Benhamed M, Herbig U, Ye T, Dejean A, Bischof O. Senescence is an endogenous trigger for microRNA-directed transcriptional gene silencing in human cells. Nat Cell Biol 2012; 14:266 - 75; http://dx.doi.org/10.1038/ncb2443; PMID: 22366686
- Ofir M, Hacohen D, Ginsberg D. MiR-15 and miR-16 are direct transcriptional targets of E2F1 that limit E2F-induced proliferation by targeting cyclin E. Mol Cancer Res 2011; 9:440 - 7; http://dx.doi.org/10.1158/1541-7786.MCR-10-0344; PMID: 21454377
- Khoshnaw SM, Green AR, Powe DG, Ellis IO. MicroRNA involvement in the pathogenesis and management of breast cancer. J Clin Pathol 2009; 62:422 - 8; http://dx.doi.org/10.1136/jcp.2008.060681; PMID: 19398594
- Smith AL, Iwanaga R, Drasin DJ, Micalizzi DS, Vartuli RL, Tan AC, et al. The miR-106b-25 cluster targets Smad7, activates TGF-β signaling, and induces EMT and tumor initiating cell characteristics downstream of Six1 in human breast cancer. Oncogene 2012; In press http://dx.doi.org/10.1038/onc.2012.11; PMID: 22286770
- Sempere LF, Christensen M, Silahtaroglu A, Bak M, Heath CV, Schwartz G, et al. Altered MicroRNA expression confined to specific epithelial cell subpopulations in breast cancer. Cancer Res 2007; 67:11612 - 20; http://dx.doi.org/10.1158/0008-5472.CAN-07-5019; PMID: 18089790
- Liu S, Patel SH, Ginestier C, Ibarra I, Martin-Trevino R, Bai S, et al. MicroRNA93 regulates proliferation and differentiation of normal and malignant breast stem cells. PLoS Genet 2012; 8:e1002751; http://dx.doi.org/10.1371/journal.pgen.1002751; PMID: 22685420
- Feng B, Dong TT, Wang LL, Zhou HM, Zhao HC, Dong F, et al. Colorectal cancer migration and invasion initiated by microRNA-106a. PLoS One 2012; 7:e43452; http://dx.doi.org/10.1371/journal.pone.0043452; PMID: 22912877
- Knudsen ES, Wang JY. Targeting the RB pathway in cancer therapy. Clin Cancer Res 2010; 16:1094 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-09-0787; PMID: 20145169
- Toogood PL, Harvey PJ, Repine JT, Sheehan DJ, VanderWel SN, Zhou H, et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem 2005; 48:2388 - 406; http://dx.doi.org/10.1021/jm049354h; PMID: 15801831
- Dean JL, McClendon AK, Hickey TE, Butler LM, Tilley WD, Witkiewicz AK, et al. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle 2012; 11:2756 - 61; http://dx.doi.org/10.4161/cc.21195; PMID: 22767154
- Nana-Sinkam SP, Fabbri M, Croce CM. MicroRNAs in cancer: personalizing diagnosis and therapy. Ann N Y Acad Sci 2010; 1210:25 - 33; http://dx.doi.org/10.1111/j.1749-6632.2010.05822.x; PMID: 20973796
- Ravi A, Gurtan AM, Kumar MS, Bhutkar A, Chin C, Lu V, et al. Proliferation and tumorigenesis of a murine sarcoma cell line in the absence of DICER1. Cancer Cell 2012; 21:848 - 55; http://dx.doi.org/10.1016/j.ccr.2012.04.037; PMID: 22698408
- Iorio MV, Croce CM. microRNA involvement in human cancer. Carcinogenesis 2012; 33:1126 - 33; http://dx.doi.org/10.1093/carcin/bgs140; PMID: 22491715
- Poliseno L, Salmena L, Riccardi L, Fornari A, Song MS, Hobbs RM, et al. Identification of the miR-106b~25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci Signal 2010; 3:ra29; http://dx.doi.org/10.1126/scisignal.2000594; PMID: 20388916
- Liu Y, Zhang Y, Wen J, Liu L, Zhai X, Liu J, et al. A genetic variant in the promoter region of miR-106b-25 cluster and risk of HBV infection and hepatocellular carcinoma. PLoS One 2012; 7:e32230; http://dx.doi.org/10.1371/journal.pone.0032230; PMID: 22393390
- Li Z, Yang CS, Nakashima K, Rana TM. Small RNA-mediated regulation of iPS cell generation. EMBO J 2011; 30:823 - 34; http://dx.doi.org/10.1038/emboj.2011.2; PMID: 21285944
- Li Y, Tan W, Neo TW, Aung MO, Wasser S, Lim SG, et al. Role of the miR-106b-25 microRNA cluster in hepatocellular carcinoma. Cancer Sci 2009; 100:1234 - 42; http://dx.doi.org/10.1111/j.1349-7006.2009.01164.x; PMID: 19486339
- Chuang TD, Luo X, Panda H, Chegini N. miR-93/106b and their host gene, MCM7, are differentially expressed in leiomyomas and functionally target F3 and IL-8. Mol Endocrinol 2012; 26:1028 - 42; http://dx.doi.org/10.1210/me.2012-1075; PMID: 22556343
- Markey MP, Bergseid J, Bosco EE, Stengel K, Xu H, Mayhew CN, et al. Loss of the retinoblastoma tumor suppressor: differential action on transcriptional programs related to cell cycle control and immune function. Oncogene 2007; 26:6307 - 18; http://dx.doi.org/10.1038/sj.onc.1210450; PMID: 17452985
- Li SS, Xue WC, Khoo US, Ngan HY, Chan KY, Tam IY, et al. Replicative MCM7 protein as a proliferation marker in endometrial carcinoma: a tissue microarray and clinicopathological analysis. Histopathology 2005; 46:307 - 13; http://dx.doi.org/10.1111/j.1365-2559.2005.02069.x; PMID: 15720416
- Ishimi Y. A DNA helicase activity is associated with an MCM4, -6, and -7 protein complex. J Biol Chem 1997; 272:24508 - 13; http://dx.doi.org/10.1074/jbc.272.39.24508; PMID: 9305914
- Poliseno L, Salmena L, Riccardi L, Fornari A, Song MS, Hobbs RM, et al. Identification of the miR-106b~25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci Signal 2010; 3:ra29; http://dx.doi.org/10.1126/scisignal.2000594; PMID: 20388916
- Kan T, Sato F, Ito T, Matsumura N, David S, Cheng Y, et al. The miR-106b-25 polycistron, activated by genomic amplification, functions as an oncogene by suppressing p21 and Bim. Gastroenterology 2009; 136:1689 - 700; http://dx.doi.org/10.1053/j.gastro.2009.02.002; PMID: 19422085
- Smith AL, Iwanaga R, Drasin DJ, Micalizzi DS, Vartuli RL, Tan AC, et al. The miR-106b-25 cluster targets Smad7, activates TGF-β signaling, and induces EMT and tumor initiating cell characteristics downstream of Six1 in human breast cancer. Oncogene 2012; In press http://dx.doi.org/10.1038/onc.2012.11; PMID: 22286770
- Ivanovska I, Ball AS, Diaz RL, Magnus JF, Kibukawa M, Schelter JM, et al. MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol Cell Biol 2008; 28:2167 - 74; http://dx.doi.org/10.1128/MCB.01977-07; PMID: 18212054
- Blenkiron C, Goldstein LD, Thorne NP, Spiteri I, Chin SF, Dunning MJ, et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol 2007; 8:R214; http://dx.doi.org/10.1186/gb-2007-8-10-r214; PMID: 17922911
- Arima Y, Inoue Y, Shibata T, Hayashi H, Nagano O, Saya H, et al. Rb depletion results in deregulation of E-cadherin and induction of cellular phenotypic changes that are characteristic of the epithelial-to-mesenchymal transition. Cancer Res 2008; 68:5104 - 12; http://dx.doi.org/10.1158/0008-5472.CAN-07-5680; PMID: 18593909
- Rizzi F, Belloni L, Crafa P, Lazzaretti M, Remondini D, Ferretti S, et al. A novel gene signature for molecular diagnosis of human prostate cancer by RT-qPCR. PLoS One 2008; 3:e3617; http://dx.doi.org/10.1371/journal.pone.0003617; PMID: 18974881
- Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010; 465:1033 - 8; http://dx.doi.org/10.1038/nature09144; PMID: 20577206
- Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene 2010; 29:4018 - 32; http://dx.doi.org/10.1038/onc.2010.154; PMID: 20473330
- Dasgupta P, Sun J, Wang S, Fusaro G, Betts V, Padmanabhan J, et al. Disruption of the Rb--Raf-1 interaction inhibits tumor growth and angiogenesis. Mol Cell Biol 2004; 24:9527 - 41; http://dx.doi.org/10.1128/MCB.24.21.9527-9541.2004; PMID: 15485920
- Suzuki S, Adachi A, Hiraiwa A, Ohashi M, Ishibashi M, Kiyono T. Cloning and characterization of human MCM7 promoter. Gene 1998; 216:85 - 91; http://dx.doi.org/10.1016/S0378-1119(98)00323-0; PMID: 9714754
- Thangavel C, Dean JL, Ertel A, Knudsen KE, Aldaz CM, Witkiewicz AK, et al. Therapeutically activating RB: reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr Relat Cancer 2011; 18:333 - 45; http://dx.doi.org/10.1530/ERC-10-0262; PMID: 21367843
- Kolachala VL, Wang L, Obertone TS, Prasad M, Yan Y, Dalmasso G, et al. Adenosine 2B receptor expression is post-transcriptionally regulated by microRNA. J Biol Chem 2010; 285:18184 - 90; http://dx.doi.org/10.1074/jbc.M109.066555; PMID: 20388705
- Thangavel C, Boopathi E, Shapiro BH. Intrinsic sexually dimorphic expression of the principal human CYP3A4 correlated with suboptimal activation of GH/glucocorticoid-dependent transcriptional pathways in men. Endocrinology 2011; 152:4813 - 24; http://dx.doi.org/10.1210/en.2011-1274; PMID: 21952236
- Djuranovic S, Zinchenko MK, Hur JK, Nahvi A, Brunelle JL, Rogers EJ, et al. Allosteric regulation of Argonaute proteins by miRNAs. Nat Struct Mol Biol 2010; 17:144 - 50; http://dx.doi.org/10.1038/nsmb.1736; PMID: 20062058
- Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci USA 2011; 108:5003 - 8; http://dx.doi.org/10.1073/pnas.1019055108; PMID: 21383194
- Chiodelli P, Urbinati C, Mitola S, Tanghetti E, Rusnati M. Sialic acid associated with αvβ3 integrin mediates HIV-1 Tat protein interaction and endothelial cell proangiogenic activation. J Biol Chem 2012; 287:20456 - 66; http://dx.doi.org/10.1074/jbc.M111.337139; PMID: 22528484
- Witkiewicz AK, Rivadeneira DB, Ertel A, Kline J, Hyslop T, Schwartz GF, et al. Association of RB/p16-pathway perturbations with DCIS recurrence: dependence on tumor versus tissue microenvironment. Am J Pathol 2011; 179:1171 - 8; http://dx.doi.org/10.1016/j.ajpath.2011.05.043; PMID: 21756866