Abstract
The illicit generation of tetraploid cells constitutes a prominent driver of oncogenesis, as it often precedes the development of aneuploidy and genomic instability. In addition, tetraploid (pre-)malignant cells display an elevated resistance against radio- and chemotherapy. Here, we report a strategy to preferentially kill tetraploid tumor cells based on the broad-spectrum kinase inhibitor SP600125. Live videomicroscopy revealed that SP600125 affects the execution of mitosis, impedes proper cell division and/or activates apoptosis in near-to-tetraploid, though less so in parental, cancer cells. We propose a novel graphical model to quantify the differential response of diploid and tetraploid cells to mitotic perturbators, including SP600125, which we baptized “transgenerational cell fate profiling.” We speculate that this representation constitutes a valid alternative to classical “single-cell fate” and “genealogical” profiling and, hence, may facilitate the analysis of cell fate within a heterogeneous population as well as the visual examination of cell cycle alterations.
Introduction
Programmed polyploidization (i.e., the acquisition of more than two sets of chromosomes) is required for the normal development and homeostasis of several tissues, including the liver, myocardium, placenta and uterus.Citation1,Citation2 Polyploidy also develops in somatic cells in response to various pathological conditions (e.g., virus-induced cell-to-cell fusion, abortive mitosis) and as a result of cellular stress (e.g., oxidative imbalances) or aging.Citation3 Unscheduled polyploidization has been linked to the development of cancer,Citation2,Citation4 and accumulating evidence indicates that one major mechanism driving tumorigenesis is the so-called “polyploidization-depolyploidization” cascade.Citation3 According to this model, tetraploid cells, which may illicitly emerge through cell-to-cell fusion,Citation5,Citation6 endoreplication (i.e., two consecutive rounds of DNA replication not separated by a mitotic divisionCitation7) or mitotic failure (for instance owing to mitotic slippage or abortive cytokinesisCitation1,Citation8,Citation9), fail to maintain a stable chromosomal content and engender aneuploidy.Citation3 Indeed, the simultaneous presence of a double number of chromosomes and centrosomes significantly interferes with the regulation and execution of mitosis in tetraploid cells, thus undermining the faithful transmission of the genome to the progeny and promoting genomic instability. The stochastic loss of one or more chromosomes during aberrant rounds of bipolar or multipolar cell division is believed to constitute the principal cause of the oncogenic progression of tetraploid cells toward aneuploidy.Citation10-Citation12 In support of the “polyploidization-depolyploidization cascade” model, tetraploidy has been observed in the early stages of multiple tumors, including colorectal, breast and cervical carcinoma,Citation2,Citation13,Citation14 as well as in pre-neoplastic lesions such as Barrett’s esophagus.Citation15 Moreover, the vast majority of solid tumors exhibit both an aneuploid DNA content (with a dominance of near-to-diploid and sub-tetraploid genomes) and high levels of chromosome mis-segregation (also known as chromosomal instability, CINCitation16).Citation17,Citation18 As proof of principle, experimentally generated Tp53−/− tetraploid cells (but not their isogenic diploid counterparts) generate tumors exhibiting high rates of CIN when transplanted into immunodeficient mice.Citation19 Accordingly, the deregulation of various mitotic factors (such as EG5,Citation20 MAD2,Citation21 and PLK1Citation22), as well as the overexpression of oncogenes (including AURKACitation22,Citation23 and MYCCitation24) or the mutations of oncosuppressor genes (including APC,Citation25 BRCA1/2Citation26 and TP53Citation27), have been shown to induce polyploidy and CIN prior to the development of neoplastic lesions.
In healthy cells, the process of polyploidization-depolyploidization is kept in check by multiple oncosuppressive mechanisms.Citation28 The first line of defense is represented by cell cycle checkpoints, a panel of surveillance mechanisms that survey genomic stability throughout the cell cycle. These include the G2-M checkpoint, which prevents the entry into mitosis of cells with unreplicated/damaged DNA,Citation29 the spindle assembly checkpoint (SAC), which halts the metaphase-anaphase transition in the presence of mitotic defects,Citation30 and a hitherto “controversial” tetraploidy checkpoint, which would arrest the proliferation of illicitly generated tetraploid cells.Citation31 Of note, tetraploidy also promotes the activation of a specific pathway of apoptosis that appears to be regulated by the tumor suppressor TP53 as well as by members of the BCL-2 protein family.Citation32 In addition, a more general oncosuppressive mechanism known as mitotic catastrophe initiates the eradication (or the long-term control) of mitosis-incompetent cells during or ensuing an aberrant mitosis.Citation28 Non-diploid cells most often are outcompeted by their diploid counterparts in the context of a proliferative disadvantage known as “aneuploid intolerance.”Citation33 Of note, the molecular mechanisms underpinning mitotic catastrophe and aneuploid intolerance have not yet been precisely elucidated. Finally, we have recently reported the existence of an immunosurveillance mechanism that ensures the elimination of hyperploid cells.Citation34
In spite of all the strategies that have evolved to prevent the establishment of tetraploidy, rare tetraploid/aneuploid cells are capable of overcoming these barriers and, hence, to initiate and/or to contribute to the tumorigenic process. In line with this notion, mutations that affect this multipronged surveillance mechanism are often associated with cancer.Citation3 In this context, the selective killing of tetraploid cells appears as a particularly appealing therapeutic aim, as it would suppress the oncogenic polyploidization-depolyploidization cascade at initial stages. During the past few years, we accumulated evidence demonstrating that tetraploid cancer cells are particularly prone to undergo apoptosis following the activation of mitotic catastrophe.Citation28 In particular, we discovered that tetraploid cells can be eradicated by the inhibition of the checkpoint kinase CHEK1Citation35 and the kinesin KIF11 (best known as EG5)Citation36 more efficiently than their diploid counterparts.
Here, we investigated the effects of SP600125, a broad-spectrum kinase inhibitor, on the survival of near-to-diploid and near-to-tetraploid human colorectal carcinoma cells. By using live videomicroscopy, we demonstrated that near-to-tetraploid cells are more sensitive to SP600125 than their parental counterparts, and that they succumb to this agent following the mitotic catastrophe-elicited activation of apoptosis. Alongside, we propose a novel, refined methodology for the visual examination of cell cycle alterations, which we baptized “transgenerational cell fate profiling.”
Results and Discussion
Preferential cytotoxic activity of SP600125 on near-to-tetraploid cells
We have recently identified SP600125 as an agent that preferentially kills TP53−/− cells.Citation37 Since the inactivation of TP53 is permissive for tetraploidization,Citation19,Citation32 we wondered whether SP600125 might exert differential effects on the survival of tetraploid and diploid cells. To explore this possibility, we took advantage of multiple parental (close-to-diploid) and close-to-tetraploid HCT 116 and RKO colon carcinoma clones that we have previously generated and characterized.Citation32,Citation35 We found that near-to-tetraploid cancer cells are more susceptible to the cytotoxic effects of SP600125 at dose 30 µM than their parental counterparts, and this in both cell lines (). This differential cytotoxicity of SP600125 could be detected by measuring the frequency of dead cells (which incorporate the vital dye propidium iodide) or that of dying cells [which manifest a reduced incorporation of the mitochondria transmembrane potential (Δψm)-sensitive dye 3,3′-dihexyloxacarbocyanine iodide, DiOC6(3)]: (). Cell death as induced by SP600125 in hyperploid colorectal carcinoma cells was mostly mediated by apoptosis, as indicated by the increased frequency of cells exhibiting a subG1 DNA content (), as well as by chromatin condensation and nuclear fragmentation (Vid. S1–7).Citation38,Citation39 Clonogenic assays confirmed that even a transient (24 h) exposure of HCT 116 cells to 30 µM SP600125 causes a dramatic decrease in cellular survival, with a tendency of parental cells to survive to higher extents than their near-to-tetraploid derivatives ().
Figure 1. Preferential killing of near-to-tetraploid colorectal carcinoma cells by SP600125. (A–C) Near-to-diploid (D) and near-to-tetraploid (T) human colorectal carcinoma HCT 116 (A and B) or RKO (C) cells were maintained in control conditions (Co) or treated for 48 and 72 h with 30 µM SP600125 (SP), then co-stained with DiOC6(3) and propidium iodide (PI) for the assessment of apoptosis-associated parameters. (A) Illustrates representative dot plots at 72 h (numbers indicate the percentage of cells found in the corresponding quadrant), while (B and C) report quantitative data. White and black columns illustrate the percentage of dying [PI- DiOC6(3)low] and dead (PI+) cells, respectively. Data are reported as means ± SEM (n = 3). *p < 0.05, as compared with D cells treated with SP. (D and E) D and T HCT 116 cells were maintained in Co conditions or incubated with 30 µM SP600125 for 48 or 72 h, followed by Hoechst 33342 staining (to quantify cells manifesting apoptotic DNA degradation, subG1). (D and E) Illustrate representative dot plots at 72 h and quantitative data (mean ± SEM; n = 3), respectively. *p < 0.05, as compared with D cells treated with SP. (F and G) D and T HCT 116 cells were left untreated (Co) or treated with 30 µM SP600125 for 24 h and then processed for clonogenic assays. Representative plates as well as quantitative data upon normalization to plating efficiency (mean ± SEM; n = 3) are shown. Scale bar = 3 cm.
![Figure 1. Preferential killing of near-to-tetraploid colorectal carcinoma cells by SP600125. (A–C) Near-to-diploid (D) and near-to-tetraploid (T) human colorectal carcinoma HCT 116 (A and B) or RKO (C) cells were maintained in control conditions (Co) or treated for 48 and 72 h with 30 µM SP600125 (SP), then co-stained with DiOC6(3) and propidium iodide (PI) for the assessment of apoptosis-associated parameters. (A) Illustrates representative dot plots at 72 h (numbers indicate the percentage of cells found in the corresponding quadrant), while (B and C) report quantitative data. White and black columns illustrate the percentage of dying [PI- DiOC6(3)low] and dead (PI+) cells, respectively. Data are reported as means ± SEM (n = 3). *p < 0.05, as compared with D cells treated with SP. (D and E) D and T HCT 116 cells were maintained in Co conditions or incubated with 30 µM SP600125 for 48 or 72 h, followed by Hoechst 33342 staining (to quantify cells manifesting apoptotic DNA degradation, subG1). (D and E) Illustrate representative dot plots at 72 h and quantitative data (mean ± SEM; n = 3), respectively. *p < 0.05, as compared with D cells treated with SP. (F and G) D and T HCT 116 cells were left untreated (Co) or treated with 30 µM SP600125 for 24 h and then processed for clonogenic assays. Representative plates as well as quantitative data upon normalization to plating efficiency (mean ± SEM; n = 3) are shown. Scale bar = 3 cm.](/cms/asset/a7e970d7-a984-4e53-9ab4-9e71d2d6f63a/kccy_a_10923046_f0001.gif)
Altogether, these results indicate that SP600125 acts more potently on near-to-tetraploid than on near-to-diploid cancer cells.
Videomicroscopic cell cycle profiling confirms the preferential sensitivity of near-to-tetraploid cells to SP600125
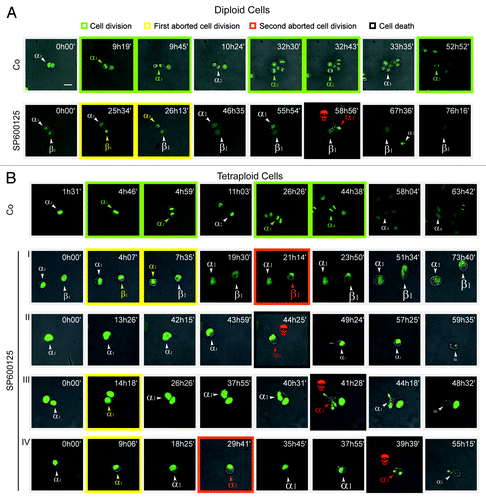
To explore the mechanisms accounting for the peculiar lethality of SP600125 for near-to-tetraploids, we analyzed the response of near-to-diploid and near-to-tetraploid HCT 116 colon carcinoma cells stably transfected with a histone 2B-green fluorescent protein (H2B-GFP) fusion protein.Citation12 To this aim, cells were cultured in the continuous presence of 30 µM SP600125 and monitored by fluorescence videomicroscopy every 13 min over more than 3 d. In this period, we observed that SP600125 alters the normal fate of both parental and near-to-tetraploid cells, generating frequent cases of abortive cell division, mitotic catastrophe and apoptotic cell death (; Vid. S1–7). In particular, the administration of SP600125 to parental and near-to-tetraploid cells dramatically perturbed the execution of mitosis and induced the appearance of aberrant (pro-)metaphases that were characterized by asymmetric and disorganized metaphase plates owing to the incomplete congression of chromosomes to the equatorial plane (; Vid. S1–7). Despite these aberrations, parental and near-to-tetraploid cells responding to SP600125 quickly progressed through mitosis, yet failed to orchestrate a correct anaphase/telophase and did not divide, often generating one single cell with an enlarged (and frequently multilobated) nucleus. In several instances, such an abortive cell division, which generated a near-to-tetraploid cell (when the parental cell was near-to-diploid) or a near-to-octoploid cell (when the parental cell was near-to-tetraploid), was followed by a second round of mitotic abortion, further increasing the ploidy of daughter cells (; Vid. S1–7). In this setting, cell death occurred either as a primary event (before mitosis) or upon one or two rounds of abortive mitosis. Of note, there was a significant difference in the behavior of parental and near-to-tetraploid cells. Indeed, while the majority of parental cells underwent a permanent cell cycle arrest upon a single round of abortive mitosis, near-to-tetraploid cells frequently underwent a second round of aberrant mitosis and/or mitotic catastrophe-initiated apoptosis (; Vid. S1–7). Quantitative analyses are required to understand the potential relevance of this finding.
Figure 2. Videomicroscopic cell cycle profiling of SP600125 effects on near-to-diploid and near-to-tetraploid cells. (A and B) Near-to-diploid (D) (A) and near-to-tetraploid (T) (B) human colorectal carcinoma HCT 116 cells engineered to express a histone 2B-GFP fusion protein were left untreated (Co) or exposed to 30 µM SP600125, and then followed by live videomicroscopy for more than 72 h. Representative snapshots are shown (scale bar = 10 μm). Greek letters refer to individual cells that underwent successful cell divisions (framed in green), generating two daughter cells; one or two rounds of abortive cell divisions (framed in yellow and orange for the first and second abortive division, respectively) leading to one single daughter cell or, alternatively, apoptosis (framed in black and labeled with a red skull). The increase in cell diameter is depicted by increased font size. In (B), two cells entering one or two consecutive rounds of abortive cell divisions (I), a cell undergoing apoptosis before entering mitosis (II) and cells undertaking one (III) or two (IV) rounds of abortive cell division followed by cell death are shown. Full-length movies are available as Video S1–7.
Transgenerational cell fate profiling of SP600125-treated cells
To illustrate the differential impact of SP600125 on the cell cycle progression of parental and near-to-tetraploid colorectal carcinoma cells, we quantified the fate of individual cells by videomicroscopy. After each successful division, we arbitrarily followed the destiny of one among the two daughter cells. Finally, we plotted cell fate as a single line in which the occurrence of productive cell divisions, abortive mitoses and apoptotic instances of cell death is color-coded (). Such a mono-dimensional “single-cell fate profiling” allows for a direct comparison between the parental and near-to-tetraploid cells responding to SP600125 (), confirming that a significant portion of SP600125-treated near-to-tetraploid cells progresses along the cell cycle even upon a round of abortive mitosis and/or dies (). However, at first sight, this type of representation may appear complex and suboptimal, as it fails to inform (1) on the destiny of both daughter cells arising from successful mitoses as well as (2) on the overall fate of each cell generation. To simplify the representation of cell fate profiles, we calculated the percentage of events affecting consecutive cell generations. To this aim, we assumed that a cell passes from one generation to the next one both (1) if it divides successfully and (2) if it duplicates its genome through an abortive mitosis and, hence, undergoes an increase in ploidy. Thus, 72 h after the administration of SP600125, the majority of parental and near-to-tetraploid cells could be assigned to the second and third generation, respectively (). However, similar to mono-dimensional cell fate profiles, this graphical representation (to which we refer as to “genealogical profiling”) is rather complex and, therefore, suboptimal (). To further simplify the representation, we designed a new model to visualize the fate of cell cultures, which we refer to as “transgenerational cell fate profiling.” In this representation, the prime cell is not represented, but its fate is followed from generation to generation, as symbolized by concentric rings that get more distant from the center of the illustration. Coupled to an appropriate color code to mark cell death and abortive mitosis, this yields a simple graph that allows for the visual appreciation of the overall composition of cell cultures as it changes from one generation to the next one as well as of cell cycle perturbations that may differentially affect distinct cell lines ().
Figure 3. Single-cell fate profiling of near-to-diploid and near-to-tetraploid HCT 116 cells treated with SP600125. (A and B) Near-to-diploid (D) (A) and near-to-tetraploid (T) (B) human colorectal carcinoma HCT 116 cells engineered to express a GFP-histone 2B fusion protein were left untreated or exposed to 30 µM SP600125, and then followed by live videomicroscopy for more than 72 h. Single-cell fate profiles of untreated and SP600125-treated cells are shown for 50 cells in each condition. Green traits depict normal cell divisions, whereas yellow and orange traits illustrate the first and the second abortive cell division, respectively. Black traits represent apoptosis induction. Darkening gray lines indicate an increase in cell ploidy.
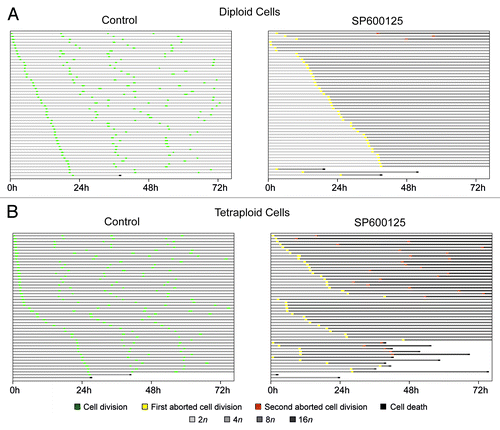
Figure 4. Genealogical cell fate profiling of parental and near-to-tetraploid HCT 116 cells treated with SP600125. (A and B) Near-to-diploid (A) and near-to-tetraploid (B) human colorectal carcinoma HCT 116 cells engineered to express a histone 2B-GFP fusion protein were left untreated or exposed to 30 µM SP600125, and then followed by live videomicroscopy for more than 72 h. Genealogical cell profiles for untreated or SP600125-treated cells are depicted for 50 cells in each condition. Green, yellow, orange and gray and illustrate the fraction of cells that underwent a successful cell division, one abortive cell division, two consecutive abortive cell divisions and cell cycle arrest, respectively. F0, F1 and F2 represent three consecutive generations. Cells were considered arrested only when cell cycle progression was blocked for more than 24 h. In the case of F2, the fraction of cells remaining inert for less than 24 h was not included in the representation. Please note that the width of arrows is proportional to the percentage of corresponding events and that the increase in ploidy is illustrated by darkening and enlarging circles.
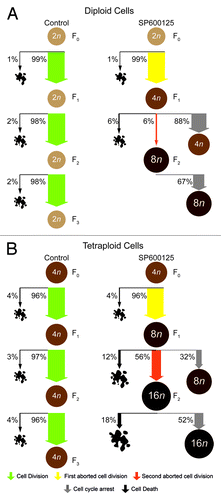
Figure 5. Transgenerational cell fate profiling of parental and near-to-tetraploid HCT 116 cells treated with SP600125. (A–C) Near-to-diploid (A and C) and near-to-tetraploid (B) human colorectal carcinoma HCT 116 cells engineered to express a histone 2B-GFP fusion protein were left untreated or exposed to 30 µM SP600125 (A and B) or, alternatively, treated with 5 nM pacitaxel or 1 µM dimethylenastron (DIMEM) (C), and then followed by live videomicroscopy for more than 72 h. Transgenerational cell profiles are illustrated for 50 cells in each condition. Green, yellow, orange and black sections illustrate the fraction of cells that underwent successful cell division (symmetric and asymmetric divisions all confounded), one abortive cell division, two consecutive abortive cell divisions and cell death, respectively. Concentric circles depict three consecutive events, starting from E1 (inner circle). Cell divisions were considered to be successful only when daughter cells were clearly separated
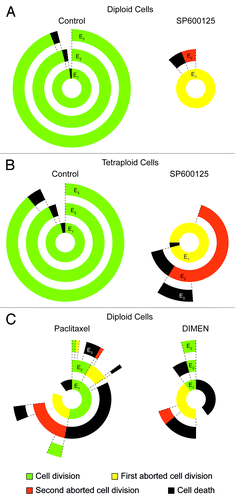
In our experimental setting, this illustration facilitates a rapid interpretation and quantification of the response of parental vs. hyperploid colorectal carcinoma cells to SP600125, which we could summarize as follows: (1) SP600125 acts as an efficient polyploidizing agent in both parental and near-to-tetraploid cells; (2) the proliferation of SP600125-treated parental cells is mainly arrested after one round of abortive cell division; and (3) SP600125-treated near-to-tetraploid cells present a more heterogeneous behavior than their parental counterparts, encompassing cells that die before mitosis as well as cells that enter one or two rounds of abortive division followed by cell cycle arrest or apoptosis (). Of note, this representation simplifies the analysis of complex cell cycle alterations, such as those induced by the administration of the antimitotic agents paclitaxel (a microtubule hyperpolymerizing compound) and dimethylenastron (a pharmacological inhibitor of the kinesin KIF11) ().
Concluding Remarks
In this paper, we analyzed the complex effects of the broad-spectrum kinase inhibitor SP600125, a cell cycle-perturbing and apoptosis-inducing agent, on human colorectal carcinoma cells. In particular, by applying live videomicroscopy coupled with multiple analytical approaches, we dissected the mechanisms accounting for the accrued sensitivity of near-to-tetraploid, as compared with parental, HCT 116 and RKO cells to SP600125. Thanks to GFP-labeled chromatin components (in our case histone 2B), it is possible to follow the fate of individual cells over time as they undergo normal or prolonged mitoses, productive or abortive cell divisions or, alternatively, as they die by apoptosis or necrosis.Citation40,Citation41 In this setting, one particular difficulty consists in representing the individual fate of multiple cells that compose a population, so that the human eye can instantaneously capture all the information that is required for an easy and efficient comparison of distinct data sets. Here, we proposed three alternative graphical representations of the cellular response to SP600125, each of which can be applied to other cell cycle-interfering agents, and each of which is associated with specific advantages and disadvantages. The juxtaposition of single-cell fate analyses () yields temporal information on the length of mitosis and interphase, as well as on the latency between abortive mitoses and subsequent cell death, yet renders difficult the appreciation of the percentage of a cell population that undergoes either fate. Genealogical profiling () provides immediate information on the proportion of cells that either pass from one generation to the next (as a result of nuclear division), either undergo a cell cycle arrest or die. However, this representation fails to inform on the duration of each process and appears rather complex. Transgenerational profiling () provides an attractive way to summarize how populations behave with respect to successful cell divisions, abortive mitoses and cell death, providing information on the overall (often heterogeneous) destiny of multiple individual cells aggregated in subpopulations. Such subpopulations are symbolized in concentric circles, radiating from an initial event ideally located at the center of the graph to successive events in ever more peripheral circles. We are convinced that this graphic representation facilitates the comparison of cell populations as they respond to various experimental interventions.
Materials and Methods
All experiments were performed in triplicate instances and independently repeated at least twice, yielding similar results. Data were analyzed with Microsoft Excel (Microsoft) and are reported as means ± SEM. Statistical significance was assessed by means of two-tailed unpaired Student’s t-tests (*p < 0.05).
Cell lines, culture conditions and chemicals
Unless otherwise indicated, media and supplements for cell culture were obtained from Gibco-Invitrogen and plasticware from Corning B.V. Life Sciences. Parental and near-to-tetraploid HCT 116 and RKO colorectal carcinoma clones were grown in McCoy's 5A medium supplemented with 10% fetal calf serum (FCS). Parental and near-to-tetraploid HCT 116 cells stably transfected with a cDNA coding for a histone 2B-green fluorescent (H2B-GFP) fusion protein were cultured in McCoy's 5A medium supplemented as above plus 20 mg/mL blasticidine. Cell lines were routinely maintained at 37°C under 5% CO2 in T175 flasks and seeded into 12- or 96-well plates 24 h before the beginning of the experiment. SP600125 [anthra (1,9-cd) pyrazole-6 (2H)-one] and paclitaxel were purchased from Sigma-Aldrich, while dimethylenastron (Eg5 Inhibitor III, DIMEN) was obtained from Calbiochem-Merck.
Cytofluorometric studies
For the simultaneous quantification of plasma membrane integrity and mitochondrial transmembrane potential (Δψm) or, alternatively, for the quantification of DNA content (cell cycle profiling), cells were collected and stained with 1 μg/mL propidium iodide (PI, which only incorporates into dead cells, from Sigma-Aldrich) plus 40 nM 3,3′-dihexyloxacarbocyanine iodide [DiOC6(3), a Δψm-sensitive dye] (Molecular Probes-Invitrogen) or with 2 μM Hoechst 33342 (a nucleophilic dye, from Molecular Probes-Invitrogen), respectively. Upon staining, cells were incubated for 30 min at 37°C.Citation37 Cytofluorometric acquisitions were performed by means of a Gallios cytofluorometer (Beckman Coulter) or a FACScan cytofluorometer (from BD Biosciences) equipped with a 70 μm nozzle. Data were statistically evaluated using the Kaluza (Beckman Coulter) or CellQuestTM (Becton Dickinson) software. Only the events characterized by normal forward scatter (FSC) and side scatter (SSC) parameters were gated for inclusion in the statistical analysis.
Clonogenic survival assays
To evaluate clonogenic survival, parental and near-to-tetraploid HCT 116 cells were seeded at different concentrations (from 100–5,000 cells/well) in 6-well plates, allowed to adapt overnight and then treated with SP600125 for 24 h. Upon a drug washout, cells were cultured in standard conditions for up to 12 d. Colonies were then fixed and stained with an aqueous solution containing 0.25% (w/v) crystal violet, 70% (v/v) methanol and 3% (v/v) formaldehyde (Carlo Erba Reagents) and counted as previously described.Citation42 Only colonies made of > 30 cells were included in the quantification. For each treatment, the surviving fraction (SF) was estimated according to the formula: SF = (number of colonies formed/number of cells seeded). The SF in untreated conditions represented plating efficiency.
Videomicroscopy
For videomicroscopy, parental and near-to-tetraploid HCT 116 cells stably expressing a H2B-GFP fusion protein were grown in 96-well imaging plates (BD Biosciences) under standard culture conditions and subjected to pulsed observations (every 13 min for up to 72 h) with a BD pathway 855 automated live-cell microscope (BD Biosciences). Images were analyzed with the open source software ImageJ (freely available from the National Institute of Health, at the address www.rsb.info.nih.gov/ij/).
Additional material
Download Zip (8.4 MB)Acknowledgments
L.G. is funded by the LabEx ImmunoOncology. G.K. is supported by the Ligue Nationale contre le Cancer (Equipe labelisée), Agence Nationale pour la Recherche (ANR), European Commission (Active p53, Artforce, ChemoRes, ApopTrain), Fondation pour la Recherche Médicale (FRM), Institut National du Cancer (INCa), Cancéropôle Ile-de-France, AXA Research Fund and the Labex Immuno-Oncology.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Lee HO, Davidson JM, Duronio RJ. Endoreplication: polyploidy with purpose. Genes Dev 2009; 23:2461 - 77; http://dx.doi.org/10.1101/gad.1829209; PMID: 19884253
- Davoli T, de Lange T. The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol 2011; 27:585 - 610; http://dx.doi.org/10.1146/annurev-cellbio-092910-154234; PMID: 21801013
- Vitale I, Galluzzi L, Senovilla L, Criollo A, Jemaà M, Castedo M, et al. Illicit survival of cancer cells during polyploidization and depolyploidization. Cell Death Differ 2011; 18:1403 - 13; http://dx.doi.org/10.1038/cdd.2010.145; PMID: 21072053
- Storchova Z, Kuffer C. The consequences of tetraploidy and aneuploidy. J Cell Sci 2008; 121:3859 - 66; http://dx.doi.org/10.1242/jcs.039537; PMID: 19020304
- Duelli DM, Padilla-Nash HM, Berman D, Murphy KM, Ried T, Lazebnik Y. A virus causes cancer by inducing massive chromosomal instability through cell fusion. Curr Biol 2007; 17:431 - 7; http://dx.doi.org/10.1016/j.cub.2007.01.049; PMID: 17320392
- Ogle BM, Cascalho M, Platt JL. Biological implications of cell fusion. Nat Rev Mol Cell Biol 2005; 6:567 - 75; http://dx.doi.org/10.1038/nrm1678; PMID: 15957005
- Davoli T, Denchi EL, de Lange T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell 2010; 141:81 - 93; http://dx.doi.org/10.1016/j.cell.2010.01.031; PMID: 20371347
- Brito DA, Rieder CL. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol 2006; 16:1194 - 200; http://dx.doi.org/10.1016/j.cub.2006.04.043; PMID: 16782009
- Lacroix B, Maddox AS. Cytokinesis, ploidy and aneuploidy. J Pathol 2012; 226:338 - 51; http://dx.doi.org/10.1002/path.3013; PMID: 21984283
- Gisselsson D, Håkanson U, Stoller P, Marti D, Jin Y, Rosengren AH, et al. When the genome plays dice: circumvention of the spindle assembly checkpoint and near-random chromosome segregation in multipolar cancer cell mitoses. PLoS One 2008; 3:e1871; http://dx.doi.org/10.1371/journal.pone.0001871; PMID: 18392149
- Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009; 460:278 - 82; http://dx.doi.org/10.1038/nature08136; PMID: 19506557
- Vitale I, Senovilla L, Jemaà M, Michaud M, Galluzzi L, Kepp O, et al. Multipolar mitosis of tetraploid cells: inhibition by p53 and dependency on Mos. EMBO J 2010; 29:1272 - 84; http://dx.doi.org/10.1038/emboj.2010.11; PMID: 20186124
- Levine DS, Rabinovitch PS, Haggitt RC, Blount PL, Dean PJ, Rubin CE, et al. Distribution of aneuploid cell populations in ulcerative colitis with dysplasia or cancer. Gastroenterology 1991; 101:1198 - 210; PMID: 1936790
- Olaharski AJ, Sotelo R, Solorza-Luna G, Gonsebatt ME, Guzman P, Mohar A, et al. Tetraploidy and chromosomal instability are early events during cervical carcinogenesis. Carcinogenesis 2006; 27:337 - 43; http://dx.doi.org/10.1093/carcin/bgi218; PMID: 16123119
- Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, Blount PL, et al. The combination of genetic instability and clonal expansion predicts progression to esophageal adenocarcinoma. Cancer Res 2004; 64:7629 - 33; http://dx.doi.org/10.1158/0008-5472.CAN-04-1738; PMID: 15492292
- Geigl JB, Obenauf AC, Schwarzbraun T, Speicher MR. Defining ‘chromosomal instability’. Trends Genet 2008; 24:64 - 9; http://dx.doi.org/10.1016/j.tig.2007.11.006; PMID: 18192061
- Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol 2009; 10:478 - 87; http://dx.doi.org/10.1038/nrm2718; PMID: 19546858
- Knutsen T, Padilla-Nash HM, Wangsa D, Barenboim-Stapleton L, Camps J, McNeil N, et al. Definitive molecular cytogenetic characterization of 15 colorectal cancer cell lines. Genes Chromosomes Cancer 2010; 49:204 - 23; PMID: 19927377
- Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005; 437:1043 - 7; http://dx.doi.org/10.1038/nature04217; PMID: 16222300
- Castillo A, Morse HC 3rd, Godfrey VL, Naeem R, Justice MJ. Overexpression of Eg5 causes genomic instability and tumor formation in mice. Cancer Res 2007; 67:10138 - 47; http://dx.doi.org/10.1158/0008-5472.CAN-07-0326; PMID: 17974955
- Sotillo R, Hernando E, Díaz-Rodríguez E, Teruya-Feldstein J, Cordón-Cardo C, Lowe SW, et al. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 2007; 11:9 - 23; http://dx.doi.org/10.1016/j.ccr.2006.10.019; PMID: 17189715
- Meraldi P, Honda R, Nigg EA. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. EMBO J 2002; 21:483 - 92; http://dx.doi.org/10.1093/emboj/21.4.483; PMID: 11847097
- Wang X, Zhou YX, Qiao W, Tominaga Y, Ouchi M, Ouchi T, et al. Overexpression of aurora kinase A in mouse mammary epithelium induces genetic instability preceding mammary tumor formation. Oncogene 2006; 25:7148 - 58; http://dx.doi.org/10.1038/sj.onc.1209707; PMID: 16715125
- Li Q, Dang CV. c-Myc overexpression uncouples DNA replication from mitosis. Mol Cell Biol 1999; 19:5339 - 51; PMID: 10409725
- Dikovskaya D, Schiffmann D, Newton IP, Oakley A, Kroboth K, Sansom O, et al. Loss of APC induces polyploidy as a result of a combination of defects in mitosis and apoptosis. J Cell Biol 2007; 176:183 - 95; http://dx.doi.org/10.1083/jcb.200610099; PMID: 17227893
- Daniels MJ, Wang Y, Lee M, Venkitaraman AR. Abnormal cytokinesis in cells deficient in the breast cancer susceptibility protein BRCA2. Science 2004; 306:876 - 9; http://dx.doi.org/10.1126/science.1102574; PMID: 15375219
- Aylon Y, Oren M. p53: guardian of ploidy. Mol Oncol 2011; 5:315 - 23; http://dx.doi.org/10.1016/j.molonc.2011.07.007; PMID: 21852209
- Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol 2011; 12:385 - 92; http://dx.doi.org/10.1038/nrm3115; PMID: 21527953
- Sperka T, Wang J, Rudolph KL. DNA damage checkpoints in stem cells, ageing and cancer. Nat Rev Mol Cell Biol 2012; 13:579 - 90; http://dx.doi.org/10.1038/nrm3420; PMID: 22914294
- Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 2007; 8:379 - 93; http://dx.doi.org/10.1038/nrm2163; PMID: 17426725
- Margolis RL, Lohez OD, Andreassen PR. G1 tetraploidy checkpoint and the suppression of tumorigenesis. J Cell Biochem 2003; 88:673 - 83; http://dx.doi.org/10.1002/jcb.10411; PMID: 12577301
- Castedo M, Coquelle A, Vivet S, Vitale I, Kauffmann A, Dessen P, et al. Apoptosis regulation in tetraploid cancer cells. EMBO J 2006; 25:2584 - 95; http://dx.doi.org/10.1038/sj.emboj.7601127; PMID: 16675948
- Thompson SL, Compton DA. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol 2010; 188:369 - 81; http://dx.doi.org/10.1083/jcb.200905057; PMID: 20123995
- Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, et al. An immunosurveillance mechanism controls cancer cell ploidy. Science 2012; 337:1678 - 84; http://dx.doi.org/10.1126/science.1224922; PMID: 23019653
- Vitale I, Galluzzi L, Vivet S, Nanty L, Dessen P, Senovilla L, et al. Inhibition of Chk1 kills tetraploid tumor cells through a p53-dependent pathway. PLoS One 2007; 2:e1337; http://dx.doi.org/10.1371/journal.pone.0001337; PMID: 18159231
- Rello-Varona S, Vitale I, Kepp O, Senovilla L, Jemaá M, Métivier D, et al. Preferential killing of tetraploid tumor cells by targeting the mitotic kinesin Eg5. Cell Cycle 2009; 8:1030 - 5; http://dx.doi.org/10.4161/cc.8.7.7950; PMID: 19270519
- Jemaà M, Vitale I, Kepp O, Berardinelli F, Galluzzi L, Senovilla L, et al. Selective killing of p53-deficient cancer cells by SP600125. EMBO Mol Med 2012; 4:500 - 14; http://dx.doi.org/10.1002/emmm.201200228; PMID: 22438244
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012; 19:107 - 20; http://dx.doi.org/10.1038/cdd.2011.96; PMID: 21760595
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al, Nomenclature Committee on Cell Death 2009. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 2009; 16:3 - 11; http://dx.doi.org/10.1038/cdd.2008.150; PMID: 18846107
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 2010; 11:700 - 14; http://dx.doi.org/10.1038/nrm2970; PMID: 20823910
- Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G. Cell death assays for drug discovery. Nat Rev Drug Discov 2011; 10:221 - 37; http://dx.doi.org/10.1038/nrd3373; PMID: 21358741
- Zhang P, Castedo M, Tao Y, Violot D, Métivier D, Deutsch E, et al. Caspase independence of radio-induced cell death. Oncogene 2006; 25:7758 - 70; http://dx.doi.org/10.1038/sj.onc.1209744; PMID: 16862186