In the popular tale “Ozma of Oz,” Lyman Frank Baum describes the princess Langwidere, who is capable of interchanging her currently worn head with another one, using a collection of heads stored in a cabinet. Remarkably, she changes her character as well when putting a new head on her neck.
In the December 15, 2012 issue of Cell Cycle, Kurt Engeland and colleagues report a similar phenomenonCitation1 with regard to a DNA-associated, transcription-regulatory complex of proteins, the DREAM/MMB complex. A central core of this complex, MuvB, binds to a DNA motif called CHR for cell cycle genes homology region.Citation2 In addition, the complex has facultative members. Either the B-Myb oncoprotein joins to activate transcription from the adjacent gene—this composition is termed MMB for Myb-MuvB. Alternatively, the retinoblastoma protein homolog p130, along with the prototype-repressive member of the E2F family, E2F4 and the auxiliary DNA-binding partner protein DP1, associate with MuvB to form an entity called DREAM for DP, RB-like, E2F and MuvB complex. This complex was first purified from Drosophila embryosCitation3 and then characterized in mammalian cells.Citation4 Exchanging the “head” of the complex in this way also changes its “character,” converting the transactivator MMB to the repressive DREAM complex.
Strikingly, the new report shows how this re-association of the MMB/DREAM complex can be induced. Activating the tumor suppressor p53 drives the expression of the cyclin-dependent kinase p21/Cip1/Waf1/CDKN1A. This, in turn, leads to the hypophosphorylation of p130 and thereby enables its association with MuvB, replacing B-Myb and mediating the repression of at least one cell cycle regulatory gene, i.e., CCNB2 encoding cyclin B2. Thus, by interchanging the association partners of MuvB, p53 indirectly represses a cell cycle regulatory gene to mediate G2 arrest.
p53 carries out most (though not all) of its tumor suppressive functions by regulating transcription; it activates target genes by DNA binding. However, p53 also represses genes, the underlying mechanisms being much less obvious.
It was previously reported that p53 binds to the promoters of repressed genes. However, unlike for transactivation, p53 did not seem to directly bind the DNA of repressed promoters, but rather associate with the DNA-bound transcription factor complex NF-Y.Citation5 In the current report, however, the authors did not observe an association of p53 with repressed promoters,Citation1 arguing against a general need for this interactions in repression.
A second possibility is that p53 may induce genes that encode repressors. Accordingly, preventing protein synthesis by cycloheximide abolishes p53-mediated repression but not activation.Citation6 Strikingly, cells that lack p21 no longer show gene repression by p53.Citation7,Citation8 p21 is an inhibitor of cyclin-dependent kinases, thus contributing to a hypophosphorylated state of the retinoblastoma protein family members pRb, p107 and p130. In turn, hypophosphorylated Rb proteins bind to members of the E2F family of transcription factors, often turning transactivators into repressors. Many p53-repressible genes contain promoter elements that bind E2F. Thus, it appeared conceivable that gene repression by p53 is largely performed through E2F-binding DNA.
The new report shows that this scenario is unlikely to reflect the full truth. The authors show that it is a CHR site, rather than E2F-responsive elements, that confers p53-mediated repression. Hence, while still involving p21 and members of the Rb and E2F families, the new model () suggests that the association of a repressive DREAM complex with the CHR site is the major route of negative gene regulation by p53. This does not exclude that the other two mechanisms (p53-NF-Y and pRb-E2F) and their respective promoter DNA motifs still contribute to the repression of a different set of promoters; however, the work by Quaas et al. clarifies a necessary role of converting MMB to DREAM by p53 and p21 for the repression of CCNB2 and for G2 arrest.
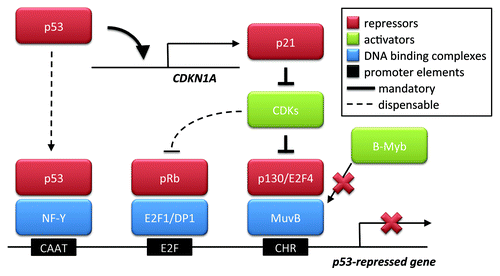
Figure 1. p53-mediated gene repression. p53 reportedly can act as a repressor of genes by at least three mechanisms. The first one involves the association of p53 with the transcription factor complex NF-Y, which, in turn, binds the CAAT box of promoter DNA. The two other mechanisms each depend on the transactivation of p21 by p53. p21 blocks cyclin-dependent kinases (CDKs), leading to the hypophosphorylation of retinoblastoma (Rb) family members. These then associate with E2F proteins. E2Fs can bind their cognate DNA elements, in cooperation with DP1, and the associated Rb proteins then mediate repression. However, p130, while binding E2F4 but independent of an E2F-binding DNA element, associates with the MuvB-complex, replacing B-Myb. MuvB binds the CHR element of DNA. As a consequence of E2F4 and p130 being tethered to the CHR, the promoter is repressed. The first two mechanisms were reported earlier, but the article in this issue of Cell CycleCitation1 argues against a direct association of p53 with the cyclin B promoter; nor did the authors observe a need for E2F binding sites in repression. Only the last mechanism, driven by MuvB and the CHR element, appears indispensable for gene repression in these experiments. This mandatory mechanism is therefore indicated by bold arrows in the scheme, whereas the other two seem dispensable, reflected by dashed lines.
If this concept can be generalized to other p53-repressed promoters, the outlined mechanism may explain how p53 and p21 prevent premature mitosis, especially upon DNA damage. In the context of cancer chemotherapy, it may prove helpful to deliberately interfere with this route to G2 arrest, thus sensitizing tumor cells by promoting mitotic failure. Putting a new head on MuvB may thereby determine the sensitive or resistant character of a cell.
References
- Quaas M, et al. Cell Cycle 2012; 11:4661 - 72; http://dx.doi.org/10.4161/cc.22917; PMID: 23187802
- Müller GA, et al. Nucleic Acids Res 2012; 40:1561 - 78; http://dx.doi.org/10.1093/nar/gkr793; PMID: 22064854
- Korenjak M, et al. Cell 2004; 119:181 - 93; http://dx.doi.org/10.1016/j.cell.2004.09.034; PMID: 15479636
- Litovchick L, et al. Mol Cell 2007; 26:539 - 51; http://dx.doi.org/10.1016/j.molcel.2007.04.015; PMID: 17531812
- Imbriano C, et al. Mol Cell Biol 2005; 25:3737 - 51; http://dx.doi.org/10.1128/MCB.25.9.3737-3751.2005; PMID: 15831478
- Böhlig L, et al. Nucleic Acids Res 2011; 39:440 - 53; http://dx.doi.org/10.1093/nar/gkq796; PMID: 20833636
- Gottifredi V, et al. Mol Cell Biol 2001; 21:1066 - 76; http://dx.doi.org/10.1128/MCB.21.4.1066-1076.2001; PMID: 11158294
- Löhr K, et al. J Biol Chem 2003; 278:32507 - 16; http://dx.doi.org/10.1074/jbc.M212517200; PMID: 12748190