Macroautophagy, hereafter referred as autophagy, is a lysosomal degradation process that is initiated from phagophore assembly sites (PAS), which expand to recruit Atg8/LC3 for the formation of isolation membranes/phagophores. The processing of Atg8/LC3 is required for the elongation and sealing of the phagophore to generate the completed autophagosome. Recent studies have identified SNARE proteins, evolutionarily conserved mediators of intracellular membrane fusion, as key regulators for the expansion of PAS and recruitment of Atg8/LC3 in both yeastCitation1 and mammalian cells.Citation2 Loss of Sec18 and Sec17, the yeast orthologs of the mammalian AAA-ATPase NSF and its cofactor αSNAP, respectively, which regulate the activation of SNARE complexes,Citation3 results in a defect in autophagy,Citation1 demonstrating that SNARE-mediated membrane fusion plays a key role in autophagosome biogenesis.
In stark contrast with these findings, Naydenov et al. report within the December 15, 2012 issue of Cell Cycle that knockdown of αSNAP promotes autophagic flux in cultured human epithelial cells under nutrient-rich culture conditions ().Citation4 They found that αSNAP siRNA (siαSNAP)-induced autophagy is accompanied by Golgi fragmentation, and that the fragmented Golgi membranes (fGMs) co-localize with an autophagosomal marker, GFP-LC3. Moreover, loss of the membrane curvature-inducer Bif-1 attenuates siαSNAP-induced autophagy. Notably, Bif-1 promotes the fission of Golgi membranes that contain the autophagy-essential transmembrane protein Atg9 to promote autophagosome formation in mammalian cells during starvation,Citation5 suggesting that Bif-1 may serve as a key regulator for the PAS formation and expansion during the suppression of αSNAP. Intriguingly, deletion of a secretory SNARE gene, Sec22, in yeast suppresses the PAS translocation of Atg9 and abrogates autophagosome biogenesis,Citation1 whereas knockdown of Sec22B in HeLa cells has minimal effects on the formation and expansion of PAS,Citation2 suggesting that the membrane supply process for autophagosome biogenesis in mammalian cells may differ from that in yeast.
Figure 1. Knockdown of αSNAP promotes autophagic flux through the inhibition of mTOR-related signaling and the fragmentation of Golgi apparatus to provide Atg9-containing membranes for the formation and expansion of PAS during autophagosome biogenesis.
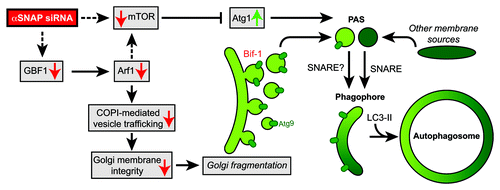
As siαSNAP-induced LC3 lipidation and GFP-LC3 foci formation are observed in the absence of Beclin 1, the authors propose that siαSNAP-induced autophagy occurs through a non-canonical pathway that bypasses the Beclin 1-dependent phagophore nucleation step.Citation4 However, it is worth noting that LC3 processing can be induced in several autophagy-defective cells.Citation6 Moreover, while siαSNAP-induced autophagy requires Bif-1, Bif-1-mediated Golgi fission and autophagosome formation during nutrient starvation require the Beclin 1-UVRAG complex.Citation5 Further analyses are warranted to clarify whether and how the autophagy induced in response to αSNAP knockdown occurs in a Beclin 1-independent manner.
Although it remains unclear if the dispersion of fGMs is in itself sufficient for the induction of autophagy, the authors demonstrate that siαSNAP diminishes the expression of several Golgi-associated proteins, including the guanine nucleotide exchange factor GBF1,Citation4 which acts upstream of Arf to regulate COPI-mediated vesicle trafficking. Dysregulation of Arf1 has been shown to inhibit mTORC1,Citation7 a negative regulator of the autophagy-essential Atg1/ULK kinase complex.Citation6 Consistently, the mTOR signaling pathway is suppressed by αSNAP knockdown. As the dispersion of fGMs by the knockdown of Rab1 is not sufficient for inducing autophagosome formation,Citation8 the activation of Atg1/ULK may be a critical factor for the induction of Golgi fragmentation-induced autophagy.
SNARE-mediated membrane fusion is indispensable not only for the expansion of PAS but also for the formation and maturation of completed autophagosomes. This raises a question, how can fGMs form degradative autophagosomes in the absence of αSNAP? Unlike yeast Sec17/Sec18 deletion mutants, the siRNA-mediated gene silencing system used by Naydenov et al. did not completely deplete αSNAP.Citation4 Thus, it is possible to speculate that a trace amount of αSNAP in the siRNA-treated cells is sufficient for activating SNAREs to mediate autophagic membrane fusion events. In addition, as the SNAP family is composed of two ubiquitously expressed α- and γ-SNAPs and neuronal cell-specific β-SNAP,Citation3 the expression of β-/γ-SNAPs may compensate for the loss of αSNAP. In this scenario, depletion of β-/γ-SNAPs or NSF would abrogate siαSNAP-induced GFP-LC3 foci formation and cause the accumulation of LC3-negative nascent autophagosomal membranes, as SNAREs are required for the expansion of PAS prior to the recruitment of Atg8/LC3.Citation1,Citation2 Indeed, unlike αSNAP depletion, knockdown of NSF fails to induce the autophagic processing of LC3.Citation4 Further investigation of the mechanism behind the regulation of autophagic membrane fusion in siαSNAP-treated cells is anticipated in the future.
References
- Nair U, et al. Cell 2011; 146:290 - 302; http://dx.doi.org/10.1016/j.cell.2011.06.022; PMID: 21784249
- Moreau K, et al. Cell 2011; 146:303 - 17; http://dx.doi.org/10.1016/j.cell.2011.06.023; PMID: 21784250
- Jahn R, et al. Nat Rev Mol Cell Biol 2006; 7:631 - 43; http://dx.doi.org/10.1038/nrm2002; PMID: 16912714
- Naydenov NG, et al. Cell Cycle 2012; 11; PMID: 23187805
- Takahashi Y, et al. Autophagy 2011; 7:61 - 73; http://dx.doi.org/10.4161/auto.7.1.14015; PMID: 21068542
- Mizushima N, et al. Annu Rev Cell Dev Biol 2011; 27:107 - 32; http://dx.doi.org/10.1146/annurev-cellbio-092910-154005; PMID: 21801009
- Li L, et al. J Biol Chem 2010; 285:19705 - 9; http://dx.doi.org/10.1074/jbc.C110.102483; PMID: 20457610
- Razi M, et al. J Cell Biol 2009; 185:305 - 21; http://dx.doi.org/10.1083/jcb.200810098; PMID: 19364919