Abstract
The therapeutic potential of pharmacologic inhibition of bromodomain and extraterminal (BET) proteins has recently emerged in hematological malignancies and chronic inflammation. We find that BET inhibitor compounds (JQ1, I-Bet, I-Bet151 and MS417) reactivate HIV from latency. This is evident in polyclonal Jurkat cell populations containing latent infectious HIV, as well as in a primary T-cell model of HIV latency. Importantly, we show that this activation is dependent on the positive transcription elongation factor p-TEFb but independent from the viral Tat protein, arguing against the possibility that removal of the BET protein BRD4, which functions as a cellular competitor for Tat, serves as a primary mechanism for BET inhibitor action. Instead, we find that the related BET protein, BRD2, enforces HIV latency in the absence of Tat, pointing to a new target for BET inhibitor treatment in HIV infection. In shRNA-mediated knockdown experiments, knockdown of BRD2 activates HIV transcription to the same extent as JQ1 treatment, while a lesser effect is observed with BRD4. In single-cell time-lapse fluorescence microscopy, quantitative analyses across ~2,000 viral integration sites confirm the Tat-independent effect of JQ1 and point to positive effects of JQ1 on transcription elongation, while delaying re-initiation of the polymerase complex at the viral promoter. Collectively, our results identify BRD2 as a new Tat-independent suppressor of HIV transcription in latently infected cells and underscore the therapeutic potential of BET inhibitors in the reversal of HIV latency.
Introduction
The main obstacle to eradicating HIV from patients is post-integration latency.Citation1 Antiretroviral treatments target only actively replicating virus, while latent infections that have low or no transcriptional activity remain untreated.Citation2 A combination of antiretroviral treatments with latency-purging strategies may accelerate the depletion of latent reservoirs and lead to a cure.Citation3 Current strategies to reactivate HIV from latency include use of prostratin, a non-tumor-promoting phorbol ester,Citation4 disulfiramCitation5 and histone deacetylase (HDAC) inhibitors, such as suberoylanilidehydroxamic acid (i.e., SAHA or Vorinostat).Citation6-Citation9 As the mechanisms of HIV latency are diverse, effective reactivation may require combinatorial strategies.Citation10
Recently, the therapeutic potential of pharmacologic inhibition of members of the bromodomain and extraterminal (BET) family of human bromodomain proteins has been described.Citation11-Citation13 The BET protein family is a well-conserved class of transcriptional regulators that are distinguished by the presence of tandem bromodomains, conserved domains that recognize and bind acetyl-lysine residues, and a so-called extraterminal (ET) domain.Citation14 The best-studied BET family member is BRD4, which contains a third functional domain, termed the positive transcription elongation factor b (P-TEFb)-interacting domain (PID), that binds the HIV cofactor P-TEFb.Citation15,Citation16 The recently reported specific BET bromodomain inhibitors (JQ1, I-BET) identified these proteins, most notably BRD4, as functionally relevant to the pathogenesis of hematological malignancies, chronic inflammation and possibly viral infections.Citation11-Citation13
The HIV promoter after integration into host chromatin is uniquely dependent on the activity of P-TEFb.Citation17 In its active form, P-TEFb comprises cyclin T1 and the serine/threonine kinase CDK9,Citation18,Citation18 which phosphorylates the negative transcription elongation factors NELFCitation19 and DSIFCitation20,Citation21 and the C-terminal domain (CTD) of the cellular RNA polymerase II complex,Citation22 a process that greatly enhances elongation of HIV transcripts. P-TEFb is recruited to the HIV promoter by the viral transactivator Tat, which cooperatively interacts with the cyclin T1 and CDK9 components of P-TEFb and an RNA stem loop structure called TAR that forms spontaneously at the 5′ ends of HIV transcripts.Citation17,Citation23 In the absence of Tat, P-TEFb interacts with the BET-containing protein BRD4 and is recruited to the HIV LTR via interactions of the BRD4 bromodomains and acetylated histones.Citation15,Citation16 BRD4 and recently described MLL-fusion partners are part of active P-TEFb complexes,Citation24 while inactive P-TEFb is stored in cells in complex with 7SK snRNA and the P-TEFb inhibitor Hexim1.Citation25,Citation26 Notably, the interaction of Tat or BRD4 with P-TEFb is mutually exclusive, as the PID in BRD4 interacts with the same region in P-TEFb as Tat.Citation27 Interestingly, the BRD4 locus was previously identified as a hotspot of integration for latent HIV in cell lines, indicating that manipulating BRD4 expression or function may cause or reverse latency.Citation27,Citation28
Tat and P-TEFb are the subjects of acetylationCitation29-Citation32 and engage in bromodomain-dependent interactions. Tat acetylated at lysine 50 interacts with the bromodomain of the histone acetyltransferase PCAF/KAT2B, a process that terminates the interaction of Tat with P-TEFb and TAR RNA and recruits the Tat/PCAF complex to the elongating polymerase complex at the HIV LTR.Citation33-Citation36 In addition, cyclin T1 is acetylated at four distinct lysine residues in its predicted coil-coil domain, and three of these lysines (K380, K386, K390) interact with the second bromodomain of BRD4, generating a second modification-specific interaction domain besides the PID.Citation37 While this acetylation-dependent interaction is relevant for P-TEFb function at the HIV LTR and on cellular genes, it is not required for Tat activity, supporting the model that Tat recruits P-TEFb in the absence of BRD4 potentially directly from inactive P-TEFb storage complexes.
Here, we show that BET inhibitors JQ1,Citation12 I-BET,Citation11 I-BET151Citation13 and MS417Citation38 effectively reactivate HIV from latency in cultured cells and primary T-cell models of latency. While this is expected given the restrictive function of BRD4 on Tat transcriptional activity, we show that this process is independent from Tat and occurs with the same efficiency in cells lacking Tat. Furthermore, our data identify another BET protein, BRD2 as a new Tat-independent suppressor of HIV transcription in latent cells. Our results, together with recently published reports from colleagues showing reactivation of HIV from latency after treatment with JQ1,Citation39-Citation43 indicate that targeting bromodomain interactions at the HIV promoter may be a promising strategy to complement the existing repertoire of latency-purging compounds and to develop an efficient “anti-latency” cocktail.
Results
JQ1 activates HIV transcription in a Tat-independent manner
As BRD4 competes with Tat for P-TEFb binding,Citation27 we speculated that treatment with BET inhibitors may activate Tat transcriptional activity and reactivate HIV from latency. To test this hypothesis, we treated a polyclonal population of Jurkat T cells containing latent HIV (clone R7/E-/GFP)Citation44 with increasing amounts of JQ1. This viral clone contains a frame shift mutation in the viral Env gene to prevent viral spread and expresses GFP in the Nef open reading frame, which allows separation of actively infected GFP+ from GFP− cells by cell sorting.Citation44 GFP− cells, which are mostly uninfected but contain a small fraction of latently infected cells with silenced HIV transcription, were treated with JQ1. Activation of transcription was measured by flow cytometry of GFP. JQ1, but not the stereoisomer control (R)-JQ1, reactivated HIV-1 in a dose-dependent manner (). Stimulation of cells with JQ1 produced up to 5-fold more GFP-expressing cells than control-treated cells. Similar results were obtained with another viral clone (NL4-3/E-/GFP-IRES-nef), which also expresses GFP in the Nef position and also has Nef expressed under the control of an IRES elementCitation45 ().
Figure 1. JQ1 activates latent HIV. HIV clones R7/E-/GFP and NL4–3/E-/GFP-IRES-nef were derived from pR7-GFP and pNLENG1-EGFP by mutating the Env gene by inserting an early stop codon in the NdeI site. Viral stocks were produced and VSV-G-pseudotyped in 293T cells and titered for p24. Jurkat cells were spininfected with 25 ng of p24 per 106 cells, and GFP− cells were collected in two rounds of cell sorting 5 and 15 d after infection. The expanded population of GFP– cells, composed of uninfected and latently infected cells, were seeded in 96-well plates and treated with the indicated concentration of drugs in duplicates. The percentage of GFP+ cells was detected after 24 h at the MACSQuant VYB FACS analyzer. Data are expressed as the mean percentage of GFP+ cells, subtracting the average percentage of spontaneous GFP-reactivation in the untreated samples. (A) Jurkat cells containing latent R7/E-/GFP virus were treated with JQ1 or (R)-JQ1 in combination with Prostratin (0.5 μM), SAHA (2.5 μM), TNFα (1 ng/μl) or control at the indicated concentrations for 18 h, followed by flow cytometry analysis. Alone or in combination with Prostratin or TNFα, the BET inhibitor JQ1, but not the stereoisomer control (R)-JQ1, reactivated HIV-1 in a dose-dependent manner. Similar results were seen in Jurkat cells containing latent NL4–3/E-/GFP-IRES -nef (B). Results represent average of two independent experiments.
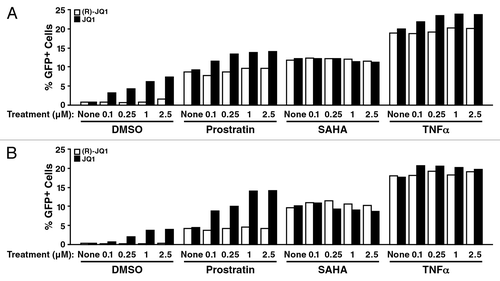
Next, we tested JQ1 reactivation in combination with HDAC inhibitor suberoylanilidehydroxamic acid (SAHA), the protein kinase C (PKC) activator prostratin or the proinflammatory cytokine TNFα. We observed enhanced activation when JQ1 was added with prostratin, while no additive or synergistic effects were observed with SAHA (). Co-treatment with TNFα led to a very modest enhancement of the JQ1 effect in this system ().
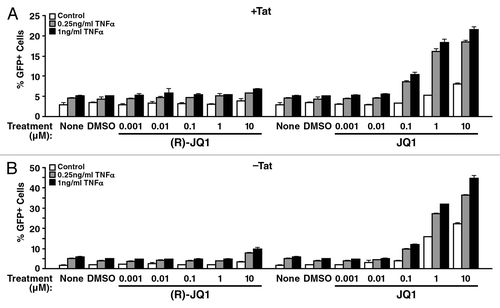
To determine if BET inhibition specifically activates Tat-dependent transcription, we utilized a J-Lat cell line harboring a latent lentiviral construct expressing Tat with GFP from the HIV LTR (clone A2; LTR-Tat-IRES-GFP).Citation44 Treatment with JQ1, but not inactive (R)-JQ1, activated HIV transcription in a dose-dependent manner, as measured by flow cytometry of GFP (). Stimulation with JQ1 yielded up to 9-fold more GFP-expressing cells than control-treated cells, and a 22-fold increase was observed when cells were co-treated with JQ1 and low doses of TNFα (). However, this effect was not specific for Tat: we observed the same effect in A72 cells containing a latent LTR-GFP construct lacking Tat.Citation44 Here, an up to 22-fold increase in GFP+ cells resulted from JQ1 treatment alone, and a 45-fold increase resulted when TNFα was added with JQ1 (). We also treated both cell lines with prostratin and SAHA (Fig. S1). As observed with the polyclonal cell populations, adding prostratin to JQ1 enhanced the JQ1 effect, while only a very modest increase was observed with SAHA, indicating that SAHA and JQ1 target a similar cellular pathway. Collectively, these results establish the effectiveness of JQ1 to reverse HIV latency in a Tat-independent manner.
Figure 2. The JQ1 effect is Tat-independent. Two latent J-Lat cell lines A2 (containing a LTR-Tat-IRES-GFP construct) or A72 (containing a LTR-GFP construct) were treated with JQ1 or (R)-JQ1 in combination with TNFα or control at the indicated concentrations for 18 h, followed by flow cytometry analysis. (A) In A2 cells JQ1, but not the control (R)-JQ1, reactivated HIV-1 in a dose-dependent manner. Similar results were seen in the Tat-deficient A72 Jurkat cell line (B). Data represent average (± SD) of three independent experiments.
Activating potential of known BET inhibitors in cell lines and a primary T-cell model of HIV latency
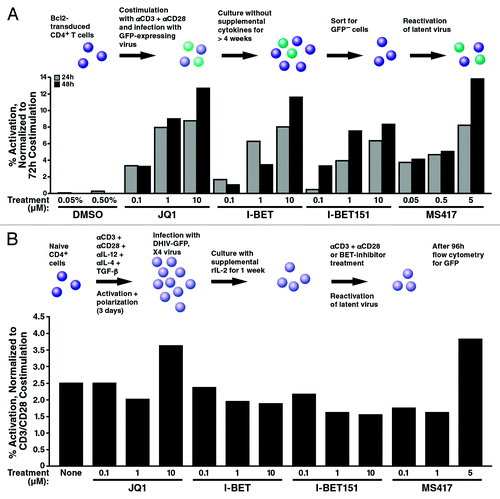
The activating effect was not unique to JQ1 but was also observed with I-Bet151Citation13 and MS417,Citation38 two recently reported small-molecule bromodomain inhibitors with similar binding affinities to BET proteins (). Both compounds effectively activated HIV from latency in A2 and A72 cell lines, underscoring the notion that the BET inhibitor effect on HIV latency is independent of Tat. We also tested these compounds in a primary T-cell model of latency.Citation46-Citation48 In this model, Bcl-2-transduced CD4+ T cells were infected in a single-round infection with HIV clone NL4-3-Δnef-Δpol-EGFPCitation46 to generate robust latent infection in vitro (). To reactivate latent HIV-1, cells were treated with the indicated compounds or a combination of αCD3 and αCD28 antibodies as a control for maximal activation. JQ1 reactivated latent HIV-1 at ~14% of the rate achieved by co-stimulation with αCD3 and αCD28 antibodies (). The same activation was observed in cells activated with I-Bet, I-Bet151 and MS417, supporting the model that BET inhibition has the potential to reverse latency in primary T cells. However, when we analyzed a second primary T-cell model of latency using ex vivo differentiated nonpolarized CD4+ T cells,Citation49 we were unable to significantly reverse latency with any of the BET inhibitor compounds, with only a minor activatory effect observed with the highest doses of JQ1 and MS417 (). Notably, current primary T-cell models of latency are diverse, and it is unknown which one faithfully reproduces the in vivo situation of latently infected cells. Interestingly, the nonpolarized T helper cell model of HIV latency is also resistant to reactivation by SAHA,Citation50 underlining that BET inhibitors and SAHA may target common mechanistic pathways.
Figure 3. Reactivation of latent HIV with other bromodomain-targeting compounds. J-Lat cell lines A2 (A) and A72 (B) were treated with JQ1 or two other bromodomain-targeting compounds, I-BET151 and MS417, at the indicated concentrations for 18 h and analyzed by flow cytometry. As indicated, in both A2 and A72 cells, stimulation with all three compounds increased GFP expression. Data represent average (± SD) of three independent experiments.
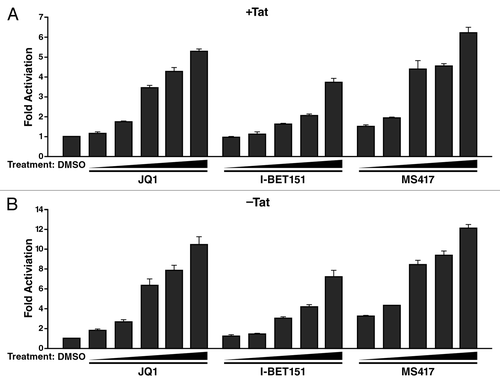
Figure 4. Effect of bromodomain-targeting compounds in primary T-cell models of HIV latency. (A) Detection of latently infected cells in sorted GFP-negative Bcl-2-transduced cells. The sorted GFP-Bcl-2-transduced resting CD4+ T cells were treated with stimuli for 24–72 h at 37 þC. Cells treated with 2.5 µg/ml αCD3 and 1 µg/ml αCD28 antibodies were used as positive controls. Reactivation of latent HIV-1 was determined by quantifying % GFP+ cells with a MACSQuant flow cytometer (Milteny Biotech GmbH). Results are expressed as percentage of reactivation in response to αCD3 plus αCD28 activation and represent the average of two independent donors. (B) Effect of bromodomain-targeting compounds in latently infected primary nonpolarized T helper cells. Latently infected T cells were generated using healthy, uninfected CD4+ T cells (DONOR 144) that were ex vivo differentiated into nonpolarized T cells and infected with DHIV-GFP, X4 virus as previously described.Citation49,Citation50 Reactivation was monitored by analysis of GFP by flow cytometry 96 h after compound addition. Beads coated with αCD3/αCD28 antibodies were used as positive controls. All compounds were also tested in non-infected cells to distinguish between HIV-1 reactivation and compound fluorescence. Similar results were observed in two independent donors.
Involvement of P-TEFb in the JQ1 effect on HIV latency
To biophysically characterize the Tat-independent JQ1 effect on the HIV LTR, single-cell time-lapse fluorescence microscopy was performed on ~2,000 Jurkat cells, where each cell carried a different integration site of the LTR promoter driving a destabilized GFP reporter (). Mean fluorescence intensity (MFI) and the magnitude of intensity fluctuations (i.e., the “noise”) were quantifiedCitation51 for sub-clusters of polyclonal cells in response to JQ1 treatment (). The data was analyzed using an established two-state model of episodic transcription,Citation52-Citation54 in which the LTR switches between a transcriptional OFF state, where RNA polymerase II is stalled, and a transcriptional ON state, where elongation occurs and multiple mRNA transcripts are produced at a rate = T (). In this model, promoter switching occurs with rates kon and koff, which generates pulses or bursts of transcription.Citation51,Citation54 Previous measurements show that the LTR typically exhibits koff > > kon, such that large bursts in expression are punctuated by long dwell times in the OFF state.
Figure 5. JQ1 enhances transcription burst size. (A and B) Depiction of the experimental model with polyclonal LTR-GFP-containing Jurkat cells. (C and D) Cells treated with JQ1 can change the mean expression level of GFP (hypothesis 1), the variability (or coefficient of variation, CV, defined as the standard deviation over the mean; hypothesis 2) or both, compared with the untreated basal expression state. (E) Genome-wide signatures of JQ1 exposure by time-lapse microscopy. Over 2,000 cells were accounted for and imaged for durations of 12–18 h. JQ1 displays a similar abundance range with elevated noise magnitude compared with the untreated cell population. (F) Histogram/bar representation of the quantified shifts in burst size (or # of mRNA per pulse, T/koff) and the average dwell time in the OFF state (of 1/kon) with and without JQ1 treatment. JQ1 increases burst size and 1/kon.
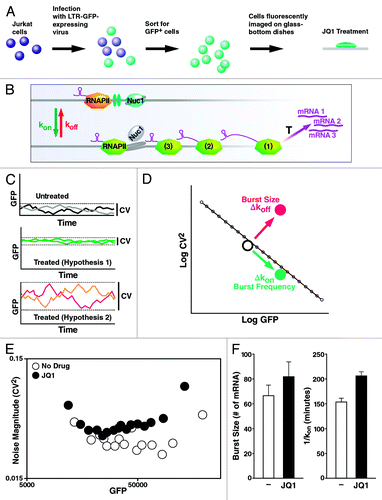
To determine how JQ1 influences these biophysical parameters (koff, kon, T), we examined changes in LTR MFI and noise (measured by the coefficient of variation, CV, which is defined as the standard deviation over the mean), compared with the untreated LTR (). By qualifying MFI and CV in time-lapse trajectories, changes in both burst frequency (kon) and burst size or the number of mRNA produced per activity pulse (T/koff) can be determined.
The results show that across thousands of integration sites, JQ1 treatment increases LTR noise without a significant shift in MFI (). It is important to note that the lack of change in MFI change is only relative to cells already expressing high levels of GFP. Quantitative analysis of the increase in variability shows a JQ1-specific enhancement in transcription burst size (T/koff) that occurs in parallel with an increase in the average dwell time in the OFF state (1/kon) (). On average, these JQ1-induced changes are equivalent to ~50 min delays in transcriptional initiation coupled to a concomitant increase of ~15 mRNAs per pulse of transcriptional activity. These results indicate that JQ1 enhances transcription elongation from the LTR, in the absence of Tat, while delaying re-initiation of the polymerase complex.
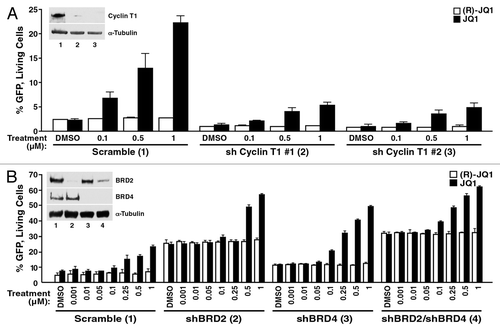
As transcription elongation at the HIV promoter uniquely depends on P-TEFb, we determined if the JQ1 effect in latent cells requires intact P-TEFb. We introduced short hairpin RNAs (shRNAs) directed against cyclin T1, an important component of P-TEFb, into A72 cells with lentiviral vectors. Knockdown of cyclin T1 yielded lower basal levels in GFP+ cells than control cells expressing non-targeting shRNAs (). Importantly, cyclin T1 knockdown also decreased the ability of the cells to respond to JQ1 treatment. Only half as many cells responded with GFP expression when JQ1 was added than in control cells (). Similar results were observed with two independent cyclin T1-targeting shRNAs, confirming that P-TEFb is involved in the reactivating effect of JQ1 in the absence of Tat.
Figure 6. The JQ1 effect in A72 cells is dependent on P-TEFb and BRD2. (A) A72 cells were infected with virus containing shRNA constructs targeting cyclin T1 or a non-targeting control. Knockdown of cyclin T1 protein levels are shown by immunoblotting with cyclin T1 or the control α-tubulin antibody. At 4 d after infection, cells were treated with JQ1 or DMSO at the indicated concentrations for 18 h and analyzed by flow cytometry. As indicated, knockdown of cyclin T1 resulted in a decrease in GFP expression under basal condition and in JQ1-treated cells. Average (± SD) of three experiments is shown. (B) A72 cells were infected with virus containing shRNA constructs targeting BRD2, BRD4 or a non-targeting control. At 4 d after infection, cells were treated with JQ1 or DMSO at the indicated concentrations for 18 h and examined by flow cytometry. As indicated, knockdown of BRD2 and BRD4 resulted in an increase in GFP expression. JQ1 treatment enhanced this effect. Results represent average (± SD) of three experiments. Knockdown of BRD2 and BRD4 protein levels were confirmed by immunoblotting with BRD2 and BRD4 antibodies or the control α-tubulin.
BRD2 suppresses HIV transcription in the absence of Tat
To test the functional relevance of BET proteins in HIV latency, we performed lentiviral shRNA knockdown studies of endogenous BRD2 and BRD4 proteins in A72 cells lacking Tat. BRD2 is a close relative of BRD4, but lacks a C-terminal PID domain. However, in the nuclear complexosome identified by Malovannaya et al., BRD2 was found to co-immunoprecipitate with CDK9/cyclin T1 or cyclin T2.Citation55 BRD2 binds JQ1 and other BET inhibitors, albeit with lower binding affinities than BRD4.Citation12 Knockdown of BRD2 resulted in a robust activation of the HIV LTR, and this effect was only slightly enhanced in response to JQ1 (2-fold as compared with 3.3-fold activation in control cells) (). BRD4 knockdown also resulted in spontaneous activation of the HIV LTR, albeit to a lesser extent than BRD2 (2.4-fold vs. 5.4-fold), and the response to JQ1 was not affected (). No significant additive or synergistic effects were observed when both factors were knocked down together, indicating that the two factors work in the same biological pathway (). Similar results were obtained when different sets of shRNAs against BRD2 and BRD4 were used (Fig. S2). These results identify BRD2 as a new factor involved in HIV transcription in latent cells.
Discussion
In this study, we demonstrate that JQ1 and other recently published bromodomain inhibitors partially reverse HIV latency. While this manuscript was in preparation, similar results were published by Banerjee et al., Bartholomeeusen et al., Li et al. and Zhu et al.Citation39,Citation41-Citation43 Banerjee et al. show that JQ1 reactivates latent HIV in Ach2 and U1 cell lines, two clonal cell lines carrying HIV proviruses defective in the Tat/TAR axis of HIV transcriptionCitation56-Citation58 and in J-Lat clone 10.6. This clone carries full-length replication-competent HIV without inactivating mutations.Citation44 They also show that JQ1 activates HIV-1 infection in acutely infected primary CD4+ T cells and observe an expression profile of upregulated chromatin modification genes and genes associated with HIV transcription, including cyclin T1, CDK9 and MLL fusion partners, all part of active P-TEFb complexes, in cells treated with JQ1. Bartholomeeusen et al. demonstrate the JQ1 effect in J∆K cells, a Jurkat clonal cell line carrying HIV that lacks both NFκB binding sites.Citation58 They further show that JQ1 treatment releases P-TEFb from inactive Hexim1-containing cellular complexes, a process that transiently promotes assembly of P-TEFb with MLL fusion partners in the so-called superelongation complex SEC.Citation41 Most recently, Li et al. and Zhu et al. identified BRD4 as a negative regulator of HIV-1 replication.Citation42,Citation43 Li et al. showed that JQ1 activates latent HIV in J-Lat A2 cells but, unlike us, found that this effect is Tat-dependent in Jurkat 1G5 and HeLa-based NH1 and NH2 cells.Citation42 Zhu et al. report modest stimulation of an HIV-LTR reporter by JQ1 in the absence of Tat, but JQ1 in combination with Tat resulted in more cells reactivating latent viruses and with greater magnitude.Citation43
We partially confirm and extend these studies by showing that the BET inhibitor effect is not confined to clonal cell lines, but is also observed in polyclonal Jurkat cells infected with latent HIV proviruses. Similar to Banerjee et al.Citation39 and Zhu et al.,Citation43 we also found activating effects of JQ1 in a primary T-cell model of latency, underscoring the therapeutic potential of JQ1 in primary T cells. However, a latency-purging effect of BET inhibitors was not observed in non-polarized primary T cells as surrogates for central memory T cells, a major latency reservoir in patients.Citation50 As it is unclear whether any model faithfully represents the in vivo situation of latently infected cells, further studies are needed to evaluate the clinical potential of BET inhibitors in primary T cells. However, a careful comparison of the two cell models used in our studies may yield important mechanistic insights into the mechanism of action of BET inhibitors in the future. Notably, non-polarized cells are not reactivated in response to SAHA, which shows mild or no synergy with JQ1 in our system. This observation indicates that JQ1, like SAHA, may target a pathway in HIV reactivation that is not active in non-polarized cells. A cooperative action of JQ1 and SAHA in the reactivation of HIV in J∆K cells was noted in the study of Bartholomeeusen et al.Citation41
In contrast to Li et al.,Citation42 we find robust effects of BET inhibitors on HIV gene expression in the absence of Tat. Our hypothesis at the onset of these studies was that BET inhibitors remove the restrictive function of BRD4 from Tat. However, when we tested the A72 cell line, it became rapidly clear that this cannot be the only mode of JQ1 function in HIV latency. We observe strong Tat-independent effects on HIV gene expression in latent cell lines lacking Tat (A72), but also in Jurkat cell lines harboring non-latent HIV LTR driving luciferase expression (IG5) (data not shown). These findings are supported by our biophysical studies in cells harboring actively transcribing LTR-GFP lentiviral vectors. Treatment of these cells with JQ1 induced marked changes in noise amplitudes of GFP expression, consistent with an increase in transcription burst length induced by JQ1 in the absence of Tat. Why we observe a concomitant decrease in transcription initiation rates in response to JQ1 in this model is unclear at this point but will be further investigated in the future.
Our study, for the first time, establishes a role of BRD2 in HIV latency (). Previously, BRD2 was shown to bind to acetylated lysine residues of histone H4, where it recruits transcription factors and regulates transcription.Citation59-Citation61 BRD2 couples histone acetylation to transcriptionCitation61,Citation62 by providing a scaffold on chromatin to recruit E2F transcription factors,Citation60,Citation63 a lysine-specific demethylase,Citation64 TBP,Citation63 HDACs,Citation59 histone H4-specific acetyltransferase (HAT)Citation62 and proteins involved in chromatin remodeling.Citation65-Citation67 Given that chromatin-remodeling components and HDACs are crucial in the maintenance and reversal of HIV latency, we hypothesize that BRD2 recruits histone modification enzymes, transcriptional activator complexes and chromatin-remodeling factors to the HIV LTR promoter, thereby activating transcription.Citation68
Figure 7. Model: BET proteins restrict HIV transcription in the absence of Tat. JQ1 removes the inhibiting function of BRD2 and BRD4 proteins from latent HIV, a process that may allow both factors to turn into activators of HIV transcription in conjunction with P-TEFb. See text for details.
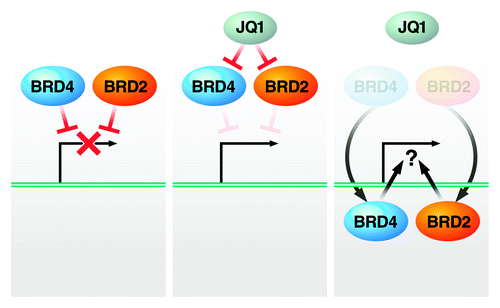
While this may explain how BRD2 could turn into an activator and enhance HIV transcription in response to JQ1, it is unclear how it acts as an inhibitory factor without the drug (). Future studies will address whether BRD2 exerts suppressive functions directly by associating with the HIV LTR or indirectly by affecting gene expression of transcriptional repressors or co-repressor. The fact that BRD2 itself binds HDACs supports a model where BRD2, by recruiting repressor complexes to the latent HIV LTR, could directly suppress HIV transcription. On the other side, the finding by Banerjee et al. that JQ1 treatment reprograms expression of chromatin modification genesCitation39 supports a model in which JQ1 activates HIV transcription by reversing a BRD2-mediated suppressive gene expression profile. Future studies will address this issue and the question whether BRD2 regulates HIV transcription via P-TEFb.
Materials and Methods
Materials
HEK293T and Jurkat cells were obtained from the American Type Culture Collection. J-Lat (clones A2 and A72) cell lines were described.Citation44 HEK293T cells were cultured in DMEM supplemented with 10% FBS, 1% L-glutamine and 1% penicillin-streptomycin (Life Technologies). Jurkat and J-Lat cells were cultured in RPMI supplemented with 10% FBS, 1% L-glutamine and 1% penicillin-streptomycin. TNFα (Sigma-Aldrich) was used at concentrations of 0.25 or 1 ng/ml. Human αCD3 and αCD28 (BD Biosciences) were used at concentrations of 3 and 1 µg/ml, respectively. Prostratin (Sigma) was used at a suboptimal concentration of 0.5 µM, and SAHA (NCI Chemical Carcinogen Repository, Midwest Research Institute) was used at 2.5 µM.
Primary T-cell model of HIV latency in Bcl2-transduced T cells
Primary CD4+ T cells from healthy donors were isolated to generate HIV-1 latent infection in vitro as described.Citation47 In brief, primary CD4+ T cells were co-stimulated with αCD3 and αCD28 antibodies and then transduced with a Bcl-2-expressing lentiviral vector to allow long-term culture. After 3 wk of culture in absence of supplemental cytokines, Bcl-2-transduced primary CD4+ T cells return to a quiescent state.Citation47 Bcl-2-transduced cells were then co-stimulated and infected with NL4-3-Δ6-drEGFP virus. The infected cells were cultured for several weeks without supplemental cytokines to allow establishment of latency in surviving cells.Citation47 Flow cytometric cell sorting was used to remove residual GFP+ cells. This approach produces cultures, in which 1–3% of the cells are latently infected, with the remaining cells (> 97%) being uninfected.Citation46,Citation47 After cell sorting, the purified GFP− Bcl-2-transduced resting CD4+ T cells, including latently infected cells, were plated at 5 × 104 cells/well, in 200 µL of RPMI 1640 + 10% FBS in U-bottomed 96-well plates and treated with the indicated compounds for 24–72 h at 37 þC. Cells treated with 2.5 µg/ml αCD3 plus 1 µg/ml αCD28 antibodies were used as positive controls. At the indicated times, the fraction of GFP+CD4+ T cells was measured by FACS.
ShRNA-mediated knockdown experiments and flow cytometry analysis
ShRNA-expressing lentiviral vectors were purchased from Open Biosystems. The plasmids TRCN0000006308 and TRCN0000006310 were used to deplete BRD2; plasmids TRCN0000021424 and TRCN0000021428 were used to deplete BRD4; and plasmids TRCN0000013673 and TRCN0000013675 were used to deplete cyclin T1. The pLKO.1 vector containing scramble shRNA was used as control. Pseudotyped viral stocks were produced in 2 × 106 HEK293T cells by the calcium phosphate method by co-transfection 10 µg of shRNA-expressing lentiviral vectors, together with 6.5 µg of the lentiviral packaging construct pCMVdelta R8.91Citation69 and 3.5 µg of VSV-G glycoprotein-expressing vector,Citation69 and titered for p24 content. J-Lat A72 cells (containing a LTR-GFP construct) were spininfected with virus (1 ng of p24 per 106 cells) containing shRNAs against BRD2, BRD4, cyclin T1 or non-targeting control shRNAs and were selected with puromycin (2 µg/ml; Sigma). After 4 d of selection, cells were treated with the indicated concentration of drugs. The percentage of GFP+ cells was determined after 18 h using a MACSQuant VYB FACS analyzer (Miltenyi Biotech GmbH). Cell viability was monitored by forward and side scatter analysis. Analysis was conducted on 3 × 10,000 live cells per condition, and all experiments were independently repeated at least three times. Data were analyzed using FlowJo 9.4 (Tree Star).
Single-cell analysis of JQ1-treated cells
Lentiviral vectors expressing the LTR-GFP cassette in the absence of Tat were describedCitation70 and used to infect 5 × 105 Jurkat cells at a multiplicity of infection < 0.1, resulting in 25,000–50,000 infected cells each presumably with a unique integration site. Cells were then sorted by FACS to isolate green fluorescent protein-labeled (GFP+) cells and fluorescently imaged on glass-bottom dishes in RPMI 1640 with 10% fetal calf serum and 1% penicillin-streptomycin and 1μM JQ1 at t = 0 h for the treated population. The imaging took place in humidified conditions at 37°C and 5% CO2 for 12–24 h with a 40X (1.2 NA) oil-immersion objective on a Zeiss Observer Z1 microscope equipped with an automated linear-encoded X-Y stage, as described.Citation71,Citation72 Image processing and cell tracking were performed in Matlab™ with an in-house algorithm,Citation51,Citation72 and a single 12-h experiment could generate up to 1,000 single-cell trajectories for analysis.
For each trajectory, noise autocorrelation [Φ(t)] and magnitude (CV2) were calculated using an established noise-processing algorithm.Citation51,Citation72,Citation73 A published theoryCitation53,Citation74 of the two-state transcriptional bursting model yields analytical expressions for the autocorrelation of the noise, Φ(τ), and noise magnitude. Derivations and calculated burst size and frequency have been reported.Citation51,Citation54 Exogenous JQ1 addition can change either mean fluorescence, variability of expression (defined by the coefficient of variation, CV) or both. Modeling of two-state transcription enables the differentiation between modulations in transcriptional initiation (∆kon) or in the burst size (∆T/koff).
| Abbreviations: | ||
| BRD2 | = | bromodomain 2 |
| BRD4 | = | bromodomain 4 |
| CTD | = | carboxyl-terminal domain |
| GFP | = | green fluorescent protein |
| HATs | = | histone acetyltransferases |
| HDACs | = | histone deacetylases |
| HIV | = | human immunodeficiency virus |
| LTR | = | long terminal repeat |
| PBS | = | phosphate-buffered saline |
| P-TEFb | = | positive transcription elongation factor b |
| SAHA | = | suberoylanilidehydroxamic acid |
| RNAPII | = | RNA polymerase II |
| SEC | = | super elongation complex |
| TNFα | = | tumor necrosis factor alpha |
Additional material
Download Zip (279.6 KB)Acknowledgments
We gratefully acknowledge support from the NIH (R01 AI083139 to M.O.). M.O., E.V., V.P., R.F.S. and L.W. are members of the CARE Collaboratory (U19AI096113). We thank Ryan Conrad, Hendrik Sy and members of the Ott, Verdin, Weinberger and Greene laboratories for sharing their reagents and for helpful discussions. We also thank John Carroll and Teresa Roberts for assistance with graphics. We thank Gary Howard and Anna Lisa Lucido for editorial and Veronica Fonseca for administrative assistance.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authorship Contributions
D.B. and R.D. designed and performed research, analyzed data and wrote the paper; V.C., P.C.L., S.X., K.A. and L.M. designed and performed research; S.S. analyzed data; M.M.Z. and J.E.B. contributed new reagents; V.P. and R.F.S. designed research and analyzed data; L.W., E.V. and M.O. designed research, analyzed data and wrote the paper.
Related Research Data
References
- Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med 1999; 5:512 - 7; http://dx.doi.org/10.1038/8394; PMID: 10229227
- Sedaghat AR, Siliciano JD, Brennan TP, Wilke CO, Siliciano RF. Limits on replenishment of the resting CD4+ T cell reservoir for HIV in patients on HAART. PLoS Pathog 2007; 3:e122; http://dx.doi.org/10.1371/journal.ppat.0030122; PMID: 17784786
- Geeraert L, Kraus G, Pomerantz RJ. Hide-and-seek: the challenge of viral persistence in HIV-1 infection. Annu Rev Med 2008; 59:487 - 501; http://dx.doi.org/10.1146/annurev.med.59.062806.123001; PMID: 17845138
- Williams SA, Chen LF, Kwon H, Fenard D, Bisgrove D, Verdin E, et al. Prostratin antagonizes HIV latency by activating NF-kappaB. J Biol Chem 2004; 279:42008 - 17; http://dx.doi.org/10.1074/jbc.M402124200; PMID: 15284245
- Xing S, Bullen CK, Shroff NS, Shan L, Yang HC, Manucci JL, et al. Disulfiram reactivates latent HIV-1 in a Bcl-2-transduced primary CD4+ T cell model without inducing global T cell activation. J Virol 2011; 85:6060 - 4; http://dx.doi.org/10.1128/JVI.02033-10; PMID: 21471244
- Archin NM, Espeseth A, Parker D, Cheema M, Hazuda D, Margolis DM. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res Hum Retroviruses 2009; 25:207 - 12; http://dx.doi.org/10.1089/aid.2008.0191; PMID: 19239360
- Contreras X, Schweneker M, Chen CS, McCune JM, Deeks SG, Martin J, et al. Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J Biol Chem 2009; 284:6782 - 9; http://dx.doi.org/10.1074/jbc.M807898200; PMID: 19136668
- Edelstein LC, Micheva-Viteva S, Phelan BD, Dougherty JP. Short communication: activation of latent HIV type 1 gene expression by suberoylanilide hydroxamic acid (SAHA), an HDAC inhibitor approved for use to treat cutaneous T cell lymphoma. AIDS Res Hum Retroviruses 2009; 25:883 - 7; http://dx.doi.org/10.1089/aid.2008.0294; PMID: 19689202
- Van Lint C, Emiliani S, Ott M, Verdin E. Transcriptional activation of the HIV-1 promoter in response to histone acetylation.. EMBO J 1996; 15:1112 - 20; PMID: 8605881
- Quivy V, Adam E, Collette Y, Demonte D, Chariot A, Vanhulle C, et al. Synergistic activation of human immunodeficiency virus type 1 promoter activity by NF-kappaB and inhibitors of deacetylases: potential perspectives for the development of therapeutic strategies. J Virol 2002; 76:11091 - 103; http://dx.doi.org/10.1128/JVI.76.21.11091-11103.2002; PMID: 12368351
- Nicodeme E, Jeffery KI, Schaefer U, Beinke S, Dewell S, Chung CW, et al. Suppression of inflammation by a synthetic histone mimic.. Nature 2010; 468:1119 - 23; http://dx.doi.org/10.1038/nature09589; PMID: 21068722
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature 2010; 468:1067 - 73; http://dx.doi.org/10.1038/nature09504; PMID: 20871596
- Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011; 478:529 - 33; http://dx.doi.org/10.1038/nature10509; PMID: 21964340
- Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem 2007; 282:13141 - 5; http://dx.doi.org/10.1074/jbc.R700001200; PMID: 17329240
- Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell 2005; 19:535 - 45; http://dx.doi.org/10.1016/j.molcel.2005.06.029; PMID: 16109377
- Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein BRD4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transactivation.. Mol Cell 2005; 19:523 - 34; PMID: 16109376
- Zhu Y, Pe'ery T, Peng J, Ramanathan Y, Marshall N, Marshall T, et al. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro.. Genes Dev 1997; 11:2622 - 32; http://dx.doi.org/10.1101/gad.11.20.2622; PMID: 9334325
- Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell 2006; 23:297 - 305; http://dx.doi.org/10.1016/j.molcel.2006.06.014; PMID: 16885020
- Yamaguchi Y, Takagi T, Wada T, Yano K, Furuya A, Sugimoto S, et al. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 1999; 97:41 - 51; http://dx.doi.org/10.1016/S0092-8674(00)80713-8; PMID: 10199401
- Wada T, Takagi T, Yamaguchi Y, Ferdous A, Imai T, Hirose S, et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev 1998; 12:343 - 56; http://dx.doi.org/10.1101/gad.12.3.343; PMID: 9450929
- Wada T, Takagi T, Yamaguchi Y, Watanabe D, Handa H. Evidence that P-TEFb alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. EMBO J 1998; 17:7395 - 403; http://dx.doi.org/10.1093/emboj/17.24.7395; PMID: 9857195
- Price DH. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol Cell Biol 2000; 20:2629 - 34; http://dx.doi.org/10.1128/MCB.20.8.2629-2634.2000; PMID: 10733565
- Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 1998; 92:451 - 62; http://dx.doi.org/10.1016/S0092-8674(00)80939-3; PMID: 9491887
- Mueller D, Bach C, Zeisig D, Garcia-Cuellar MP, Monroe S, Sreekumar A, et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 2007; 110:4445 - 4454; http://dx.doi.org/10.1182/blood-2007-05-090514; PMID: 17855633
- Yik JH, Chen R, Jennings JL, Link AJ, Zhou Q. Inhibition of p_TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol Cell 2003; 12:971 - 82; PMID: 14580347
- Michels AA, Fraldi A, Li Q, Adamson TE, Bonnet F, Nguyen VT, et al. Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J 2004; 23:2608 - 19; http://dx.doi.org/10.1038/sj.emboj.7600275; PMID: 15201869
- Bisgrove DA, Mahmoudi T, Henklein P, Verdin E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc Natl Acad Sci USA 2007; 104:13690 - 5; http://dx.doi.org/10.1073/pnas.0705053104; PMID: 17690245
- Bisgrove D, Lewinski M, Bushman F, Verdin E. Molecular mechanisms of HIV-1 proviral latency. Expert Rev Anti Infect Ther 2005; 3:805 - 14; http://dx.doi.org/10.1586/14787210.3.5.805; PMID: 16207172
- Fu J, Yoon HG, Qin J, Wong J. Regulation of P-TEFb Elongation Complex Activity by CDK9 Acetylation. Mol Cell Biol 2007; 27:4641 - 51; http://dx.doi.org/10.1128/MCB.00857-06
- Cho S, Schroeder S, Kaehlcke K, Kwon HS, Pedal A, Herker E, et al. Acetylation of cyclin T1 regulates the equilibrium between active and inactive P-TEFb in cells. EMBO J 2009; 28:1407 - 17; http://dx.doi.org/10.1038/emboj.2009.99; PMID: 19387490
- Ott M, Schnölzer M, Garnica J, Fischle W, Emiliani S, Rackwitz HR, et al. Acetylation of the HIV-1 Tat protein by p300 is important for its transcriptional activity. Curr Biol 1999; 9:1489 - 92; http://dx.doi.org/10.1016/S0960-9822(00)80120-7; PMID: 10607594
- Kiernan RE, Vanhulle C, Schiltz L, Adam E, Xiao H, Maudoux F, et al. HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J 1999; 18:6106 - 18; http://dx.doi.org/10.1093/emboj/18.21.6106; PMID: 10545121
- Dorr A, Kiermer V, Pedal A, Rackwitz HR, Henklein P, Schubert U, et al. Transcriptional synergy between Tat and PCAF is dependent on the binding of acetylated Tat to the PCAF bromodomain. EMBO J 2002; 21:2715 - 23; http://dx.doi.org/10.1093/emboj/21.11.2715; PMID: 12032084
- Mujtaba S, He Y, Zeng L, Farooq A, Carlson JE, Ott M, et al. Structural basis of lysine-acetylated HIV-1 Tat recognition by PCAF bromodomain. Mol Cell 2002; 9:575 - 86; http://dx.doi.org/10.1016/S1097-2765(02)00483-5; PMID: 11931765
- Kaehlcke K, Dorr A, Hetzer-Egger C, Kiermer V, Henklein P, Schnoelzer M, et al. Acetylation of Tat defines a cyclinT1-independent step in HIV transactivation. Mol Cell 2003; 12:167 - 76; http://dx.doi.org/10.1016/S1097-2765(03)00245-4; PMID: 12887902
- Schröder S, Cho S, Zeng L, Zhang Q, Kaehlcke K, Mak L, et al. Two-pronged binding with bromodomain-containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J Biol Chem 2012; 287:1090 - 9; http://dx.doi.org/10.1074/jbc.M111.282855; PMID: 22084242
- Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010; 468:1119 - 23; http://dx.doi.org/10.1038/nature09589; PMID: 21068722
- Zhang G, Liu R, Zhong Y, Plotnikov AN, Zhang W, Zeng L, et al. Down-regulation of NF-κB transcriptional activity in HIV-associated kidney disease by BRD4 inhibition. J Biol Chem 2012; 287:28840 - 51; http://dx.doi.org/10.1074/jbc.M112.359505; PMID: 22645123
- Banerjee C, Archin N, Michaels D, Belkina AC, Denis GV, Bradner J, et al. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J Leukoc Biol 2012; 92:1147 - 54; http://dx.doi.org/10.1189/jlb.0312165; PMID: 22802445
- Ott CJ, Kopp N, Bird L, Paranal RM, Qi J, Bowman T, et al. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood 2012; 120:2843 - 52; http://dx.doi.org/10.1182/blood-2012-02-413021; PMID: 22904298
- Bartholomeeusen K, Xiang Y, Fujinaga K, Peterlin BM. Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. J Biol Chem 2012; 287:36609 - 16; http://dx.doi.org/10.1074/jbc.M112.410746; PMID: 22952229
- Li Z, Guo J, Wu Y, Zhou Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res 2013; 41:277 - 87; http://dx.doi.org/10.1093/nar/gks976; PMID: 23087374
- Zhu J, Gaiha GD, John SP, Pertel T, Chin CR, Gao G, et al. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep 2012; 2:807 - 16; http://dx.doi.org/10.1016/j.celrep.2012.09.008; PMID: 23041316
- Jordan A, Bisgrove D, Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J 2003; 22:1868 - 77; http://dx.doi.org/10.1093/emboj/cdg188; PMID: 12682019
- Kutsch O, Benveniste EN, Shaw GM, Levy DN. Direct and quantitative single-cell analysis of human immunodeficiency virus type 1 reactivation from latency. J Virol 2002; 76:8776 - 86; http://dx.doi.org/10.1128/JVI.76.17.8776-8786.2002; PMID: 12163598
- Xing S, Bhat S, Shroff NS, Zhang H, Lopez JA, Margolick JB, et al. Novel structurally related compounds reactivate latent HIV-1 in a bcl-2-transduced primary CD4+ T cell model without inducing global T cell activation. J Antimicrob Chemother 2012; 67:398 - 403; http://dx.doi.org/10.1093/jac/dkr496; PMID: 22160146
- Yang HC, Xing S, Shan L, O’Connell K, Dinoso J, Shen A, et al. Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J Clin Invest 2009; 119:3473 - 86; PMID: 19805909
- Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012; 36:491 - 501; http://dx.doi.org/10.1016/j.immuni.2012.01.014; PMID: 22406268
- Bosque A, Planelles V. Studies of HIV-1 latency in an ex vivo model that uses primary central memory T cells. Methods 2011; 53:54 - 61; http://dx.doi.org/10.1016/j.ymeth.2010.10.002; PMID: 20970502
- Bosque A, Planelles V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 2009; 113:58 - 65; http://dx.doi.org/10.1182/blood-2008-07-168393; PMID: 18849485
- Dar RD, Razooky BS, Singh A, Trimeloni TV, McCollum JM, Cox CD, et al. Transcriptional burst frequency and burst size are equally modulated across the human genome. Proc Natl Acad Sci USA 2012; 109:17454 - 9; http://dx.doi.org/10.1073/pnas.1213530109; PMID: 23064634
- Kepler TB, Elston TC. Stochasticity in transcriptional regulation: origins, consequences, and mathematical representations. Biophys J 2001; 81:3116 - 36; http://dx.doi.org/10.1016/S0006-3495(01)75949-8; PMID: 11720979
- Simpson ML, Cox CD, Sayler GS. Frequency domain chemical Langevin analysis of stochasticity in gene transcriptional regulation. J Theor Biol 2004; 229:383 - 94; http://dx.doi.org/10.1016/j.jtbi.2004.04.017; PMID: 15234205
- Singh A, Razooky B, Cox CD, Simpson ML, Weinberger LS. Transcriptional bursting from the HIV-1 promoter is a significant source of stochastic noise in HIV-1 gene expression. Biophys J 2010; 98:L32 - 4; http://dx.doi.org/10.1016/j.bpj.2010.03.001; PMID: 20409455
- Malovannaya A, Lanz RB, Jung SY, Bulynko Y, Le NT, Chan DW, et al. Analysis of the human endogenous coregulator complexome. Cell 2011; 145:787 - 99; http://dx.doi.org/10.1016/j.cell.2011.05.006; PMID: 21620140
- Antoni BA, Rabson AB, Kinter A, Bodkin M, Poli G. NF-kappa B-dependent and -independent pathways of HIV activation in a chronically infected T cell line. Virology 1994; 202:684 - 94; http://dx.doi.org/10.1006/viro.1994.1390; PMID: 7913275
- Folks TM, Justement J, Kinter A, Dinarello CA, Fauci AS. Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science 1987; 238:800 - 2; http://dx.doi.org/10.1126/science.3313729; PMID: 3313729
- Han Y, Wind-Rotolo M, Yang HC, Siliciano JD, Siliciano RF. Experimental approaches to the study of HIV-1 latency. Nat Rev Microbiol 2007; 5:95 - 106; http://dx.doi.org/10.1038/nrmicro1580; PMID: 17224919
- Denis GV, McComb ME, Faller DV, Sinha A, Romesser PB, Costello CE. Identification of transcription complexes that contain the double bromodomain protein Brd2 and chromatin remodeling machines. J Proteome Res 2006; 5:502 - 11; http://dx.doi.org/10.1021/pr050430u; PMID: 16512664
- Denis GV, Vaziri C, Guo N, Faller DV. RING3 kinase transactivates promoters of cell cycle regulatory genes through E2F. Cell Growth Differ 2000; 11:417 - 24; PMID: 10965846
- LeRoy G, Rickards B, Flint SJ. The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol Cell 2008; 30:51 - 60; http://dx.doi.org/10.1016/j.molcel.2008.01.018; PMID: 18406326
- Sinha A, Faller DV, Denis GV. Bromodomain analysis of Brd2-dependent transcriptional activation of cyclin A. Biochem J 2005; 387:257 - 69; http://dx.doi.org/10.1042/BJ20041793; PMID: 15548137
- Peng J, Dong W, Chen L, Zou T, Qi Y, Liu Y. Brd2 is a TBP-associated protein and recruits TBP into E2F-1 transcriptional complex in response to serum stimulation. Mol Cell Biochem 2007; 294:45 - 54; http://dx.doi.org/10.1007/s11010-006-9223-6; PMID: 17111193
- Benevolenskaya EV, Murray HL, Branton P, Young RA, Kaelin WG Jr.. Binding of pRB to the PHD protein RBP2 promotes cellular differentiation. Mol Cell 2005; 18:623 - 35; http://dx.doi.org/10.1016/j.molcel.2005.05.012; PMID: 15949438
- Jiang YW, Veschambre P, Erdjument-Bromage H, Tempst P, Conaway JW, Conaway RC, et al. Mammalian mediator of transcriptional regulation and its possible role as an end-point of signal transduction pathways. Proc Natl Acad Sci USA 1998; 95:8538 - 43; http://dx.doi.org/10.1073/pnas.95.15.8538; PMID: 9671713
- Kuras L, Borggrefe T, Kornberg RD. Association of the Mediator complex with enhancers of active genes. Proc Natl Acad Sci USA 2003; 100:13887 - 91; http://dx.doi.org/10.1073/pnas.2036346100; PMID: 14623974
- Kornberg RD. Mediator and the mechanism of transcriptional activation. Trends Biochem Sci 2005; 30:235 - 9; http://dx.doi.org/10.1016/j.tibs.2005.03.011; PMID: 15896740
- Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer 2012; 12:465 - 77; http://dx.doi.org/10.1038/nrc3256; PMID: 22722403
- Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996; 272:263 - 7; http://dx.doi.org/10.1126/science.272.5259.263; PMID: 8602510
- Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, Schaffer DV. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell 2005; 122:169 - 82; http://dx.doi.org/10.1016/j.cell.2005.06.006; PMID: 16051143
- Weinberger LS, Shenk T. An HIV feedback resistor: auto-regulatory circuit deactivator and noise buffer. PLoS Biol 2007; 5:e9; http://dx.doi.org/10.1371/journal.pbio.0050009; PMID: 17194214
- Weinberger LS, Dar RD, Simpson ML. Transient-mediated fate determination in a transcriptional circuit of HIV. Nat Genet 2008; 40:466 - 70; http://dx.doi.org/10.1038/ng.116; PMID: 18344999
- Austin DW, Allen MS, McCollum JM, Dar RD, Wilgus JR, Sayler GS, et al. Gene network shaping of inherent noise spectra. Nature 2006; 439:608 - 11; http://dx.doi.org/10.1038/nature04194; PMID: 16452980
- Cox CD, McCollum JM, Allen MS, Dar RD, Simpson ML. Using noise to probe and characterize gene circuits. Proc Natl Acad Sci USA 2008; 105:10809 - 14; http://dx.doi.org/10.1073/pnas.0804829105; PMID: 18669661