Abstract
Activation of a cellular senescence program is a common response to prolonged oncogene activation or tumor suppressor loss, providing a physiological mechanism for tumor suppression in premalignant cells. The link between senescence and tumor suppression supports the hypothesis that a loss-of-function screen measuring bona fide senescence marker activation should identify candidate tumor suppressors. Using a high-content siRNA screening assay for cell morphology and proliferation measures, we identify 12 senescence-regulating kinases and determine their senescence marker signatures, including elevation of senescence-associated β-galactosidase, DNA damage and p53 or p16INK4a expression. Consistent with our hypothesis, SNP array CGH data supports loss of gene copy number of five senescence-suppressing genes across multiple tumor samples. One such candidate is the EPHA3 receptor tyrosine kinase, a gene commonly mutated in human cancer. We demonstrate that selected intracellular EPHA3 tumor-associated point mutations decrease receptor expression level and/or receptor tyrosine kinase (RTK) activity. Our study therefore describes a new strategy to mine for novel candidate tumor suppressors and provides compelling evidence that EPHA3 mutations may promote tumorigenesis only when key senescence-inducing pathways have been inactivated.
Keywords: :
Introduction
Cancer genomic profiling studies have revealed that the majority of drivers for malignant tumor progression are mutations in tumor suppressors,Citation1,Citation2 illustrating the need to accurately catalog cancer-specific tumor suppressors and their impact on treatment efficacy. Reverse genomics approaches using RNA interference (RNAi) libraries allow for rapid identification of novel gene functions and, ideally, monitor cellular parameters tightly linked to the function in question. Common to many potent oncogenic signals is the activation of built-in checkpoint mechanisms that halt aberrant proliferation, instead triggering irreversible cellular fates, including apoptosis and senescence.Citation3 It is well-established that senescence can result not only from prolonged oncogene activation (e.g., oncogenic Ras, cyclin E), but also loss of tumor suppression (e.g., loss of PTEN, VHL or NF1 genes).Citation4 A physiological role for senescence in suppressing malignant progression is supported by its in vivo detection during early stages cancer development.Citation5,Citation6
The program of cellular senescence comprises permanent cell cycle arrest associated with cell morphological changes, notably cell and nuclear flattening and an increase in senescence-associated β-galactosidase (SA-β-Gal) activity.Citation7 Senescence in vitro and in vivo coincides with the occurrence of ATM/ATR and p53/p21Cip1 DNA damage response (DDR) pathways, and previous research, including our own, suggests this to be a consequence of unbalanced DNA replication vs. mitotic cell cycle stages.Citation8-Citation10 Senescence is also commonly triggered by activation of the Ets/p16INK4a/Rb tumor suppressor pathway. Although the role of DDR and p16INK4a pathways in mediating senescence is well-established, their relative contributions remain ill-defined, and multiple pathways may act cooperatively.Citation4
Since senescence is commonly detected in cells containing intact cellular DDR and p16INK4a pathways, we reasoned that a siRNA screen in untransformed cells would potentially identify putative tumor suppressor genes. Our screen contrasts with previous studies, which focused on senescence bypass events, and biased for discovery of genes intrinsically linked to or required for the specific arrest pathway.Citation11-Citation13 Furthermore, the proposed pro-tumorigenic effect of a senescence-associated inflammatory responseCitation14 warrants a closer study to assess whether senescence can be induced upon loss of relevant gene function. Here we identify 12 kinases as newly identified regulators of cellular senescence using quantitative senescence β-galactosidase staining, and show that senescence frequently correlates with DNA damage induction and is mostly p53- and sometimes p16INK4A-dependent. In support of our hypothesis to reveal candidate tumor suppressor genes, analysis of SNP aCGH data showed that a significant number of candidate genes displayed gene copy loss in sets of tumor samples.
Interestingly, one novel senescence regulator, the EPHA3 receptor tyrosine kinase (RTK) gene, is among the most frequently mutated genes in human lung adenocarcinomas and colorectal cancers.Citation15-Citation18 EPHA receptors have traditionally been assigned oncogenic roles due to their overexpression in a variety of cancers including carcinomas, melanoma and gliomas.Citation19,Citation20 However, in support of additional tumor suppressor functions, certain ephrin-EPHs are suggested to show temporally bi-phasic roles during tumor progression,Citation21 and EPHA7 receptor was recently shown to act as a tumor suppressor in follicular lymphoma.Citation22 We here show that senescence upon loss of EPHA3 is regulated by p16INK4a and p53 and, in agreement with recent data from the Pasquale and Zhuang labs,Citation23,Citation24 show that selected EPHA3 tumor-associated mutations decrease receptor expression levels and overall kinase activity. Our results suggests that concomitant loss of EPHA3 and key senescence-inducing tumor suppressor functions may cooperatively stimulate tumorigenesis, and conclude that our senescence screen successfully identified strong candidates for tumor suppressor genes.
Results
Identification of senescence-inducing kinase siRNAs
We utilized a cell-based screen to identify regulators of premature senescence in untransformed cells, hypothesizing that a loss-of-function screen may identify new tumor suppressor genes. We chose to screen the hTERT-immortalized retinal pigmented epithelial line hTERT-RPE1, because it models key aspects of untransformed cells, including quiescence with a high efficiency of G0 markers, notably the presence of primary cilia.Citation25 Unlike quiescent cells, senescent cells exhibit an irreversible proliferative arrest, followed by formation of enlarged nuclei and a flattened cytoplasmic morphology, with rare exceptions.Citation4 These features were used to analyze a multiparametric HCS siRNA screen of hTERT-RPE1 cells engineered to express a doxycycline-inducible p53 shRNA (from here on called hTERT-RPE1 p53shRNA), designed to identify proteins that impinge on the p53 pathway (Corson LB, et al., manuscript in preparation). Our analysis focused on loss of the proliferation marker Ki67 and decreased nuclei counts, combined with an average increase in nuclear size (). This approach identified a group of 16 kinase siRNAs, which we labeled as “senescence-like” ().
Figure 1. Identification of senescence-inducing kinase siRNAs. (A) Schematic overview of the high-content senescence screen. (B) Scatterplot analysis of data from the kinome HCS screen in p53+ cells. Cells were stained for Ki67, p53, p21CIP1 and Hoechst at day 3, and multiparametric analysis was performed to determine siRNAs displaying decreased proliferation (< 0.5 StDev Ki67%) and an increased nuclear size (> 2.0 StDev Hoechst mean area). High, medium and low cell number indicators are assigned as the third parameter. (C) Senescence scores per gene are the sum of the senescence scores of its siRNA pool and the four individual siRNAs, omitting siRNAs conferring less than 50% specific gene knockdown. Scoring details are depicted in Table S2. References to studies establishing bona fide tumor suppressor functions for human (Hs) or mouse (Mm) genes are indicated.
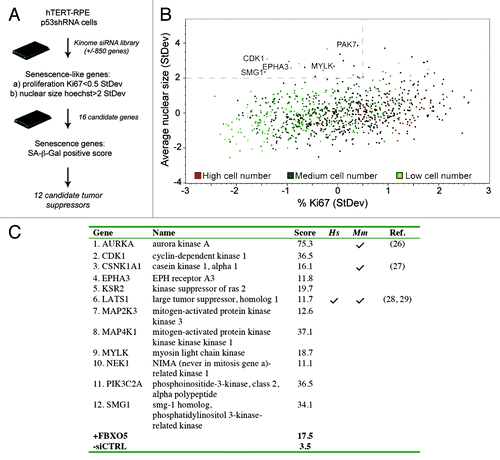
We next tested hits as bona fide senescence regulators in a secondary screen detecting the widely studied SA-β-Gal senescence biomarker, on parallel transfections using siRNA pools plus the four individual siRNAs per gene. Knockdown of Emi1, previously reported to elicit pronounced DNA damage-induced senescence in hTERT-RPE1 cells, was included as a positive controlCitation8 (Fig. S1A). Normalized senescence scores were calculated by quantitation of the staining intensities per single oligonucleotide or pool (Fig. S1B and Table S2). To correlate the phenotype with knockdown efficiency, we performed TaqMan RNA expression analysis (Fig. S2). Genuine hits were assigned if both the siRNA pool, plus at least two oligonuceotides conferring > 50% knockdown efficiency, scored positive (Table S3). This approach validated 12 of 16 senescence-like kinome siRNAs as genuine senescence hits ().
Delineation of senescence signatures
Activation of a p53-dependent DNA damage response (DDR) and increased p16INK4a CKI expression have been causally linked to senescence. hTERT-RPE1 cells contain an intact p53 checkpoint, and p53 stabilization and nuclear accumulation are seen upon sustained damage signaling.Citation8,Citation26 A two- to 4-fold increase in cells expressing threshold p53 and p21CIP1 protein levels was measured in the HCS analysis (). In all cases, total cell numbers and percentages of proliferating cells were significantly increased in p53 knockdown cells compared with p53+ cells (; Fig. S3A), suggesting a p53-dependent growth arrest. Furthermore, we measured increased DNA damage by γ-H2AX foci formation upon senescence induction in all but one case (PIK3C2A), with strongest hits showing foci in 40–50% of cells (Fig. S3B). Finally, a decreased senescence score was measured after transfection of six of 12 senescence hit siRNAs in p53 knockdown cells (), implying that senescence typically required activation of a p53-dependent DDR.
Figure 2. Delineation of senescence signatures. (A) hTERT-RPE1 p53 shRNA cells were transfected with kinome siRNAs in the absence of doxycycline and immunostained for p53 and p21CIP1 following 72 h culture. Percentages of cells expressing p53, p21CIP1 above threshold levels were calculated, and averages from four 384 wells are shown. (B) hTERT-RPE1 p53 shRNA cells were transfected with kinome siRNAs in the absence or presence of doxycycline and immunostained for Ki67. Percentages of cells expressing Ki67 protein above threshold levels were calculated, and averages from four 384 wells are shown. * indicates p ≤ 0,05 and ** indicates p ≤ 0,01. (C) Senescence scores per gene are the sum of the senescence scores of the siRNA pool plus four individual siRNAs, omitting siRNAs conferring less than 50% knockdown. Detailed scoring information is depicted in Table S3. (D) Quantitative p16INK4A mRNA expression analyses during senescence induction. hTERT-RPE1 p53 shRNA cells were transfected with pooled siRNAs in the absence of doxycycline. RNA expression was quantitated using TaqMan analyses after 3 days of transfection. (E) Schematic model summarizing kinome screen data, using data depicted in (A) and (D). In an incipient tumor, modeled by hTERT-RPE1 cells, loss of selected tumor suppressors activates p53- and/or p16INK4A-dependent senescence, and overt DNA damage. In a premalignant tumor, senescence may serve as a cell-intrinsic tumor suppressor mechanism to subvert oncogenic transformation. Loss of p16INK4A and/or p53 promotes malignancy.
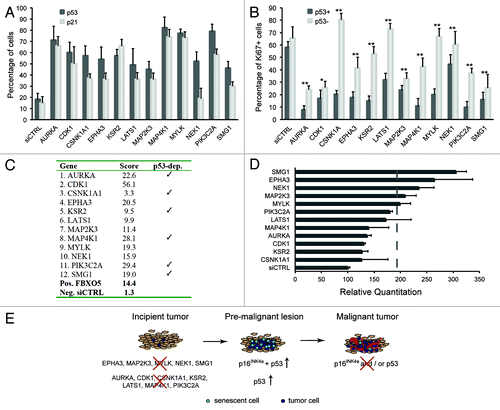
Next, we assessed activation of the p16INK4a tumor suppressor pathway following senescence suppressor siRNA transfection. In hTERT-RPE1 cells, classic senescence induction following oncogenic RAS expression did not result in a significant increase in p16INK4a mRNA (data not shown). Contrary to p53 pathway activation, significant increases in p16INK4a mRNA expression were less frequently detected upon senescence hit siRNA transfection, and most clearly upon knockdown of MYLK, MAP2K3, NEK1, EPHA3 and SMG1 (). We conclude that in a majority of cases senescence induction is associated with activation of well-known cell cycle arrest pathways, schematically summarized in . As expected, this corresponds to a delayed cell cycle progression, as measured by DNA replication analysis in synchronized cells (Fig. S4).
Genome copy number and mutational analysis to appoint putative tumor suppressors
To establish whether senescence hits encompass tumor suppressors, we combined a cancer genomic analysis approach with cancer mutation and literature surveys. Validating our hypothesis, three hits identified have previously been shown to display bona fide tumor suppressor functions in murine models (). Specifically, AurkA heterozygous mice present haploinsufficient tumor suppression with increased incidence of lymphoma, lung and liver tumorsCitation27; conditional Csnk1a1 ablation triggers colorectal carcinogenesis and LOHCitation28; and Lats1-deficient mice develop soft tissue sarcoma and ovarian tumors.Citation29 Interestingly, all three models exhibit signs of chromosomal instability or DDR activation, and a p53-dependent DDR-senescence response is proposed to halt carcinogenesis in the Csnk1a1-null or heterozygous animals.Citation28 This verifies that individual findings from our in vitro study have a capacity to broaden our knowledge of physiological disease progression.
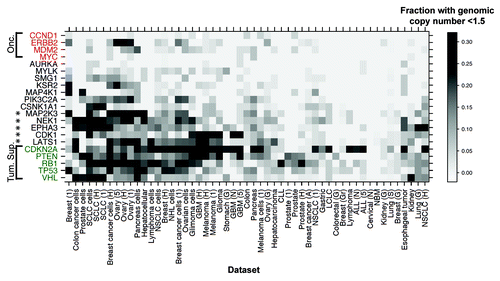
Among the candidates, only LATS1 is suggested to act as a tumor suppressor in human disease, supported by its conserved role in growth control from flies to manCitation30 (). We therefore next asked if we could observe an increased propensity for genome copy number loss of our senescence genes in human tumor samples. Validating our strategy, loci encompassing known tumor suppressors showed significant copy number loss across data sets, defining a “tumor suppressor copy number signature”. Importantly, clustering analysis of this signature identified a set of five screen candidates with a similar genomic profile, namely LATS1, CDK1, EPHA3, NEK1 and MAP2K3 (). Locus inspection showed none to be in the vicinity of known tumor suppressors, supporting the deduction that these constitute candidate tumor suppressor genes.
Figure 3. Senescence gene loss evidenced by genomic copy number analysis. Heatmap clustering analysis of SNP array-CGH data from 2654 samples from 53 panels of human tumors and cell lines, to identify sets with a genome copy number lower than 1.5. A sliding scale density gradient was applied. Loci encompassing tumor suppressors CDKN2A, RB1, TP53 and VHL were included as positive controls and oncogenes CCND1, ERBB2, MDM2 and MYC as negative controls. Stars indicate genes closely resembling the tumor suppressor signature.
As a final in silico approach, we surveyed the increasing collection of cancer-specific mutations as reported in the COSMIC database. Indeed, out of the 12 genes, seven genes were mutated in 1–4% of tumors. Of outstanding interest, with a reported mutation frequency of 4–6%, the EPHA3 RTK gene was shown to be among the most frequently mutated genes in human colorectal, lung and ovarian carcinomas.Citation15-Citation18,Citation31 Furthermore, EPHA RTK family kinases have pleiotropic functions and EPHA3 was originally assigned oncogenic properties in lymphomas.Citation21,Citation32 We thus decided to key in on a putative new role for EPHA3 as a tumor suppressor.
Loss of EPHA3 receptor signaling confers p16INK4A and p53-dependent senescence
Since the EPHA RTK family consists of nine members, we first asked if EPHA3 uniquely induced cellular senescence using SA-β-Gal staining upon siRNA transfection of additional EPHA receptors expressed in hTERT-RPE1 (EPHA1, A2, A4, and A5 mRNAs, data not shown). Knockdown of Emi1 was included as a positive control.Citation8 Our results showed that senescence was uniquely detected after knockdown of EPHA3 and not measurably apparent after knockdown of other EPHA RTKs expressed in hTERT-RPE1 cells (Fig. S5).
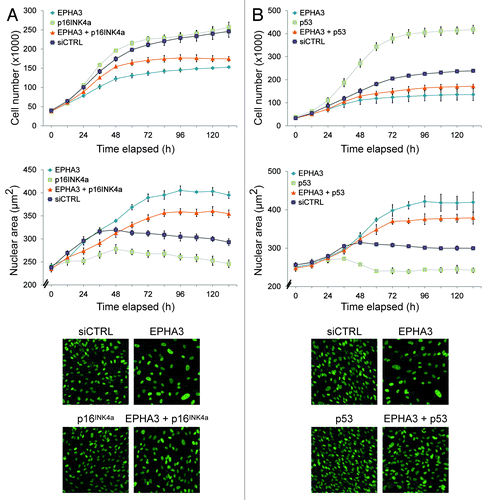
Characterization of the senescence signature showed that siRNA-mediated knockdown of EPHA3 leads to increased p16INK4a and p53 expression ( and ). We next asked if the senescence phenotype was mediated by p16INK4a or p53 activation using in vivo imaging. Time-lapse imaging of siRNA-treated hTERT-RPE1 cells stably expressing GFP-tagged H2B protein showed a rescue of EPHA3 knockdown-induced senescence when p16INK4a (; Fig. S5E) or p53 (; Fig. S5F) were co-depleted. A rescue of senescence was both measured by an increase in total cell number, as well as a decrease in average nuclear size. We therefore conclude that EPHA3 knockdown-induced senescence requires p16INK4a and p53.
Figure 4. Loss of EPHA3 confers p16INK4A- and p53-dependent senescence. Live cell analysis of (A) EPHA3 plus p16INK4a siRNA-treated and (B) EPHA3 plus p53 siRNA-treated hTERT-RPE1 cells. The average nuclear area off all cells per image is shown. Representative 20x images from each condition are shown in the lower panels.
Selected EPHA3 gene mutations confer loss of kinase function
Similar to other EPHA RTKs, activation of EPHA3 occurs via autophosphorylation of conserved tyrosine residuesCitation33 and ephrin ligand binding triggers receptor oligomerisation and consequent receptor activation.Citation34 To find how tumor-associated EPHA3 point mutations affect receptor activity we assayed for kinase activity monitoring autophosphorylation. HEK 293T and hTERT-RPE1 cells expressing a select set of EPHA3-LAP tumor-associated mutation variants were generated to assess relative cellular receptor activities (; Fig. S6A and B). As expected, ephrin-A5 ligand activation leads to receptor internalisation and trafficking via the early endosomal compartment labeled with EEA1Citation35 (Fig S7). Of note, a decreased steady-state expression of the R728L variant was detected in both cell lines, and the T933M variant in hTERT-RPE1 cells (Fig. S6A and B).
Figure 5. Selected EPHA3 cancer gene mutations confer decreased kinase function. (A) Schematic representation of EPHA3 protein domains and selected cancer point mutations pertinent to this study. (B) Cellular kinase activities of selected EPHA3 tumor variants. 293T cells expressing EPHA3-LAP variants were treated with preclustered ephrin-A5 and immunoprecipitates were immunoblotted. Anti-GFP was used to detect EPHA3-LAP. Relative cellular kinase activities were determined by normalization of the PY (phosphotyrosine) against the EPHA3 signal, and normalized values relative to WT are shown in arbitrary units. (C) In vitro kinase assays of selected EPHA3 tumor variants. Immunoprecipitates from kinase assays or input lysate samples from 293T EPHA3-LAP lines were immunoblotted with indicated antibodies, and normalized values were calculated as in (B). (D, E and F) Structural analyses of EPHA3 tumor variants R728L and D806N and structural alignment of the EPHA3 and LKB1 kinase domains. PDB structure files were analyzed using the PyMOL molecular visualization package. Notable alterations are encircled. (G) In vitro kinase assay of EPHA3 D806Y variant. 293T cells expressing EPHA3-LAP proteins or control lysate were treated with preclustered ephrin-A5. GFP immunoprecipitates from kinase assays were immunoblotted with the indicated antibodies.
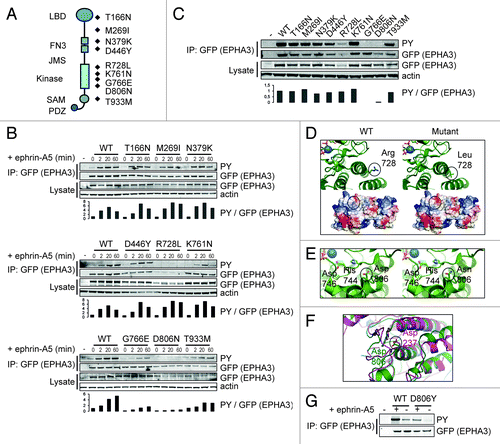
Wild type EPHA3 shows a transient activation curve following ephrin-A5 ligand-induced activation, peaking at 20 min (), consistent with published data.Citation35 Decreased normalized cellular kinase activities were detected for two receptor variants mutated in the cytoplasmic region (G766E and D806N) and one variant mutated in the cytoplasmic region showed a decrease in overall receptor level (R728L) (). We next asked how point mutations affected absolute kinase activity using in vitro immunoprecipitation kinase assays. In this assay, EPHA3 variants G766E and D806N showed a clear loss of kinase activity (; Fig. S6C). We conclude that selected intracellular EPHA3 tumor-associated point mutations lead to a decrease in receptor expression level and/or normalized tyrosine kinase activity.
Structural analysis of EPHA3 kinase domain cancer point mutations
Next, we examined the crystal structure of the juxtamembrane and kinase domain (JMKIN) of wild type EPHA3Citation34 to explain possible functional deficiencies of the tumor-associated mutations. The mutated residues K761, G766 and D806 are situated within the kinase domain, while R728 is situated on the surface area between two α-helices (Fig. S6D). We were unable to assess the G766 variant as the crystal structure surrounding this residue is disordered. Arginine 728 to Leucine alteration causes a shift in the surface charge from positive to non-charged (), which may affect the steady-state expression level of the receptor via altered protein-protein interactions and/or increased receptor degradation, explaining its reduced expression level (; Fig. S6A and B). Mutation of residue 806 from Aspartic acid to Asparagine may alter the interaction between residues Aspartic acid 746 and Histidine 744, which face the ATP binding pocket and may therefore directly affect the EPHA3 catalytic activity ().
Interestingly, structural alignment of EPHA3 JMKIN with the tumor suppressor LKB1 kinase domain showed that LKB1 lung cancer-associated point mutation D237Y is homologous to mutated residue D806 in EPHA3.Citation15 We therefore tested EPHA3 D806Y mutation for kinase activity, and show that this mutant is kinase-defective (). We conclude that selected cancer-associated point mutations appear to affect EPHA3 kinase activity either through altering protein-protein interactions, or through direct effects on kinase domain structural determinants, decreasing EPHA3 receptor tyrosine kinase activity. This supports our hypothesis that EPHA3 acts as a tumor suppressor gene.
Discussion
Cancer genomics initiatives have identified numerous often low-frequency mutations that can prove predictive for clinical response to targeted therapy, creating new opportunities for personalized cancer care and biomarker discovery. Not surprisingly, clinical response and therapy resistance can be critically modified by the landscape of tumor suppressor mutations,Citation36 which constitute in fact the majority of cancer drivers.Citation1 Yet, the detailed consequences of tumor suppressor mutations are often unknown. We developed a “bottom-up” senescence-based strategy to functionally identify tumor suppressor candidates. Validating our approach, we identified three genes with known tumor suppressor function in human or mouse (AURKA, CSNK1A1 and LATS1), and SNP aCGH data showed that half of the candidates displayed increased genome copy loss in tumor sample sets (). Clearly, our study is not exhaustive and would be enhanced by parallel assessment of DNA methylation and gene expression changes. Nonetheless, it illustrates the power of combining functional genomics with delineation of somatic mutations. We propose that detection of senescence in cells harbouring intact damage checkpoint pathways, such as hTERT-RPE1, can be employed as a shortcut to identify cancer-promoting events.
Our senescence gene hits can readily be grouped in three functional categories: (1) regulators of accurate mitotic cell cycle progression and cytokinesis, DNA damage signaling and the spindle assembly checkpoint (SAC): AURKA, CDK1, CSNK1A1, LATS1, NEK1, SMG1 (2) regulators of adhesion and migration: EPHA3, MYLK, PIK3C2A and (3) regulators of growth factor-induced mitogenic signaling via p38 SAPK and JNK: KSR2, MAP2K3, MAP4K1. Hence, a compendium of genes that intersect on accurate spindle assembly checkpoint control and fine-tuning of cell adhesion as well as cytokinesis machineries describe a likely set of candidate tumor suppressors. This is further supported by recent sequencing efforts that reveal frequent mutations in mitotic kinases including Aurora, Polo and LATS kinases as well as Nek family kinases.Citation17,Citation37 Although conceptually perhaps unsurprising, functional data mining also reveals unexpected dual functions for previously established oncogenic proteins, as exemplified by the AURKA oncogene, which indeed was shown to be haploinsufficient for tumor suppression in mice.Citation27
Senescence can be viewed as a cell-intrinsic fail-safe response to overt oncogenic proliferative signals,Citation3 a key effector of cancer chemotherapy response,Citation38,Citation39 or a barrier to tumor initiation in incipient human tumors.Citation4,Citation5 Contrary to such anti-proliferative responses, senescence in the stromal microenvironment may also promote paracrine epithelial tumor growth via secretion of inflammatory molecules, akin to a tissue repair response.Citation40 Furthermore, an elegant recent study suggested that inflammatory signals themselves can subvert a senescence barrier.Citation41 Its precise physiological role thus is likely complex and possibly context-dependent, yet common to a multi-step carcinogenesis paradigm, wherein loss of p53 and/or p16INK4a affords proliferation of damaged cells. We confirm that senescence correlates with activation of a pronounced p53-dependent DDR, consistent with previous studies describing p53 activation and/or senescence upon loss or inhibition of AURKA,Citation42 CDK1,Citation43 CSNK1A1,Citation28 LATS1,Citation44 MAP2K3Citation45 and SMG1.Citation46 This further illustrates the importance of perturbations in both surveillance systems and checkpoint response pathways as a frequent driver of tumor progression.
Contrary to p53 activation, significant increases in p16INK4a mRNA expression were less frequently detected upon senescence hit siRNA transfection, and most clearly upon loss of MYLK, MAP2K3, SMG1, NEK1 and EPHA3 (). None of these genes were previously known to activate the p16INK4A-dependent senescence program. Of the candidate tumor suppressors, the EPHA3 RTK gene was shown to be frequently mutated in human lung adenocarcinomas and colorectal cancers.Citation15-Citation18 Consistent with its candidacy as a tumor suppressor, array CGH data showed frequent loss of the chromosome 3 arm encoding EPHA3 in the lung cancer data set in particular. This is confirmed by recent data reporting a striking loss of the EPHA3 gene in 42% of primary lung adenocarcinomas.Citation24 We found that senescence upon EPHA3 loss is partially rescued by concomitant p16INK4a or p53 depletion, and that this is a function unique to EPHA3. Our data therefore suggests that loss of EPHA3 activity may promote tumorigenesis only when key senescence-inducing tumor suppressor pathways are absent. We however did not detect an enrichment for p16INK4a mutations in lung cancer samples that display EPHA3 copy number loss, using the CONAN CGP copy number analysis tool available via the Sanger Institute. The in vivo functional cooperation of EPHA3 and p16INK4a loss of function is, however, unlikely straightforward and uniform, warranting closer inspection of their co-dependencies during tumorigenesis.
Previous EPH receptor studies have revealed dual opposing roles during tumor initiation vs progression,Citation47,Citation48 and EPHA3 has been ascribed both oncogenicCitation49,Citation50 and tumor-suppressive functions.Citation51 In support of its role as a tumor suppressor, EPHA3 gene mutations are distributed along the gene rather than clustered in hot spots. Importantly, we show that cancer-associated point mutations G766E and D806N are kinase-defective, as well as a mutation analogous to D237Y in the well-established LKB1 tumor suppressor. Furthermore, we show that the R728L variant decreases overall receptor levels and cellular receptor activity. Our results agree with recent reports by Lisabeth and Zhuang and colleagues that describe a disruption of RTK functions for selected EPHA3 tumor variants,Citation23,Citation24 likely acting to decrease apoptotic susceptibility in a dominant-negative fashion.Citation24 In conclusion, our results strengthen evidence for a role for EPHA3 as a candidate tumor suppressor gene.
It is well-established that the interplay between different EPH receptors influences their signaling outcome.Citation22 Furthermore, EPH receptors cross signal to other tyrosine kinase families, with evidence for negative regulation of c-MET by EPHA signaling.Citation52 Interestingly, a secreted form of EPHA7 was recently shown to act as a tumor suppressor in follicular lymphoma via inhibition of EPHA2 in a dominant-negative fashion.Citation22 Since also EPHA3 is expressed as an alternative isoform, it will be interesting to study how it may influence the EPH-RTK signaling network. Furthermore, while no increased tumor growth is detected in a constitutive EPHA3 murine null model,Citation53,Citation54 our study warrants further comprehensive in vivo loss-of-function analyses. Taken together, we developed a successful cell-based strategy to functionally mine for novel candidate tumor suppressors, and provide compelling evidence that loss-of-function EPHA3 variants may promote tumorigenesis when common senescence-inducing pathways are inactive.
Materials and Methods
Cell lines, culture and treatment
hTERT-RPE1 cells (Clontech) were maintained in DMEM:F-12 (Invitrogen) containing 10% FBS, 2mM L-Glutamine and 0,348% sodium bicarbonate, following manufacturer recommendations. 293T cells were maintained in DMEM (Lonza) containing 10% FBS and 2 mM L-Glutamine, following manufacturer recommendations. Inducible p53 shRNA hTERT-RPE1 cells were constructed using the pHUSH tetracycline-inducible retrovirus gene transfer vectorCitation55 with a previously described p53 shRNA,Citation56 and labeled hTERT-RPE1 p53 shRNA. Knockdown in the selected subclone was validated by absence of p53 and p21 induction upon etoposide treatment, and p53 protein was absent after 72h treatment with 1 μg/ml doxycycline. A stable H2B-GFP hTERT-RPE1 clonal cell line was generated by transfection with GFP-tagged human H2B plasmid (pEGFP-H2B-N1) followed by clonal selection using G418 (Roche).
High-content kinase siRNA screen
hTERT-RPE1 p53 shRNA cells were treated +/− 1 µg/ml doxycycline for three days prior to transfection, also called “p53+” or “p53-“ cells. A Dharmacon siGENOME library, composed of 862 siRNA pools targeting kinases and proteins predicted to influence kinase signaling was used (Table S1; Corson et al. manuscript in preparation). Following three days of culture, cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 and blocked in 5% fish gelatin (Sigma). Cells were immunostained with primary antibodies in 5% fish gelatin during overnight incubation at 4°C and incubated with secondary antibodies for 1 h at ambient temperature. Primary antibodies were rabbit anti-p53 (Cell Signaling 928; 1:1500), goat anti-p21 (R&D Systems AF1047; 1:20.000) and mouse-anti-Ki67 (BD PharMingen 55600; 1:1500). Secondary antibodies were Cy5 anti-mouse (Jackson 715–495–150), Cy3 anti-rabbit (Jackson 711–165–152), and AF488 anti-goat (Invitrogen A11055). Hoechst 33342 was used at 1:50,000. For primary screen data analysis depicted in , the data set in which p53 was functional (p53+) was used.
HCS imaging, quantitation and statistics
384 Multiwell plates were imaged with a 10X objective and 2X binning on the ImageXpress Micro high content imaging system (Molecular Devices). Four sites were acquired per well with duplicate wells for every condition. Analysis using MetaXpress Cell Scoring and Multiwavelength Modules included (1) total number of nuclei, (2) percentage cells positive for p53, p21 and Ki67, and (3) average nuclear size. An image analysis example is shown in Figure S1C. Data was collected in an Oracle database managed through Molecular Devices MDCStore. Images, measurements, annotations, normalizations, and statistical analysis were managed through AcuityXpress software.
Senescence-associated β-galactosidase (SA-β-gal) staining and quantitation
Cellular senescence was detected in cells transfected in 96-well cellBIND plates (Corning), by staining for acidic β-galactosidase as described,Citation7 followed by DNA counterstaining with Hoechst 33342. Cells were imaged on a Nikon Eclipse Ti microscope equipped with a monochrome camera, applying a DAPI scan and sequential brightfield RGB images. Composite color images were generated using a NIS Elements AR (Nikon) colorcombine journal. Image analysis was performed using a MetaMorph journal (Molecular Devices) following a scoring system detailed in the Supplementary Materials and Methods.
SNP array-CGH tumor sample analysis
SNP/CHIP array-CGH data available at Genentech (CARFOG, June 2009) for 2654 samples from 53 human tumor and cell line panels was mined using the “genoset” package from BioconductorCitation57 and plotted as a heatmap to show the fraction of samples per data set containing a genome locus copy number lower than 1.5 copies. For visualization, the scale was trimmed at a value of 0.3: data sets with this value or greater were shown with the maximum density of the scale.
Immunoprecipitation and western blotting
Cells expressing stable LAP-tagged EPHA3 proteins were serum starved for 1 h, and then incubation with 1,5 µg/ml recombinant human ephrin-A5-Fc or IgG1-Fc proteins (R&D Systems) preclustered for 20 min with anti-human Fc antibody (Jackson ImmunoResearch) at 10:1 molar ratio. Ligand-activated 293T cells expressing LAP-tagged EPHA3 proteins were lysed in RIPA buffer containing Protein Inhibitor Cocktail (Roche) and Phosphatase Inhibitor Cocktail (Roche). 500 μg of protein was rotated for 16h with 1 μg of anti-GFP antibody (Invitrogen). Antigen was captured using Protein G/A Sepharose beads (Sigma). Alternatively, specific antigen was captured using S-protein agarose beads (Novagen). Samples were immunoblotted using anti-phosphotyrosine (PY 4G10; Upstate Biotechnology Inc.) or in-house affinity-purified anti-GFP antibodies.
In vitro kinase assay
293T and hTERT-RPE1 cells expressing LAP-tagged EPHA3 proteins were lysed in RIPA buffer containing protease inhibitors. 500 μg of total protein was incubated for 1h with 1 μg of antibodies to GFP (Invitrogen). Specific antigen was captured using Protein A/G Sepharose beads (Thermo Scientific) or, alternatively, with S-protein agarose beads (Novagen), and washed using RIPA and kinase buffer (50 mM NaCl, 20 mM Hepes pH 7.2, 10 mM MgCl2, 2 mM EDTA, 0.02% Triton X-100). Beads were next incubated in kinase buffer supplemented with 20 μM Adenosine 5′-triphosphate disodium salt hydrate (Sigma). Samples were immunoblotting using anti-phosphotyrosine or anti-GFP antibodies.
Live cell imaging
hTERT-RPE1 cells expressing H2B-GFP protein were treated with 50 nM pooled siRNA using forward transfection with Oligofectamine reagent (Invitrogen) on 24-well plates. Transfections were in replicates of four and imaging was performed using the IncuCyte™ FLR instrument (Essen BioScience) for five days. A duplicate plate was harvested at day three to confirm siRNA knockdown. IncuCyte object analysis was performed with default parameters.
| Abbreviations: | ||
| DDR | = | DNA damage response |
| SA-β-Gal | = | senescence associated-β-galactosidase |
| CGH | = | comparative genomic hybridization |
| EPHA3 | = | EPH receptor A3 |
| RTK | = | receptor tyrosine kinase |
Additional material
Download Zip (2.6 MB)Acknowledgments
We wish to acknowledge Alex Loktev, Mindan Sfakianos, Chris Westlake, David Davis and Seija Hackl for providing expert advice and technical support. We are grateful to Meredith Sagolla and Jeff Eastham-Anderson for advanced light microscopy support, Denis Kainov for structural analysis support and thank members of the Jackson and Verschuren labs for discussions. pEGFP-H2B-N1 was a gift from Jorge Torres (UCLA).
Disclosure of Potential Conflicts of Interest
The authors disclose no potential conflicts of interest.
Author Contributions
Jenni L., L.B.C., A.H., S.K., T.L.H. and E.W.V. conducted aspects of the experimental design, performed experiments, and data interpretation. James L. managed the siRNA library and M.J.B. performed statistical data analysis. P.K.J., L.B.C., Jenni L. and E.W.V. interpreted results, and Jenni L. and E.W.V. wrote the manuscript.
Financial Disclosures
Research was supported by an EU-FP7 Marie Curie Grant PIRG06-GA-2009–256485 (E.W.V.), the Sigrid Juselius and Orion-Farmos Foundations (E.W.V.), and a Graduate Program scholarship to Jenni L.
Supplemental Materials
Supplemental materials may be found here:
http://www.landesbioscience.com/journals/cc/article/23515/
Related Research Data
References
- Bozic I, Antal T, Ohtsuki H, Carter H, Kim D, Chen S, et al. Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci U S A 2010; 107:18545 - 50; http://dx.doi.org/10.1073/pnas.1010978107; PMID: 20876136
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 2011; 39:Database issue D945 - 50; http://dx.doi.org/10.1093/nar/gkq929; PMID: 20952405
- Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature 2004; 432:307 - 15; http://dx.doi.org/10.1038/nature03098; PMID: 15549092
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev 2010; 24:2463 - 79; http://dx.doi.org/10.1101/gad.1971610; PMID: 21078816
- Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 2010; 10:51 - 7; http://dx.doi.org/10.1038/nrc2772; PMID: 20029423
- Ohtani N, Mann DJ, Hara E. Cellular senescence: its role in tumor suppression and aging. Cancer Sci 2009; 100:792 - 7; http://dx.doi.org/10.1111/j.1349-7006.2009.01123.x; PMID: 19302284
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995; 92:9363 - 7; http://dx.doi.org/10.1073/pnas.92.20.9363; PMID: 7568133
- Verschuren EW, Ban KH, Masek MA, Lehman NL, Jackson PK. Loss of Emi1-dependent APC/C inhibition deregulates E2F target expression and elicits DNA damage-induced senescence. Mol Cell Biol 2007; 27:7955 - 65; http://dx.doi.org/10.1128/MCB.00908-07; PMID: 17875940
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006; 444:633 - 7; http://dx.doi.org/10.1038/nature05268; PMID: 17136093
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006; 444:638 - 42; http://dx.doi.org/10.1038/nature05327; PMID: 17136094
- Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M, et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 2004; 428:431 - 7; http://dx.doi.org/10.1038/nature02371; PMID: 15042092
- Mullenders J, Fabius AW, Madiredjo M, Bernards R, Beijersbergen RL. A large scale shRNA barcode screen identifies the circadian clock component ARNTL as putative regulator of the p53 tumor suppressor pathway. PLoS One 2009; 4:e4798; http://dx.doi.org/10.1371/journal.pone.0004798; PMID: 19277210
- Rovillain E, Mansfield L, Lord CJ, Ashworth A, Jat PS. An RNA interference screen for identifying downstream effectors of the p53 and pRB tumour suppressor pathways involved in senescence. BMC Genomics 2011; 12:355; http://dx.doi.org/10.1186/1471-2164-12-355; PMID: 21740549
- Campisi J. Cellular senescence: putting the paradoxes in perspective. Curr Opin Genet Dev 2011; 21:107 - 12; http://dx.doi.org/10.1016/j.gde.2010.10.005; PMID: 21093253
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008; 455:1069 - 75; http://dx.doi.org/10.1038/nature07423; PMID: 18948947
- Wood LD, Calhoun ES, Silliman N, Ptak J, Szabo S, Powell SM, et al. Somatic mutations of GUCY2F, EPHA3, and NTRK3 in human cancers. Hum Mutat 2006; 27:1060 - 1; http://dx.doi.org/10.1002/humu.9452; PMID: 16941478
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature 2007; 446:153 - 8; http://dx.doi.org/10.1038/nature05610; PMID: 17344846
- Bardelli A, Parsons DW, Silliman N, Ptak J, Szabo S, Saha S, et al. Mutational analysis of the tyrosine kinome in colorectal cancers. Science 2003; 300:949; http://dx.doi.org/10.1126/science.1082596; PMID: 12738854
- Easty DJ, Bennett DC. Protein tyrosine kinases in malignant melanoma. Melanoma Res 2000; 10:401 - 11; http://dx.doi.org/10.1097/00008390-200010000-00001; PMID: 11095400
- Wykosky J, Debinski W. The EphA2 receptor and ephrinA1 ligand in solid tumors: function and therapeutic targeting. Mol Cancer Res 2008; 6:1795 - 806; http://dx.doi.org/10.1158/1541-7786.MCR-08-0244; PMID: 19074825
- Lackmann M, Boyd AW. Eph, a protein family coming of age: more confusion, insight, or complexity?. Sci Signal 2008; 1:re2; http://dx.doi.org/10.1126/stke.115re2; PMID: 18413883
- Oricchio E, Nanjangud G, Wolfe AL, Schatz JH, Mavrakis KJ, Jiang M, et al. The Eph-receptor A7 is a soluble tumor suppressor for follicular lymphoma. Cell 2011; 147:554 - 64; http://dx.doi.org/10.1016/j.cell.2011.09.035; PMID: 22036564
- Lisabeth EM, Fernandez C, Pasquale EB. Cancer somatic mutations disrupt functions of the EphA3 receptor tyrosine kinase through multiple mechanisms. Biochemistry 2012; 51:1464 - 75; http://dx.doi.org/10.1021/bi2014079; PMID: 22242939
- Zhuang G, Song W, Amato K, Hwang Y, Lee K, Boothby M, et al. Effects of cancer-associated EPHA3 mutations on lung cancer. J Natl Cancer Inst 2012; 104:1182 - 97; http://dx.doi.org/10.1093/jnci/djs297; PMID: 22829656
- Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peränen J, Merdes A, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 2007; 129:1201 - 13; http://dx.doi.org/10.1016/j.cell.2007.03.053; PMID: 17574030
- Loewer A, Batchelor E, Gaglia G, Lahav G. Basal dynamics of p53 reveal transcriptionally attenuated pulses in cycling cells. Cell 2010; 142:89 - 100; http://dx.doi.org/10.1016/j.cell.2010.05.031; PMID: 20598361
- Lu LY, Wood JL, Ye L, Minter-Dykhouse K, Saunders TL, Yu X, et al. Aurora A is essential for early embryonic development and tumor suppression. J Biol Chem 2008; 283:31785 - 90; http://dx.doi.org/10.1074/jbc.M805880200; PMID: 18801727
- Elyada E, Pribluda A, Goldstein RE, Morgenstern Y, Brachya G, Cojocaru G, et al. CKIα ablation highlights a critical role for p53 in invasiveness control. Nature 2011; 470:409 - 13; http://dx.doi.org/10.1038/nature09673; PMID: 21331045
- St John MA, Tao W, Fei X, Fukumoto R, Carcangiu ML, Brownstein DG, et al. Mice deficient of Lats1 develop soft-tissue sarcomas, ovarian tumours and pituitary dysfunction. Nat Genet 1999; 21:182 - 6; http://dx.doi.org/10.1038/5965; PMID: 9988269
- Pan D. The hippo signaling pathway in development and cancer. Dev Cell 2010; 19:491 - 505; http://dx.doi.org/10.1016/j.devcel.2010.09.011; PMID: 20951342
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011; 474:609 - 15; http://dx.doi.org/10.1038/nature10166; PMID: 21720365
- Janes PW, Adikari S, Lackmann M. Eph/ephrin signalling and function in oncogenesis: lessons from embryonic development. Curr Cancer Drug Targets 2008; 8:473 - 9; http://dx.doi.org/10.2174/156800908785699315; PMID: 18781894
- Binns KL, Taylor PP, Sicheri F, Pawson T, Holland SJ. Phosphorylation of tyrosine residues in the kinase domain and juxtamembrane region regulates the biological and catalytic activities of Eph receptors. Mol Cell Biol 2000; 20:4791 - 805; http://dx.doi.org/10.1128/MCB.20.13.4791-4805.2000; PMID: 10848605
- Davis TL, Walker JR, Loppnau P, Butler-Cole C, Allali-Hassani A, Dhe-Paganon S. Autoregulation by the juxtamembrane region of the human ephrin receptor tyrosine kinase A3 (EphA3). Structure 2008; 16:873 - 84; http://dx.doi.org/10.1016/j.str.2008.03.008; PMID: 18547520
- Nievergall E, Janes PW, Stegmayer C, Vail ME, Haj FG, Teng SW, et al. PTP1B regulates Eph receptor function and trafficking. J Cell Biol 2010; 191:1189 - 203; http://dx.doi.org/10.1083/jcb.201005035; PMID: 21135139
- Chen Z, Cheng K, Walton Z, Wang Y, Ebi H, Shimamura T, et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature 2012; 483:613 - 7; http://dx.doi.org/10.1038/nature10937; PMID: 22425996
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009; 9:153 - 66; http://dx.doi.org/10.1038/nrc2602; PMID: 19238148
- Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev 2011; 25:2125 - 36; http://dx.doi.org/10.1101/gad.17276711; PMID: 21979375
- Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002; 109:335 - 46; http://dx.doi.org/10.1016/S0092-8674(02)00734-1; PMID: 12015983
- Salminen A, Kauppinen A, Kaarniranta K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal 2012; 24:835 - 45; http://dx.doi.org/10.1016/j.cellsig.2011.12.006; PMID: 22182507
- Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernández-Porras I, Cañamero M, et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011; 19:728 - 39; http://dx.doi.org/10.1016/j.ccr.2011.05.011; PMID: 21665147
- Huck JJ, Zhang M, McDonald A, Bowman D, Hoar KM, Stringer B, et al. MLN8054, an inhibitor of Aurora A kinase, induces senescence in human tumor cells both in vitro and in vivo. Mol Cancer Res 2010; 8:373 - 84; http://dx.doi.org/10.1158/1541-7786.MCR-09-0300; PMID: 20197380
- Zhang W, Peng G, Lin SY, Zhang P. DNA damage response is suppressed by the high cyclin-dependent kinase 1 activity in mitotic mammalian cells. J Biol Chem 2011; 286:35899 - 905; http://dx.doi.org/10.1074/jbc.M111.267690; PMID: 21878640
- Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, Nakayama K, et al. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol 2006; 8:1291 - 7; http://dx.doi.org/10.1038/ncb1491; PMID: 17028578
- Jia M, Souchelnytskyi N, Hellman U, O”Hare M, Jat PS, Souchelnytskyi S. Proteome profiling of immortalization-to-senescence transition of human breast epithelial cells identified MAP2K3 as a senescence-promoting protein which is downregulated in human breast cancer. Proteomics Clin Appl 2010; 4:816 - 28; http://dx.doi.org/10.1002/prca.201000006; PMID: 21137025
- Brumbaugh KM, Otterness DM, Geisen C, Oliveira V, Brognard J, Li X, et al. The mRNA surveillance protein hSMG-1 functions in genotoxic stress response pathways in mammalian cells. Mol Cell 2004; 14:585 - 98; http://dx.doi.org/10.1016/j.molcel.2004.05.005; PMID: 15175154
- Nievergall E, Lackmann M, Janes PW. Eph-dependent cell-cell adhesion and segregation in development and cancer. Cell Mol Life Sci 2012; 69:1813 - 42; http://dx.doi.org/10.1007/s00018-011-0900-6; PMID: 22204021
- Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer 2010; 10:165 - 80; http://dx.doi.org/10.1038/nrc2806; PMID: 20179713
- Vecchi M, Confalonieri S, Nuciforo P, Viganò MA, Capra M, Bianchi M, et al. Breast cancer metastases are molecularly distinct from their primary tumors. Oncogene 2008; 27:2148 - 58; http://dx.doi.org/10.1038/sj.onc.1210858; PMID: 17952122
- Xi HQ, Wu XS, Wei B, Chen L. Aberrant expression of EphA3 in gastric carcinoma: correlation with tumor angiogenesis and survival. J Gastroenterol 2012; 47:785 - 94; http://dx.doi.org/10.1007/s00535-012-0549-4; PMID: 22350700
- Guan M, Liu L, Zhao X, Wu Q, Yu B, Shao Y, et al. Copy number variations of EphA3 are associated with multiple types of hematologic malignancies. Clin Lymphoma Myeloma Leuk 2011; 11:50 - 3; http://dx.doi.org/10.3816/CLML.2011.n.006; PMID: 21454190
- Miao H, Nickel CH, Cantley LG, Bruggeman LA, Bennardo LN, Wang B. EphA kinase activation regulates HGF-induced epithelial branching morphogenesis. J Cell Biol 2003; 162:1281 - 92; http://dx.doi.org/10.1083/jcb.200304018; PMID: 14517207
- Vaidya A, Pniak A, Lemke G, Brown A. EphA3 null mutants do not demonstrate motor axon guidance defects. Mol Cell Biol 2003; 23:8092 - 8; http://dx.doi.org/10.1128/MCB.23.22.8092-8098.2003; PMID: 14585969
- Stephen LJ, Fawkes AL, Verhoeve A, Lemke G, Brown A. A critical role for the EphA3 receptor tyrosine kinase in heart development. Dev Biol 2007; 302:66 - 79; http://dx.doi.org/10.1016/j.ydbio.2006.08.058; PMID: 17046737
- Gray DC, Hoeflich KP, Peng L, Gu Z, Gogineni A, Murray LJ, et al. pHUSH: a single vector system for conditional gene expression. BMC Biotechnol 2007; 7:61; http://dx.doi.org/10.1186/1472-6750-7-61; PMID: 17897455
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002; 296:550 - 3; http://dx.doi.org/10.1126/science.1068999; PMID: 11910072
- Haverty PM. genoset: Provides classes similar to ExpressionSet for copy number analysis. R package version 1.2.0. http://www.bioconductor.org. 2011.