Abstract
Aurora B kinase is an integral regulator of cytokinesis as it stabilizes the intercellular canal within the midbody to ensure proper chromosomal segregation during cell division. Here we identified an E3 ligase subunit, F box protein FBXL2, that by recognizing a calmodulin binding signature within Aurora B, ubiquitinates and removes the kinase from the midbody. Calmodulin, by competing with the F box protein for access to the calmodulin binding signature, protected Aurora B from FBXL2. Calmodulin co-localized with Aurora B on the midbody, preserved Aurora B levels in cells, and stabilized intercellular canals during delayed abscission. Genetic or pharmaceutical depletion of endogenous calmodulin significantly reduced Aurora B protein levels at the midbody resulting in tetraploidy and multi-spindle formation. The calmodulin inhibitor, calmidazolium, reduced Aurora B protein levels resulting in tetraploidy, mitotic arrest, and apoptosis of tumorigenic cells and profoundly inhibiting tumor formation in athymic nude mice. These observations indicate molecular interplay between Aurora B and calmodulin in telophase and suggest that calmodulin acts as a checkpoint sensor for chromosomal segregation errors during mitosis.
Keywords: :
Introduction
A cardinal event during mitosis is proper chromosomal segregation.Citation1,Citation2 During this process, spindle checkpoint proteins ensure the correct distribution of sister chromatids in anaphase before the completion of cytokinesis by abscission in telophase.Citation3-Citation7 However, in the event of delayed or defective chromosome segregation, cytokinesis is unable to conclude due to cleavage furrow regression, resulting in tetraploidy and tumorigenesis.Citation8-Citation10 Recently, studies have unveiled possible mechanisms whereby cells ensure faithful abscission in response to delayed chromosome segregation by lagging or bridged chromosomes.Citation11,Citation12 In this pathway, the presence of Aurora B kinase at the midbody is essential for regulating furrow ingression and abscission.Citation13 During delayed chromosome segregation, Aurora B retains high activity at the midbody, which delays abscission and stabilizes the intercellular canal through downstream effectors such as mitotic kinesin-like protein 1 (Mklp1) until the chromosome bridge is resolved, thus providing last-minute rescue of trapped chromatin and preventing furrow regression and tetraploidy.Citation14 Aurora B is a multi-functional protein that plays many different roles in mitosisCitation15; Aurora B is also subjected to phosphorylation and polyubiquitination during mitotic progression.Citation16,Citation17 Aurora B activation also requires binding to its cofactor inner centromere protein (INCENP),Citation18 autophosphorylation at Thr232 within its activation loop is required for kinase activity, and enzyme dephosphorylation is inhibitory.Citation19,Citation20 The anaphase-promoting complex (APC) through binding to the degradation-targeting proteins Cdh1 and Cdc20 and assembly of an E3 ligase complex comprised of Cullin3 with the Bric-a-brac–Tramtrack–Broad complex Kelch proteins (KLHL21) is sufficient to ubiquitinate Aurora B.Citation21-Citation23 Nonetheless, the molecular factors that regulate activities or concentrations of Aurora B within the midbody during telophase remain limited. Hence, post-translational events within Aurora B protein might be important in governing kinase relocation or levels during early phases of the mitotic program and yet other effectors might regulate local concentrations of Aurora B within the midbody during telophase as feedback inhibitory or activating signals.
The Skp-Cullin- F box (SCF) ubiquitin E3 ligase machinery regulates cell cycle progression, centrosome stability, and mitotic fidelity.Citation24-Citation29 The SCF complex contains a catalytic core consisting of Skp1, Cullin1, and the E2 ubiquitin-conjugating (Ubc) enzyme and a F box component that acts as a receptor targeting numerous substrates via phosphodegron elicited interactions.Citation30-Citation33 The observation that Skp1 interacts with centromere-associated protein E (CENP-E) at the midbody suggests that SCF based ligase complexes might regulate cytokinesis associated proteins although identification of an F-box protein targeting the midbody has not been demonstrated.Citation34 One potential candidate is F box protein FBXL2, which in a calcium-regulated manner induces mitotic arrest, in part, by facilitating ubiquitin mediated-degradation of cyclin D3 in lung epithelia.Citation35,Citation36
Calmodulin (CaM) (16.7kD) is a highly conserved calcium-sensing protein that antagonizes some proteinases and exerts multiple modes of regulatory control during the mitotic program.Citation37-Citation40 CaM binds its targets in a calcium -dependent or calcium -independent manner targeting specific recognition motifs characterized by a basic amphipathic helix, moderate to high helical hydrophobic moment, and a net positive charge.Citation37 Molecular signatures for CaM interaction include an IQ motif (I/LQXXXRGXXXR), a 1–8-14, and 1–5-10 CaM binding motif.Citation37 CaM binds to centrosome protein CP110 to regulate the assembly of microtubules by controlling CaM kinase activities.Citation38-Citation40 Of note, initial studies using electron microscopy localized CaM on the midbody during mitosis and CaM depletion in cells induces mitotic abnormalities and impairs the latter stages of cytokinesis leading to the formation of binucleate cells.Citation41-Citation44 These observations suggest that CaM might have an integral role in modulating cytokinesis however the precise mechanisms for its activity remain elusive.
Here we provide the first evidence of molecular interplay between CaM and an F box protein to regulate mitotic abscission and cytokinesis through Aurora B. We propose an exquisite model whereby CaM, acting as a sensor of chromosome bridges, translocates from microtubules to the midbody to protect Aurora B, which in turn stabilizes the ingressed furrow for delayed abscission. The CaM inhibitor, calmidazolium, was sufficient to deplete Aurora B levels at the midbody and displayed robust anti-tumor activity. We propose that a delicate balance between an F-box protein and CaM regulates mitotic abscission through dynamic control of Aurora B abundance, thereby ensuring faithful mitotic progression.
Results
Aurora B is a CaM binding protein
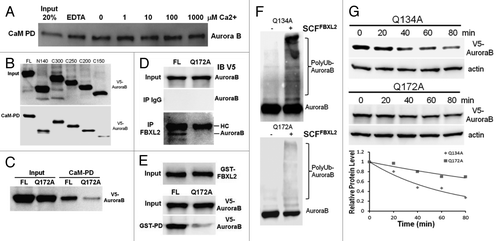
CaM binds and protects some regulatory proteins and is needed for mitosis.Citation44 Aurora B harbors two potential CaM binding IQ motifs within its NH2-terminus suggesting that these motifs may be required for CaM interaction (data not shown). Thus we first tested the hypothesis that Aurora B is a CaM binding protein. Murine lung epithelial (MLE) cell lysates were applied to CaM-sepharose beads to test protein interaction. The pull-down experiments show that Aurora B interacts with CaM in a calcium independent manner (). To map a CaM binding motif, we used a deletional approach where Aurora B constructs were synthesized and tested in the CaM binding assay. Aurora B wild-type (WT) and deletion mutants were all synthesized and expressed in vitro (, upper panel), and the Aurora B C150 mutant showed reduced ability to bind CaM (, lower panel). Sequence analysis of this region revealed a potential CaM-related IQ motif within Aurora B, with Glu172 as a molecular site essential for CaM binding. Our previous studies suggest that the SCF E3 ligase subunit FBXL2 usually docks within a CaM binding site of substrates.Citation45 Interestingly, our screening studies suggested that FBXL2 also targets Aurora B protein for ubiquitination and degradation (Fig. S1). Importantly, FBXL2 utilized this molecular site within the IQ motif to target Aurora B (), as FBXL2 was not able to interact with the Aurora BQ172A mutant (). For confirmation, in vitro ubiquitination assays demonstrated that Aurora BQ172A displayed minimal ubiquitination (), and this variant exhibited a significantly longer t1/2 compared with a related Aurora B mutation (Q134A) ().
Figure 1. Aurora B is a CaM binding protein. (A) CaM-sepharose pull-down (PD) assays showing no effects of exogenous calcium on binding between CaM and endogenous Aurora B. (B) CaM-sepharose PD assays showing binding of in vitro synthesized full-length (FL) or various Aurora B deletion mutants to CaM. (C) CaM-sepharose PD assays showing binding of in vitro synthesized FL or point mutants of Aurora B. (D) Murine lung epithelial (MLE) cells were transfected with FL or a V5-Aurora B variant harboring a point mutation within the IQ motif followed by co-immunoprecipitation of endogenous FBXL2 and V5-immunoblotting. HC = heavy chain. (E) Purified GST-FBXL2 was incubated with in vitro synthesized V5- tagged FL or a V5-Aurora B IQ mutant, followed by GST pull-down. PD products were resolved on SDS-PAGE followed by V5 immunoblotting. (F) In vitro ubiquitination assays. Purified SCF complexes were incubated with Aurora B point mutants and the full complement of ubiquitination reaction components. (G) Aurora B protein half-life determination after expression of V5-Aurora B point mutants (n = 2 experiments).
Aurora BQ172A mutant promotes abscission
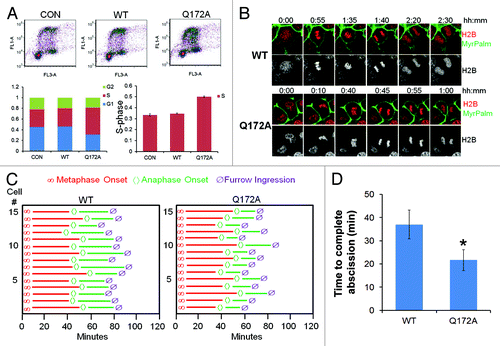
To test the function of this mutant, cells were transfected with either WT Aurora B or Aurora BQ172A, cells labeled with BrdU, and harvested for processing by two-color FACS. The results indicate an increase in a cell population within the S-phase (). Using cells co-expressing mCherry-tagged histone H2B and MyrPalm-mEGFP, we observed that expression of Aurora BQ172A significantly accelerated furrow ingression (). Specifically, while segregating sister cells transfected with WT Aurora B undergo abscission ~35 min after anaphase onset, the cells transfected with Aurora BQ172A exhibited a significantly shorter anaphase (~20min) (). These results suggest that expression of FBXL2-resistant Aurora B variant results in high concentrations within the midbody to allow for accelerated abscission in cells.
Figure 2. Aurora BQ172A mutant promotes abscission. (A) MLE cells were transfected with either wild-type (WT) or a Q172A Aurora B plasmid, transfected cells were processed by BrdU uptake and 7-AAD staining followed by FACS cell cycle analysis. (B) Expression of Aurora B Q172A mutant accellerates furrow ingression in MLE cells expressing H2B-mCherry and MyrPalm-mEGFP. (C) Quantitative analysis of mitosis in MLE cells from video imaging from (I) (n = 15 cells in each condition). (D) Quantification of furrow ingression time from (C).
CaM antagonizes SCFFBXL2 directed Aurora B ubiquitination
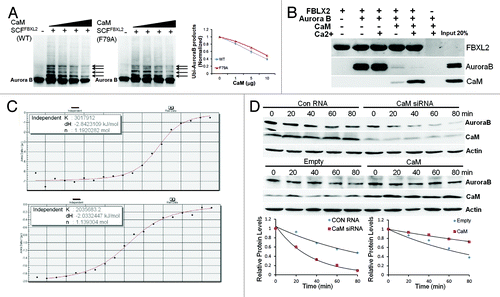
As FBXL2 docks within a CaM binding signature within Aurora B, we next tested if CaM competes with SCFFBXL2 for Aurora B binding and opposes kinase ubiquitination. When Aurora B protein was incubated with purified SCFFBXL2 with the full complement of ubiquitin-related enzymes, increasing amounts of CaM were able to antagonize FBXL2 activity evidenced by decreased ubiquitinated Aurora B species (, arrows). Interestingly, CaM directly interacts with several bulky and hydrophobic residues within the F-box protein NH2-terminus.Citation45 Thus, we tested functionality of a mutant FBXL2F79A protein that does not interact with CaM (, middle panel). The ubiquitination activities of this FBXL2F79A variant were also partially blocked by CaM suggesting that intermolecular competition at the IQ motif, rather than F box protein- CaM binding, may be more relevant to Aurora B regulation (, middle panel). To assess intermolecular competition between FBXL2 and CaM for Aurora B binding within the IQ motif, we used pull-down experiments in the presence or absence of calcium in which FBXL2 was immobilized on beads and used as bait for Aurora B and CaM (). Three negative controls were included: (1) FBXL2-agarose alone was assayed to control for Aurora B and CaM contamination; (2) Aurora B and CaM with calcium were run over empty talon beads, eluted, and proteins were resolved by SDS-PAGE followed by Aurora B and CaM immunoblotting to ensure that associations were FBXL2 specific; and (3) V5 immunoblotting was used as a loading control, to ensure that pull-down experiments had equivalent amounts of FBXL2. The results indicate that not only does FBXL2 directly interact with Aurora B and CaM, but excess CaM disrupts FBXL2 interaction with Aurora B (). Calcium also enhanced interaction between CaM and FBXL2 (). By using a synthetic Aurora B peptide (LQKSRTFEDQR) encoding the putative CaM-binding motif, we observed tight binding between CaM and Aurora B (Kd = 0.33 μM) and similar binding between FBXL2 and Aurora B (Kd = 0.49 μM) using isothermal calorimetry (). Last, CaM overexpression significantly increased Aurora B t1/2, whereas CaM knockdown decreased stability of this protein ().
Figure 3. SCFFBXL2 directed ubiquitination of Aurora B is antagonized by CaM. (A) In vitro ubiquitination assays. Purified SCF complex containing either WT FBXL2 or F79A FBXL2 were incubated with Aurora B and the full complement of ubiquitination reaction components with increasing amounts of CaM (1, 5, 10, 20 ug protein). Arrows indicate polyubiquitinated products. Ubiquitinated Aurora B products were normalized and graphed (right panel, n = 2 experiments). (B) V5-FBXL2-agarose beads were generated and used as bait and incubated with combinations of purified Aurora B or CaM with or without exogenous calcium. After washing of beads (150mM NaCl, 0.1% Triton X-100), proteins were eluted and resolved by SDS-PAGE followed by Aurora B, CaM, and V5 immunoblotting. (C) ITC binding analysis of Aurora B peptide (LQKSRTFEDQR) encoding a CaM-binding motif binding with CaM (upper panel) and FBXL2 (lower panel) in vitro (n = 2 experiments). (D) Aurora B protein half-life determination after CaM overexpression or CaM knockdown using siRNA (n = 2 experiments). Below each panel levels of each protein on immunoblots were quantified densitometrically and shown graphically.
CaM regulates mitosis
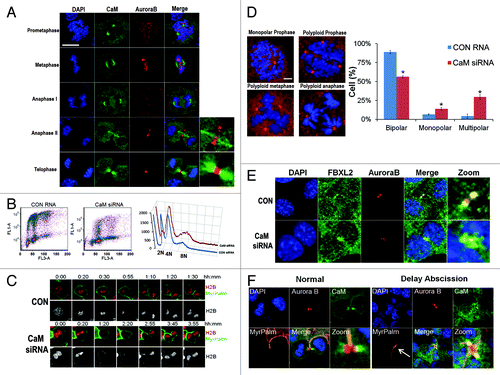
We investigated CaM subcellular localization throughout the cell cycle by co-immunostaining MLE cells (). Aurora B specifically localizes at the midzone and midbody during the late phase of mitosis, whereas CaM localization was quite distinct, residing within the centrosome from prometaphase to the early anaphase (; Fig. S2). However, beginning in late anaphase to the telophase, CaM localized on the α-tubulin bundle on either side of the central midbody (Fig. S3). To test whether CaM is an important mitotic regulator, cells were transfected with CaM siRNA, followed by FACS analysis. The results indicate a significant increase in a cell population within the G2/M phase (). CaM knockdown also tended to reduce the diploidal cell population and increase numbers of polyploidy cells (, right plot). Using cells co-expressing mCherry-tagged histone H2B and MyrPalm-mEGFP, we observed that CaM knockdown induced binucleated cell formation () and multi-polar spindle formation (Fig. S4). Further, abnormal cells with several representative mitotic abnormalities were detected after CaM knockdown suggestive of loss of centrosomal and mitotic spindle integrity (). Other findings with CaM depletion include circular prophase where chromosomal figures are concentrically arranged on monopolar spindles around large centrosomes (, upper left panel), and the appearance of polyploidy during prophase, metaphase and anaphase (). Quantitative analysis revealed a significant increase in these aberrant figures after CaM knockdown (, right). Further, CaM knockdown also induced the formation of bi-nucleate cells where Aurora B levels on the midbody significantly decreased compared with cells transfected with a control RNA (). In studies to visualize the intracellular canal during cytokinesis, segregating sister cells underwent abscission after the completion of chromosome separation with a closed intercellular canal, and CaM localized on the microtubule bundle on both sides of the remnant midbody (, bottom, , left panels). However, in the event of delayed abscission, the two sister cell chromosomes were in extremely close proximity, suggesting of presence of chromosome bridges. Importantly, the data suggest that instead of associating with the microtubule bundle on either side of the central midbody, CaM specifically co-localizes with Aurora B on the midbody, which in turn might stabilize the intercellular canals (, right panel, MyrPalm, white arrow).
Figure 4. CaM regulates mitosis. (A) MLE cells (2 × 105) were plated on 35 mm glass bottom tissue culture dishes for 48 h, cells were then washed with PBS and fixed with 4% paraformaldehyde for 20 min. Cells were co-immunostained for CaM and Aurora B. Nuclei were counterstained using DAPI. Green: CaM, Red: Aurora B, Blue: DAPI. White scale bar indicates 10 μm. (B) MLE cells were transfected with either control (CON) RNA or CaM siRNA for 48 h, transfected cells were processed by BrdU uptake and 7-AAD staining followed by FACS cell cycle analysis. (C) Expression of CaM siRNA leads to furrow regression (see Citation3:45) and binucleate cell formation in MLE cells expressing H2B-mCherry and MyrPalm-mEGFP. (D) MLE cells were transfected with either control RNA or CaM siRNA for 48 h. Cells were then immunostained for γ-tubulin and counterstained with DAPI to visualize the nucleus. Specific chromosomal anomalies are presented in the far right panel. 150 cells were counted from experiments in (D) for abnormal centrosomal phenotypes and are presented graphically. *p < 0.05 vs. con. (E) MLE cells were transfected with either control RNA or CaM siRNA for 48 h. Cells were then washed with PBS and fixed with 4% paraformaldehyde for 20 min. Cells were co-immunostained for FBXL2 and Aurora B. Nuclei were counterstained using DAPI. Green: FBXL2, Red: Aurora B, Blue: DAPI. (F) MLE cells expressing MyrPalm-mEGFP as a plasma membrane marker were plated on 35 mm glass bottom tissue culture dishes for 48 h, cells were then washed with PBS and fixed with 4% paraformaldehyde for 20 min. Cells were co-immunostained for CaM and Aurora B. Nuclei were counterstained using DAPI. Green: CaM, Red: Aurora B, Blue: DAPI. White arrow indicates a stabilized intercellular canal.
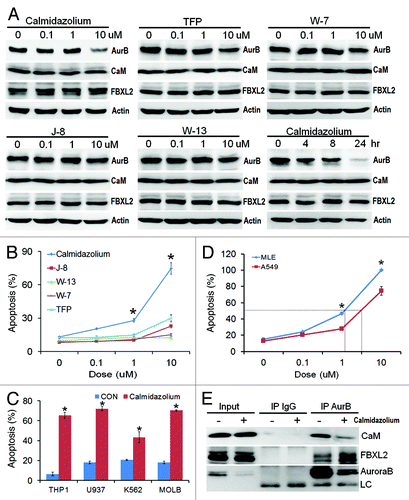
CaM inhibitors inhibit cell proliferation and induce apoptosis
As an alternative approach to modulating Aurora B stability, we tested the hypothesis that inhibition of CaM is sufficient to impair tumor growth. Of several CaM inhibitors examined, calmidazolium, significantly decreased Aurora B protein levels (). Calmidazolium is a highly potent CaM inhibitor (Kd = 0.05 μM, Fig. S5A), inhibited interaction between CaM and Aurora B (Fig. S5B) and potently induced apoptosis of A549 cell and four leukemic lines (IC50 = ~5μM, ). Interestingly, calmidazolium exerted more potent effects on rapidly growing MLE cells (double time ~8 h) (IC50 = ~1μM, ). Mechanistically, calmidazolium significantly reduced the levels of CaM associated with Aurora B in coimmunoprecipitation studies, where levels of FBXL2 associated with Aurora B increased (). Thus, CaM binding to Aurora B is essential for its protein stability and its loss would be predicted to open up occupancy within the kinase for FBXL2. ITC binding studies also confirmed that calmidazolium disrupts binding between CaM and Aurora B (Kd = ~250 μM, Fig. S5B).
Figure 5. CaM inhibitors inhibit cell proliferation and induce apoptosis. (A-B) A549 cells were treated with five different CaM inhibitors for 16 h before assaying for endogenous Aurora B (AurB), CaM, FBXL2 and actin protein by immunoblotting (A). A549 cells were also treated with calmidazolium at 10 μM concentration for 24 h before assaying for endogenous Aurora B, CaM, FBXL2 and actin protein by immunoblotting. A549 cells treated in (A) were also assayed for apoptosis. (C) Four leukemia cell lines were treated with calmidazolium (10 μM) for 16 h, before assaying for apoptosis (n = 3 experiments). (D) A549 cells and faster growing MLE cells were treated with calmidazolium with indicated dose for 16 h, before assaying for apoptosis using Annexin V. IC50 of calmidazolium induced MLE cell apoptosis is ~1 μM, whereas IC50 of calmidazolium induced A549 cell apoptosis is ~5 μM (n = 3 experiments). (E) A549 cells were also treated with calmidazolium (10 um) for 24 h; cells were then collected and subjected to Aurora B immunoprecipitation (AurB) followed by CaM, FBXL2, and Aurora B immunoblotting. LC = light chain.
CaM regulates in vivo tumorogenicity
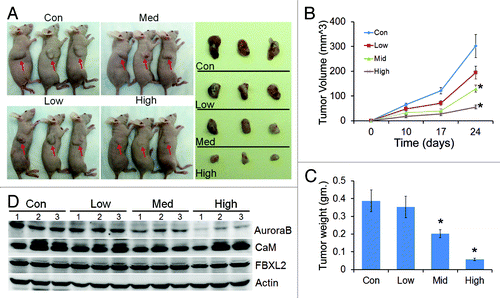
Effect of calmidazolium was also tested in vivo on growth of A549 tumor implants in nude mice. Mice were feed with drug with concentration at 2 μM (low), 10 μM (medium) and 40 μM (high) in the drinking water. Panel A shows representative images of variable sizes of xenografts in three nude mice (arrows) after drug treatment and are displayed after excision (right side panel). Calmidazolium treatment in a dose dependent manner significantly reduced tumor size compared with control implants in athymic nude mice (). Importantly, when tumor tissues were collected from control and treated mice at the end-point analyzed, immunoblotting showed significant decreases in Aurora B protein levels with calmidazolium ().
Figure 6. CaM regulates tumorogenicity. (A) Effect of calmidazolium on growth of A549 tumor implants in nude mice, n = 4 mice/group with drug concentration at 2 μM (low), 10 μM (medium) and 40 μM (high) in the drinking water. Each panel shows representative images of variable sizes of xenografts in three nude mice (arrows) after drug treatment and are displayed after excision (right side panel). (B) Tumor volume measurements over time (n = 4 mice/group, *p < 0.05 vs. con). (C) Tumor tissue from (A) were weighted and graphed (n = 4 mice/group, *p < 0.05 vs. con). (D) Tumors from three mice out of each group were collected at the end-point, and assayed for Aurora B, CaM, and FBXL2 proteins by immunoblotting.
Discussion
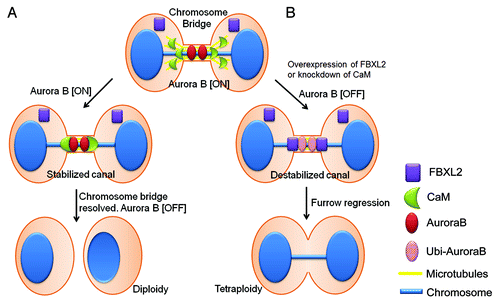
Recently, Steigemann et al. proposed an elegant model by which Aurora B is the key regulator of abscission timing, which responds to chromosome bridge formation by delaying abscission to stabilize the intercellular canal until the chromosome bridge is resolved.Citation12 In essence, this model provides a signal for resolution of trapped chromatin to prevent tetraploidy.Citation14 However, little is known about the mechanism that protects Aurora B and stabilizes the intercellular canal in the event of chromosome bridge formation. Here, we provide evidence that CaM and FBXL2 serve as key regulators of mitotic abscission through Aurora B (). CaM, acting as a sensor of chromosome bridges, protects Aurora B at the midbody to stabilize the ingressed furrow for delayed abscission. However, these activities are opposed by FBXL2, which engages in intermolecular competition with CaM for access to Aurora B within a shared recognition motif. Last, based on the molecular behavior of CaM and FBXL2, we uncovered that chemical inhibition of CaM was highly effective in reducing both Aurora B levels and tumor viability. These observations implicate a role for the F-box protein and CaM as diametric homeostatic sensors that regulate the cytokinesis program through modulation of Aurora B concentrations. This molecular model could be mechanistically significant in providing insight into interventional strategies for carcinogenesis.
Figure 7. Model for mitotic regulation of Aurora B in cells by CaM and FBXL2. (A) In the event of a chromosome bridge and delayed abscission, CaM translocates from microtubules to protect Aurora B on the midbody; this results in delayed abscission and stabilization of the intercellular canal to maintain patency for resolution of the chromosome bridge. (B) In the event of high physiologic levels of FBXL2 or CaM depletion or inhibition, CaM fails to protect Aurora B in response to chromosome bridge formation resulting in premature reduced local concentrations of Aurora B by SCFFBXL2-induced ubiquitination and degradation, leading to tetraploidization.
According to the model proposed by Steigemann et al., Aurora B localization at midbody is important and required for regulating abscission timing. In this model, Aurora B inactivation from dephosphorylation is essential for removal from the midbody and completion of abscission. Here we showed that FBXL2, acting as the receptor component of a prototypical SCF ubiquitin E3 ligase, targets a CaM binding signature within the kinase. FBXL2 recognition of CaM binding motifs is a unique property as many other F-Box proteins recognize phosphodegrons within target substrates.Citation46-Citation48 FBXL2 interacts with a canonical IQ motif within Aurora B whereas a mutant Aurora B with a disrupted IQ motif (Q172A) resists SCFFBXL2 () and its overexpression significantly decreases abscission timing in cells (). This suggests that removal of Aurora B on the midbody is not required for furrow ingression. On the contrary, a FBXL2-resistant stable Aurora B will facilitate the resolution of chromosome-bridge, and accelerate abscission timing. This was supported by our findings showing that overexpression of FBXL2 prematurely degrades Aurora B within the midbody, resulting in reduced ability to resolve the chromosome bridge causing tetraploidization (data not shown).
Overexpression of an Aurora B docking mutant for FBXL2 also produced an impressive increase in the S-phase population (), an unexpected finding that requires further investigation. Since overexpression of Aurora B kinase is associated with many types of cancer with high proliferative rates and poor prognosis,Citation49 we analyzed the SNP database to identify a naturally occurring mutation at the Aurora B Q172 site. Notably, an Aurora B mutation (T176M) was identified that resides within the IQ signature (LQKSRTFEDQR) that led us to hypothesize that this variant is also resistant to FBXL2 ubiquitination. However, this naturally occurring variant was vulnerable to SCFFBXL2 directed ubiquitination in support of other sites (Q172) being essential for protein interaction (Fig. S6).Citation45
Like FBXL2, CaM is also an Aurora B binding protein where its accesses the canonical IQ motif in calcium independent manner (). Accordingly, both FBXL2 and CaM target this signature within Aurora B and bind in the absence of calcium (). Mechanistically, CaM also interacts with FBXL2 in a calcium dependent manner ().Citation45 Thus, the molecular interplay between these effectors, FBXL2 and CaM, and their putative target Aurora B will depend on their relative binding affinities for each other. Our ITC results demonstrate a very tight interaction between CaM and Aurora B (Kd = 0.33 μM, (, upper panel)), and similar interaction between FBXL2 and Aurora B (Kd = 0.49 μM, (, lower panel)) vs. relatively lower affinity between CaM and FBXL2 (Kd = 0.81 μM, data not shown) suggesting that CaM competition with FBXL2 toward occupancy within the IQ motif of Aurora B might be a more functionally relevant mechanism in vitro. Indeed, CaM was able to inhibit SCFFBXL2 directed Aurora B ubiquitination with either wild-type FBXL2 or a FBXL2 mutant (F79A) that lacks ability to interact with CaM. However, this does not rule out CaM’s ability to act as a decoy to directly antagonize and sequester the F-box protein in vivo.
We investigated a biological role for CaM during mitosis. Because of its centrosome localization and role in microtubule assembly,Citation38-Citation40 CaM plays an important role in spindle pore formationCitation50 and chromosome segregation.Citation38,Citation44,Citation51 Consistent with previous studies, we found that CaM knockdown causes mitotic arrest, specifically causing G2/M arrest, inducing binucleate cell formation (), and multi-spindle formation (; Fig. S4). Moreover, CaM depletion significantly decreased Aurora B stability () and importantly depletes Aurora B protein within the midbody () resulting in the formation of binucleate cells. In normal dividing cells, CaM is largely associated with microtubules, and does not colocalize with Aurora B in the midbody (Fig. S3; ); however, in the event of chromosome bridge formation and delayed abscission, in which case the intercellular canal is kept open for the trapped chromatin (, white arrow), CaM specifically colocalizes with Aurora B and appears to stabilize the ingressed furrow for delayed abscission. Hence, CaM appears to act as a sensor of chromosome bridges, translocating from microtubules to protect Aurora B, in turn regulating abscission timing (). Thus, CaM inactivation or depletion in the setting of chromosome bridge formation would be predicted to result in accelerated loss of Aurora B by SCFFBXL2-induced ubiquitination leading to tetraploidization.
For years, many important mitosis proteins have been chosen as drug targets for treating cancer.Citation52 One rationale of designing small molecule inhibitors to target these proteins is that they are highly overexpressed in many type of cancer.Citation53 Interestingly, Aurora B fits perfectly in such profile, as it is overexpressed in many types of cancer with high proliferation rates linked to poor outcomes.Citation49 Investigations of small molecule Aurora B therapeutics are currently on the way, as several Aurora B protein inhibitors are entering clinical trials.Citation54-Citation59 However, all the research so far were all focused on the inhibition of Aurora B activity. When testing CaM inhibitors on human adenocarcinoma cell growth only calmidazolium displayed highly potent inhibitory activity (Kd = 46 nM, Fig. S5A) coupled with potent tumor killing activity. Importantly, we uncovered that this CaM inhibitor substantially decreases Aurora B protein levels and dissociates CaM from Aurora B in vitro and in vivo. This latter mechanism would release a subpopulation of Aurora B molecules vulnerable for SCFFBXL2 complex targeting. Hence, the dissociation of CaM from Aurora B and its targeting by the SCFFBXL2 complex during mitotic abscission on one hand might balance Aurora B levels to regulate cell division but also serve as a novel means to restrict tumor growth (). As CaM inhibition might disrupt other cellular activities resulting in secondary effects in vivo, additional studies are warranted for its therapeutic potential. Nevertheless, by optimizing drug concentrations of calmidazolium, we are able to induce apoptosis of mitotic cells without producing weight loss, systemic toxicity, or triggering behavioral abnormalities in mice (data not shown).
Materials and Methods
Materials
The sources of the transformed murine lung epithelial (MLE) cell line, CaM and GST antibodies were described previously.Citation60,Citation61 Purified SCFFBXL2 was purchased from Abnova. Purified bovine calmodulin, ubiquitin, E1, E2 and cycloheximide were purchased from Calbiochem. Aurora B rabbit polyclonal antibody, γ-tubulin and α-tubulin antibodies were purchased from Cell Signaling and Abcam. Cobalt affinity beads were purchased from Clontech. Lipofectamine 2000, mouse monoclonal V5 antibody, fluorescent labeled antibodies, DAPI nuclear staining kits, the pcDNA3.1D cloning kit, E. coli One Shot competent cells, the pENTR Directional TOPO cloning kits, and the Gateway mammalian expression system were from Invitrogen. FACS kits were purchased from BD Biosciences. The F-box proteins cDNA were purchased from OpenBiosystems. Nucleofector transfection kits were from Amaxa. Adenoviral constructs encoding CaM were generated as described.Citation61 Immobilized protein A/G beads were from Pierce. Annexin V staining kits and the complete proteasome inhibitors were from Roche. All peptides were custom made from CHI scientific. Goat polyclonal FBXL2 antibody, W7, W13, TFP, J-8, calmidazolium, scrambled RNA, and siRNAs were from Santa Cruz Biotechnology. Rabbit polyclonal FBXL2 antibody was custom made from AvivaBioscience. All DNA sequencing was performed by the University of Pittsburgh DNA Core Facility.
Cell culture
MLE cells were cultured in Dulbecco’s Modified Eagle Medium-F12 (Gibco) supplemented with 2–10% fetal bovine serum (DMEM-10). A549 cells were cultured in F12/K medium (Gibco) supplemented with 10% fetal bovine serum. THP1, MOLM, K562 and U937 cells were cultured in RPMI1640 with 10% fetal bovine serum. For half-life studies, cells were treated with cycloheximide (40 μg/ml) at different time points in blank medium. Cells lysates were prepared by brief sonication in 150 mM NaCl, 50 mM Tris, 1.0 mM EDTA, 2 mM dithiothreitol, 0.025% sodium azide, and 1 mM phenylmethylsulfonyl fluoride (Buffer A) at 4°C.
Expression of recombinant protein and RNAi
All plasmids were delivered into cells using nucleofection or lipofectamine 2000.Citation62,Citation63 Cellular expression of green fluorescent tagged plasmids using this device was achieved at > 90%. For siRNA studies, 1 x 106 cells were transfected using lipofectamine 2000 with 10 μg of RNA and harvested after an additional 48 h.
Co-immunoprecipitation and binding assays
250 μg of total protein from cell lysates was precleared with 20 μl of protein A/G beads for 1 h at 4°C. Five µg of primary antibody was added for 18 h incubation at 4°C. 40μl of protein A/G beads were added for an additional 6 h of incubation. Beads were slowly centrifuged and washed five times using 50 mM HEPES, 150 mM NaCl, 0.5 mM EGTA, 50 mM NaF, 10 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 20 µM leupeptin, and 1% (v/v) Triton X-100 (RIPA) buffer, as described.Citation64 The beads were heated at 100°C for 5 min. with 80 µl of protein sample buffer prior to SDS-PAGE and immunoblotting. For CaM binding assays, CaM sepharose beads were incubated with V5-FBXL2 or Aurora B transfected cell lysates (50 μg), with or without Ca2+ at 4°C for 2 h. Eluted products were processed for SDS-PAGE and immunoblotting as described.Citation61
Microscopy and immunostaining
All the microscopy work was done on a Nikon A1 confocal microscope using a 60 × oil objective. The microscope was equipped with Ti Perfect Focus system and Tokai Hit live cell chamber providing a humidified atmosphere at 37°C with 5% CO2. For long-term movie acquisition, sample illumination was kept to minimum to reduce the effect of photobleaching. Transfected cells (2 × 105) were plated at 70% confluence on 35mm MatTek glass bottom culture dishes. Immunofluorescent cell imaging was performed on a Nikon A1 confocal microscope using 405 nm, 458 nm, 488 nm, 514 nm or 647 nm wavelengths. All experiments were done with a 60 x oil differential interference contrast objective lens. Cells were washed with PBS and fixed with 4% paraformaldehyde for 20 min, then exposed to 2% BSA, 1:500 primary antibodies, and 1:1000 Alexa 488, Alexa 567 or Alexa 647 labeled chicken anti-mouse, Donkey anti-goat or Goat anti-rabbit secondary antibody sequentially for immunostaining. Image analysis was by Nikon NIS-element and ImageJ software.
In vitro ubiquitin conjugation assays
The ubiquitination of V5-Aurora B was performed in a volume of 25 μl containing 50 mM Tris pH 7.6, 5 mM MgCl2, 0.6 mM DTT, 2 mM ATP, 1.5 ng/μl E1, 10 ng/μl Ubc5, 10 ng/μl Ubc7, 1μg/μl ubiquitin (Calbiochem), 1 μM ubiquitin aldehyde, 4–16 μl of purified Cullin1, Skp1, Rbx1, and in vitro synthesized FBXL2. Reaction products were processed for V5 immunoblotting.
Quantitative RT–PCR, cloning and mutagenesis
Total RNA was isolated and reverse transcription was performed followed by quantitative real-time PCR with SYBR Green qPCR mixture as described.Citation65 PCR based approaches were used to clone different F-box proteins into pcDNA3.1D/v5-his (Invitrogen) for constitutive expression in cells. All mutant constructs were generated using PCR-based approaches using appropriate primers or site-directed mutagenesis.
Cell cycle and apoptosis analysis
Transfected cells were incubated with BrdU (20 μM) for 40 min, fixed, and stained following manufacturer’s protocols (BD Biosciences). FACS samples were analyzed with the AccuriC6 system. DNA content was analyzed using FCS3 express software (De Novo Software). When analyzing the cell cycle, a gate for 7AAD was set to exclude polyploidy cells. Otherwise cells were counted and the percentage of cells with 2N, 4N and 8N DNA content was expressed as a percentage of total cells. Cells were also stained with annexin V for 15 min following the manufacturer’s protocol (Roche). Apoptotic cells were counted, and apoptotic cells were expressed as a percentage of total cells.
Animal studies
Nude/Nude mice (purchased from Charles River) were acclimated at the University of Pittsburgh Animal Care Facility and maintained according to all federal guidelines and under the University of Pittsburgh Institutional Animal Care and Use Committee (IACUC) approved protocols. Mice were deeply anesthetized with ketamine (80–100 mg/kg intraperitoneally (i.p.) and xylazine (10 mg/kg i.p.), followed by i.p. injection of 5 x106 A549 cells (100μl) into the right shoulder. For calmidazolium treatment, an aliquot of calmidazolium stock solution (20mM) was added to drinking water (containing 2% sucrose) to maintain the final drug concentration at 2μM, 10μM and 40μM. Mice were closely monitored every three days; tumor volume was calculated using a formula length × width × height × π/6.
Statistical Analysis
Statistical comparisons were performed with the Prism program, version 4.03 (GraphPad Software, Inc.) using an ANOVA 1 or an unpaired 2 t-test with p < 0.05 indicative of significance.
Additional material
Download Zip (2.8 MB)Acknowledgments
We thank D.W. Gerlich for providing pH2B-mCherry-IRES-puro2 and pMyrPalm-mEGFP plasmids. This material is based upon work supported, in part, by the US Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development. This work was supported by a Merit Review Award from the US Department of Veterans Affairs and National Institutes of Health R01 grants HL116472 (to B.B.C.), HL096376, HL097376 and HL098174 (to R.K.M.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here:
http://www.landesbioscience.com/journals/cc/article/23586/
Related Research Data
References
- Margolis RL. Tetraploidy and tumor development. Cancer Cell 2005; 8:353 - 4; http://dx.doi.org/10.1016/j.ccr.2005.10.017; PMID: 16286243
- Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005; 437:1043 - 7; http://dx.doi.org/10.1038/nature04217; PMID: 16222300
- Gortchakov AA, Eggert H, Gan M, Mattow J, Zhimulev IF, Saumweber H. Chriz, a chromodomain protein specific for the interbands of Drosophila melanogaster polytene chromosomes. Chromosoma 2005; 114:54 - 66; http://dx.doi.org/10.1007/s00412-005-0339-3; PMID: 15821938
- Cimini D, Wan X, Hirel CB, Salmon ED. Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors. Curr Biol 2006; 16:1711 - 8; http://dx.doi.org/10.1016/j.cub.2006.07.022; PMID: 16950108
- Wilsker D, Chung JH, Bunz F. Chk1 suppresses bypass of mitosis and tetraploidization in p53-deficient cancer cells. Cell Cycle 2012; 11:1564 - 72; http://dx.doi.org/10.4161/cc.19944; PMID: 22433954
- Bannon JH, O’Donovan DS, Kennelly SM, Mc Gee MM. The peptidyl prolyl isomerase cyclophilin A localizes at the centrosome and the midbody and is required for cytokinesis. Cell Cycle 2012; 11:1340 - 53; http://dx.doi.org/10.4161/cc.19711; PMID: 22421161
- Vazquez-Martin A, Sauri-Nadal T, Menendez OJ, Oliveras-Ferraros C, Cufí S, Corominas-Faja B, et al. Ser2481-autophosphorylated mTOR colocalizes with chromosomal passenger proteins during mammalian cell cytokinesis. Cell Cycle 2012; 11:4211 - 21; http://dx.doi.org/10.4161/cc.22551; PMID: 23095638
- Weaver BA, Silk AD, Cleveland DW. Cell biology: nondisjunction, aneuploidy and tetraploidy. [discussion E.] Nature 2006; 442:E9 - 10, discussion E10; http://dx.doi.org/10.1038/nature05139; PMID: 16915240
- Li Y, Xu FL, Lu J, Saunders WS, Prochownik EV. Widespread genomic instability mediated by a pathway involving glycoprotein Ib alpha and Aurora B kinase. J Biol Chem 2010; 285:13183 - 92; http://dx.doi.org/10.1074/jbc.M109.084913; PMID: 20157117
- Nicholson JM, Cimini D. Doubling the deck: Tetraploidy induces chromosome shuffling and cancer. Cell Cycle 2012; 11:3354 - 5; http://dx.doi.org/10.4161/cc.21850; PMID: 22918235
- Norden C, Mendoza M, Dobbelaere J, Kotwaliwale CV, Biggins S, Barral Y. The NoCut pathway links completion of cytokinesis to spindle midzone function to prevent chromosome breakage. Cell 2006; 125:85 - 98; http://dx.doi.org/10.1016/j.cell.2006.01.045; PMID: 16615892
- Steigemann P, Wurzenberger C, Schmitz MH, Held M, Guizetti J, Maar S, et al. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 2009; 136:473 - 84; http://dx.doi.org/10.1016/j.cell.2008.12.020; PMID: 19203582
- Guse A, Mishima M, Glotzer M. Phosphorylation of ZEN-4/MKLP1 by aurora B regulates completion of cytokinesis. Curr Biol 2005; 15:778 - 86; http://dx.doi.org/10.1016/j.cub.2005.03.041; PMID: 15854913
- Chen CT, Doxsey S. A last-minute rescue of trapped chromatin. Cell 2009; 136:397 - 9; http://dx.doi.org/10.1016/j.cell.2009.01.028; PMID: 19203574
- Shao H, Ma C, Zhang X, Li R, Miller AL, Bement WM, et al. Aurora B regulates spindle bipolarity in meiosis in vertebrate oocytes. Cell Cycle 2012; 11:2672 - 80; http://dx.doi.org/10.4161/cc.21016; PMID: 22751439
- Yabuta N, Mukai S, Okada N, Aylon Y, Nojima H. The tumor suppressor Lats2 is pivotal in Aurora A and Aurora B signaling during mitosis. Cell Cycle 2011; 10:2724 - 36; http://dx.doi.org/10.4161/cc.10.16.16873; PMID: 21822051
- Archambault V, Carmena M. Polo-like kinase-activating kinases: Aurora A, Aurora B and what else?. Cell Cycle 2012; 11:1490 - 5; http://dx.doi.org/10.4161/cc.19724; PMID: 22433949
- Bolton MA, Lan W, Powers SE, McCleland ML, Kuang J, Stukenberg PT. Aurora B kinase exists in a complex with survivin and INCENP and its kinase activity is stimulated by survivin binding and phosphorylation. Mol Biol Cell 2002; 13:3064 - 77; http://dx.doi.org/10.1091/mbc.E02-02-0092; PMID: 12221116
- Yasui Y, Urano T, Kawajiri A, Nagata K, Tatsuka M, Saya H, et al. Autophosphorylation of a newly identified site of Aurora-B is indispensable for cytokinesis. J Biol Chem 2004; 279:12997 - 3003; http://dx.doi.org/10.1074/jbc.M311128200; PMID: 14722118
- Sun L, Gao J, Dong X, Liu M, Li D, Shi X, et al. EB1 promotes Aurora-B kinase activity through blocking its inactivation by protein phosphatase 2A. Proc Natl Acad Sci U S A 2008; 105:7153 - 8; http://dx.doi.org/10.1073/pnas.0710018105; PMID: 18477699
- Nguyen HG, Chinnappan D, Urano T, Ravid K. Mechanism of Aurora-B degradation and its dependency on intact KEN and A-boxes: identification of an aneuploidy-promoting property. Mol Cell Biol 2005; 25:4977 - 92; http://dx.doi.org/10.1128/MCB.25.12.4977-4992.2005; PMID: 15923616
- Sumara I, Quadroni M, Frei C, Olma MH, Sumara G, Ricci R, et al. A Cul3-based E3 ligase removes Aurora B from mitotic chromosomes, regulating mitotic progression and completion of cytokinesis in human cells. Dev Cell 2007; 12:887 - 900; http://dx.doi.org/10.1016/j.devcel.2007.03.019; PMID: 17543862
- Maerki S, Olma MH, Staubli T, Steigemann P, Gerlich DW, Quadroni M, et al. The Cul3-KLHL21 E3 ubiquitin ligase targets aurora B to midzone microtubules in anaphase and is required for cytokinesis. J Cell Biol 2009; 187:791 - 800; http://dx.doi.org/10.1083/jcb.200906117; PMID: 19995937
- Tyers M, Willems AR. One ring to rule a superfamily of E3 ubiquitin ligases. Science 1999; 284:601 - , 603-4; http://dx.doi.org/10.1126/science.284.5414.601; PMID: 10328744
- Guardavaccaro D, Frescas D, Dorrello NV, Peschiaroli A, Multani AS, Cardozo T, et al. Control of chromosome stability by the beta-TrCP-REST-Mad2 axis. Nature 2008; 452:365 - 9; http://dx.doi.org/10.1038/nature06641; PMID: 18354482
- Seki A, Coppinger JA, Du H, Jang CY, Yates JR 3rd, Fang G. Plk1- and beta-TrCP-dependent degradation of Bora controls mitotic progression. J Cell Biol 2008; 181:65 - 78; http://dx.doi.org/10.1083/jcb.200712027; PMID: 18378770
- Gusti A, Baumberger N, Nowack M, Pusch S, Eisler H, Potuschak T, et al. The Arabidopsis thaliana F-box protein FBL17 is essential for progression through the second mitosis during pollen development. PLoS One 2009; 4:e4780; http://dx.doi.org/10.1371/journal.pone.0004780; PMID: 19277118
- D’Angiolella V, Donato V, Vijayakumar S, Saraf A, Florens L, Washburn MP, et al. SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature 2010; 466:138 - 42; http://dx.doi.org/10.1038/nature09140; PMID: 20596027
- Coon TA, Glasser JR, Mallampalli RK, Chen BB. Novel E3 ligase component FBXL7 ubiquitinates and degrades Aurora A, causing mitotic arrest. Cell Cycle 2012; 11:721 - 9; http://dx.doi.org/10.4161/cc.11.4.19171; PMID: 22306998
- Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 2002; 416:703 - 9; http://dx.doi.org/10.1038/416703a; PMID: 11961546
- Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol 2004; 5:739 - 51; http://dx.doi.org/10.1038/nrm1471; PMID: 15340381
- Cenciarelli C, Chiaur DS, Guardavaccaro D, Parks W, Vidal M, Pagano M. Identification of a family of human F-box proteins. Curr Biol 1999; 9:1177 - 9; http://dx.doi.org/10.1016/S0960-9822(00)80020-2; PMID: 10531035
- Ilyin GP, Rialland M, Glaise D, Guguen-Guillouzo C. Identification of a novel Skp2-like mammalian protein containing F-box and leucine-rich repeats. FEBS Lett 1999; 459:75 - 9; http://dx.doi.org/10.1016/S0014-5793(99)01211-9; PMID: 10508920
- Liu D, Zhang N, Du J, Cai X, Zhu M, Jin C, et al. Interaction of Skp1 with CENP-E at the midbody is essential for cytokinesis. Biochem Biophys Res Commun 2006; 345:394 - 402; http://dx.doi.org/10.1016/j.bbrc.2006.04.062; PMID: 16682006
- Chen BB, Glasser JR, Coon TA, Mallampalli RK. F-box protein FBXL2 exerts human lung tumor suppressor-like activity by ubiquitin-mediated degradation of cyclin D3 resulting in cell cycle arrest. Oncogene 2012; 31:2566 - 79; http://dx.doi.org/10.1038/onc.2011.432; PMID: 22020328
- Chen BB, Glasser JR, Coon TA, Mallampalli RK. FBXL2 is a ubiquitin E3 ligase subunit that triggers mitotic arrest. Cell Cycle 2011; 10:3487 - 94; http://dx.doi.org/10.4161/cc.10.20.17742; PMID: 22024926
- Rhoads AR, Friedberg F. Sequence motifs for calmodulin recognition. FASEB J 1997; 11:331 - 40; PMID: 9141499
- Moser MJ, Flory MR, Davis TN. Calmodulin localizes to the spindle pole body of Schizosaccharomyces pombe and performs an essential function in chromosome segregation. J Cell Sci 1997; 110:1805 - 12; PMID: 9264467
- Zhu Q, Liu T, Clarke M. Calmodulin and the contractile vacuole complex in mitotic cells of Dictyostelium discoideum. J Cell Sci 1993; 104:1119 - 27; PMID: 8314896
- Sweet SC, Rogers CM, Welsh MJ. Calmodulin is associated with microtubules forming in PTK1 cells upon release from nocodazole treatment. Cell Motil Cytoskeleton 1989; 12:113 - 22; http://dx.doi.org/10.1002/cm.970120206; PMID: 2713899
- Welsh MJ, Dedman JR, Brinkley BR, Means AR. Tubulin and calmodulin. Effects of microtubule and microfilament inhibitors on localization in the mitotic apparatus. J Cell Biol 1979; 81:624 - 34; http://dx.doi.org/10.1083/jcb.81.3.624; PMID: 379022
- Liu T, Williams JG, Clarke M. Inducible expression of calmodulin antisense RNA in Dictyostelium cells inhibits the completion of cytokinesis. Mol Biol Cell 1992; 3:1403 - 13; PMID: 1493336
- Yu YY, Dai G, Pan FY, Chen J, Li CJ. Calmodulin regulates the post-anaphase reposition of centrioles during cytokinesis. Cell Res 2005; 15:548 - 52; http://dx.doi.org/10.1038/sj.cr.7290324; PMID: 16045818
- Tsang WY, Spektor A, Luciano DJ, Indjeian VB, Chen Z, Salisbury JL, et al. CP110 cooperates with two calcium-binding proteins to regulate cytokinesis and genome stability. Mol Biol Cell 2006; 17:3423 - 34; http://dx.doi.org/10.1091/mbc.E06-04-0371; PMID: 16760425
- Chen BB, Coon TA, Glasser JR, Mallampalli RK. Calmodulin antagonizes a calcium-activated SCF ubiquitin E3 ligase subunit, FBXL2, to regulate surfactant homeostasis. Mol Cell Biol 2011; 31:1905 - 20; http://dx.doi.org/10.1128/MCB.00723-10; PMID: 21343341
- Liu C, Kato Y, Zhang Z, Do VM, Yankner BA, He X. beta-Trcp couples beta-catenin phosphorylation-degradation and regulates Xenopus axis formation. Proc Natl Acad Sci U S A 1999; 96:6273 - 8; http://dx.doi.org/10.1073/pnas.96.11.6273; PMID: 10339577
- Hansen DV, Loktev AV, Ban KH, Jackson PK. Plk1 regulates activation of the anaphase promoting complex by phosphorylating and triggering SCFbetaTrCP-dependent destruction of the APC Inhibitor Emi1. Mol Biol Cell 2004; 15:5623 - 34; http://dx.doi.org/10.1091/mbc.E04-07-0598; PMID: 15469984
- Watanabe N, Arai H, Iwasaki J, Shiina M, Ogata K, Hunter T, et al. Cyclin-dependent kinase (CDK) phosphorylation destabilizes somatic Wee1 via multiple pathways. Proc Natl Acad Sci U S A 2005; 102:11663 - 8; http://dx.doi.org/10.1073/pnas.0500410102; PMID: 16085715
- Green MR, Woolery JE, Mahadevan D. Update on Aurora Kinase Targeted Therapeutics in Oncology. Expert Opin Drug Discov 2011; 6:291 - 307; http://dx.doi.org/10.1517/17460441.2011.555395; PMID: 21556291
- Stirling DA, Welch KA, Stark MJ. Interaction with calmodulin is required for the function of Spc110p, an essential component of the yeast spindle pole body. EMBO J 1994; 13:4329 - 42; PMID: 7925277
- Sundberg HA, Goetsch L, Byers B, Davis TN. Role of calmodulin and Spc110p interaction in the proper assembly of spindle pole body compenents. J Cell Biol 1996; 133:111 - 24; http://dx.doi.org/10.1083/jcb.133.1.111; PMID: 8601600
- Sanhaji M, Friel CT, Wordeman L, Louwen F, Yuan J. Mitotic centromere-associated kinesin (MCAK): a potential cancer drug target. Oncotarget 2011; 2:935 - 47; PMID: 22249213
- Blanchard Z, Malik R, Mullins N, Maric C, Luk H, Horio D, et al. Geminin overexpression induces mammary tumors via suppressing cytokinesis. Oncotarget 2011; 2:1011 - 27; PMID: 22184288
- Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med 2004; 10:262 - 7; http://dx.doi.org/10.1038/nm1003; PMID: 14981513
- Gadea BB, Ruderman JV. Aurora kinase inhibitor ZM447439 blocks chromosome-induced spindle assembly, the completion of chromosome condensation, and the establishment of the spindle integrity checkpoint in Xenopus egg extracts. Mol Biol Cell 2005; 16:1305 - 18; http://dx.doi.org/10.1091/mbc.E04-10-0891; PMID: 15616188
- Soncini C, Carpinelli P, Gianellini L, Fancelli D, Vianello P, Rusconi L, et al. PHA-680632, a novel Aurora kinase inhibitor with potent antitumoral activity. Clin Cancer Res 2006; 12:4080 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-05-1964; PMID: 16818708
- Hardwicke MA, Oleykowski CA, Plant R, Wang J, Liao Q, Moss K, et al. GSK1070916, a potent Aurora B/C kinase inhibitor with broad antitumor activity in tissue culture cells and human tumor xenograft models. Mol Cancer Ther 2009; 8:1808 - 17; http://dx.doi.org/10.1158/1535-7163.MCT-09-0041; PMID: 19567821
- Hoellein A, Pickhard A, von Keitz F, Schoeffmann S, Piontek G, Rudelius M, et al. Aurora kinase inhibition overcomes cetuximab resistance in squamous cell cancer of the head and neck. Oncotarget 2011; 2:599 - 609; PMID: 21865609
- Marxer M, Foucar CE, Man WY, Chen Y, Ma HT, Poon RY. Tetraploidization increases sensitivity to Aurora B kinase inhibition. Cell Cycle 2012; 11:2567 - 77; http://dx.doi.org/10.4161/cc.20947; PMID: 22722494
- Ray NB, Durairaj L, Chen BB, McVerry BJ, Ryan AJ, Donahoe M, et al. Dynamic regulation of cardiolipin by the lipid pump Atp8b1 determines the severity of lung injury in experimental pneumonia. Nat Med 2010; 16:1120 - 7; http://dx.doi.org/10.1038/nm.2213; PMID: 20852622
- Chen BB, Mallampalli RK. Calmodulin binds and stabilizes the regulatory enzyme, CTP: phosphocholine cytidylyltransferase. J Biol Chem 2007; 282:33494 - 506; http://dx.doi.org/10.1074/jbc.M706472200; PMID: 17804406
- Agassandian M, Chen BB, Schuster CC, Houtman JC, Mallampalli RK. 14-3-3zeta escorts CCTalpha for calcium-activated nuclear import in lung epithelia. FASEB J 2010; 24:1271 - 83; http://dx.doi.org/10.1096/fj.09-136044; PMID: 20007511
- Chen BB, Mallampalli RK. Masking of a nuclear signal motif by monoubiquitination leads to mislocalization and degradation of the regulatory enzyme cytidylyltransferase. Mol Cell Biol 2009; 29:3062 - 75; http://dx.doi.org/10.1128/MCB.01824-08; PMID: 19332566
- Mallampalli RK, Ryan AJ, Salome RG, Jackowski S. Tumor necrosis factor-alpha inhibits expression of CTP:phosphocholine cytidylyltransferase. J Biol Chem 2000; 275:9699 - 708; http://dx.doi.org/10.1074/jbc.275.13.9699; PMID: 10734122
- Butler PL, Mallampalli RK. Cross-talk between remodeling and de novo pathways maintains phospholipid balance through ubiquitination. J Biol Chem 2010; 285:6246 - 58; http://dx.doi.org/10.1074/jbc.M109.017350; PMID: 20018880