Abstract
It is well established that the p53 tumor suppressor plays a crucial role in controlling cell proliferation and apoptosis upon various types of stress. There is increasing evidence showing that p53 is also critically involved in various metabolic pathways, both in tumor and normal cells. Here, we have identified a novel p53 metabolic target pantothenate kinase-1 (PANK1) via ChIP-on-chip. PanK1 catalyzes the rate-limiting step for CoA synthesis and, therefore, controls intracellular CoA content; Pank1-knockout mice exhibit defect in β-oxidation and gluconeogenesis in the liver after starvation due to insufficient CoA levels. We demonstrated that PANK1 gene is a direct transcriptional target of p53. Although DNA damage-induced p53 upregulates PanK1 expression, depletion of PanK1 expression does not affect p53-dependent growth arrest or apoptosis. Interestingly, upon glucose starvation, PanK1 expression is significantly reduced in HCT116 p53 (−/−) but not in HCT116 p53 (+/+) cells, suggesting that p53 is required to maintain PanK1 expression under metabolic stress conditions. Moreover, by using p53-mutant mice, we observed that, similar to the case in Pank1-knockout mice, gluconeogenesis is partially impaired in p53-null mice. Together, our findings show that p53 plays an important role in regulating energy homeostasis through transcriptional control of PANK1, independent of its canonical functions in apoptosis and cell cycle arrest.
Introduction
The p53 protein is a tumor suppressor whose primary function is to maintain the integrity of the genome through cell fate control.Citation1 Classically, p53 is activated under genotoxic stress and acts as a transcription factor to regulate the expression of downstream apoptotic or growth-arrest targets.Citation2,Citation3 However, recent works have shown that functions of p53 go far beyond cell death and growth arrest, and now also include metabolism, autophagy, senescence and aging.Citation4,Citation5
Proper metabolic function is crucial to the viability of normal cells. Interestingly, metabolism also plays a key role in the adaptability of neoplastic cells, the process of tumorigenesis and genome stability, and p53 has been recently linked to these aspects of metabolic functions.Citation6 p53 has been shown to regulate the balance between glycolysis and mitochondrial respiration through transcriptional control of several metabolic genes. SCO2 gene, a transcription target of p53, codes for a protein that assembles and regulates the cytochrome C oxidase complex of the electron transport chain. Synthesis of the Sco2 protein increases cellular mitochondrial respiration as a mean of energy production while decreasing the need for glycolysis. Interestingly, loss of p53 in cancer cells correlates with decreased SCO2 expression, which leads to a shift of energy production toward glycolysis, a phenomenon known as the Warburg effect.Citation7 p53 also represses the expression of glucose transporters GLUT1/3/4, thereby decreasing cellular uptake of glucose and subsequent glycolysis.Citation8 In addition, p53 negatively regulates the transcription of the pyruvate dehydrogenase kinase PDK2, which phosphorylates and inhibits pyruvate dehydrogenase (PDH). By relieving the inhibitory effect on PDH, p53 facilitates the conversion of pyruvate into acetyl-CoA by PDH as substrate for mitochondrial respiration.Citation9
Furthermore, p53 has been implicated in metabolic pathways that regulate the synthesis of cellular antioxidant glutathione, which protects cells against reactive oxygen species (ROS). TIGAR, a p53 target, inhibits glycolysis by decreasing the level of fructose-2,6,-bisphosphate and shunts glucose into the pentose phosphate pathway for NADPH production that leads to glutathione recycling.Citation10 To a similar effect, p53 can upregulate the expression of glutaminase GLS2, which converts glutamine to glutamate for glutathione synthesis.Citation11,Citation12
Aside from genotoxic stress, cells may also activate p53 under metabolic stress. Studies have shown that p53 is induced under energy and nutrient deprivation by energy-sensing AMP-activated protein kinase (AMPK), and may lead to subsequent activation of growth arrest pathways.Citation13,Citation14 In addition, p53 also induces autophagy in the face of prolonged starvation to generate sufficient energy for survival through regulation on LC3.Citation15 Together, this evidence suggests that p53, besides being a tumor suppressor, may also play a prosurvival role of maintaining metabolic homeostasis under energy crisis.
In light of this recent evidence, which points toward the significance of p53 in metabolic regulation, we performed a chromatin-immunoprecipitation-on-chip (ChIP-on-chip) analysis using p53 as bait and identified pantothenate kinase-1 (PANK1) as a potential metabolic target of p53. PanK1 is a member of a family of proteins (PanK family) that are involved in the biosynthesis of coenzyme A (CoA).Citation16,Citation17 CoA is an essential and ubiquitous cofactor that is obligatory in over 100 different intermediary metabolic reactions, including those involved in fatty acid metabolism, tricarboxylic acid cycle and cholesterol and bile acid synthesis. The PanK family of proteins catalyze the first and rate-limiting step of CoA synthesis (phosphorylation of precursor pantothenate), and thus control the cellular content of CoA.Citation18
There are two PanK isoforms encoded by the PANK1 gene, PanK1α and PanK1β, and two other isoforms (PanK2 and PanK3) encoded by two other distinct genes. PanK1 is most highly expressed in the liver and corresponds to the liver possessing the highest concentration of CoA among tissues.Citation19 This is consistent with the fact that a plethora of tissue-specific metabolic functions of the liver require CoA, which includes β-oxidation, ketogenesis, gluconeogenesis and sterol synthesis. CoA levels in tissues, especially in the liver, change under metabolic stress in order to utilize alternative fuel sources to meet energy demands. In cases such as starvation and diabetes, increase in the liver intracellular CoA concentration is required to promote sufficient conversion of stored fatty acids and amino acids into ketone bodies and glucose to supply the rest of the body.Citation20
Here, we report that PanK1 is a transcriptional target of p53, but does not contribute to p53-dependent apoptosis or growth arrest. Instead, we observed that p53 is required to maintain PanK1 expression, suggesting a crucial role of p53 in regulating metabolic pathways that is independent of its canonical functions in apoptosis and cell growth arrest.
Results
PANK1 gene is a p53 transcriptional target
PANK1 promoter was identified in a ChIP-on-chip assay using p53-transfected H1299 cells. The potential p53 binding site (BS) on PANK1 promoter was approximated to the 5′-end of exon 1 of PANK1α isoform (). To verify the binding of p53 to the PANK1 promoter, we performed chromatin-immunoprecipitation (ChIP) in H1299 cells transfected with p53 expression vector, followed by quantitative real-time PCR (qPCR) amplification of the pulled-down DNA fragments. The relative p53 enrichment at the potential PANK1 p53 BS was similar to those at promoters of well-established p53 metabolic targets (TIGAR, SCO2, and GLS2), while no enrichment was detected at the region 2 kb upstream of the potential PANK1 p53 BS, proving that p53 indeed binds to the PANK1 promoter ().
Figure 1.PANK1 gene is a p53 transcriptional target. (A) Schematic representation of the human PANK1 gene and its promoter. The potential p53 binding sites (p53 BS) are located in the 5′ region of PANK1 exon 1α. TSS represents the transcriptional start site. Luciferase construct pLucA containing the p53 BS and construct pLucB lacking the p53 BS are derived from the potential PANK1 promoter region for luciferase reporter gene assays in (C and D). In addition, luciferase constructs containing mutations of the potential p53 consensus sequences are denoted as pLucA’1, pLucA’23, pLucA’45, and pLucA’2345. (B) The left panel shows ChIP-qPCR analysis of p53 enrichment at the promoter regions of PANK1, TIGAR, SCO2, GLS2, and p21 in H1299 cells expressing p53. GAPDH and a region located 2 kb upstream of the PANK1 p53 BS were used as negative controls. The right panel shows semi-quantitative analysis of p53 binding to the corresponding regions in the left panel. (C) p53 activates luciferase activity of reporter construct containing p53-binding elements in the PANK1 promoter. H1299 cells were transfected with control vector (pCIN4), increasing amounts of p53 expression vector, or vector expressing binding-deficient p53 mutant R175H along with luciferase constructs pLucA or pLucB for 24 h before measuring luciferase activity. (D) Mutations of p53-binding elements on the reporter construct abolished p53-induced luciferase activity. H1299 cells were co-transfected with p53 expression vector and various mutated luciferase constructs for 24 h before measuring luciferase activity. (E) Gel shift assay showing p53 binding on oligonucleotide containing p53-binding sites in the PANK1 promoter region. The DNA binding activity of purified p53 protein was enhanced with C-terminal p53 antibody pAb421. Specificity of the binding was verified by competition with non-radiolabeled wild-type and mutant probes.
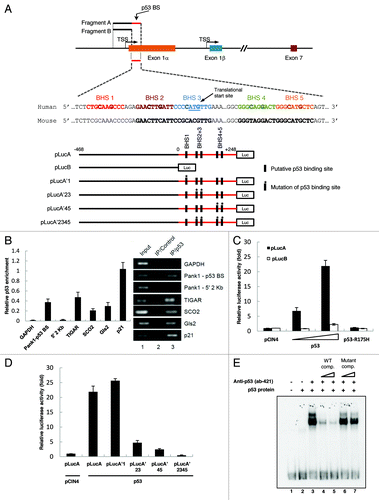
Since p53 is a transcription factor, we tested whether or not p53 is able to activate transcription through the PANK1 promoter using luciferase assay. Luciferase constructs pLucA containing fragment A of PANK1 promoter (as depicted in ) with the potential p53 BS and pLucB containing fragment B without the p53 BS were generated. Co-transfection of pLucA with WT p53 expression vector into p53-null H1299 cells increased luciferase activity in a p53 dose-dependent manner, while co-transfection with binding-deficient mutant p53-R175H failed to do so (). Interestingly, co-transfection of pLucB with p53 expression vector did not induce reporter activity, confirming that the potential p53 binding site is indeed at the predicted region.
Five potential p53 binding half-sites (BHS) were found at the 5′ region of PANK1 exon1α that overlaps with the translational start site (). Mutation of each BHS was generated by mutating the most conserved C/G to A/T (RRRCWWGYYY → RRRAWWTYYY) in pLucA. Mutation of BHS 1 did not reduce transcriptional activity in the presence of p53, while mutations of BHS 2+3 and BHS 4+5 significantly reduced p53-dependent transcriptional activity (). Furthermore, mutating all four binding half-sites completely abrogated reporter transcriptional activity by p53 ().
We next performed gel mobility shift assay to determine whether p53 binds to the four BHS. Using immunoprecipitated Flag-tagged p53, we observed a shift using a 171-bp DNA fragment containing the four WT BHS, which was supershifted and enhanced in the presence of anti-p53 antibody (ab 421) (). Furthermore, binding of p53 to the radiolabeled fragment was outcompeted by the cold WT fragment, but not by the cold mutant fragment (). Together, these results indicate that the PANK1 gene is a transcriptional target of p53 and that the two consensus p53-binding sites (four half-sites) at the 5′-end of PANK1 exon 1α are responsible for p53-dependent PANK1 gene activation.
p53 induces PanK1 expression
To confirm that PANK1 is a p53-inducible gene, we examined PANK1 mRNA and protein expression in response to p53 overexpression and DNA damage. Transfection of p53 expression vector in H1299 cells induced mRNA level of PANK1α as well as that of p21 (). Similarly, PanK1α protein level increased in a p53 dose-dependent manner in H1299 cells transfected with p53 expression vector, but no induction was observed in the presence of mutant p53 R175H (). In HCT116 cells containing endogenous WT-p53, induction of PanK1α protein level was observed upon treatment with DNA-damaging agents (Etoposide, Actinomycin D and Doxorubicin), which coincided with p53 induction. In contrast, PanK1α expression remained at the basal level in p53-deficient HCT116 cells, despite undergoing the same DNA-damaging treatments ().
Figure 2. p53 induces PanK1 expression. (A) H1299 cells were transfected with either control or p53-expressing vectors, and mRNA were extracted from cells 24 h after transfection and analyzed via RT-qPCR for PanK1α and p21 expressions. (B) western blot analysis of the protein levels of PanK1α, p21, p53, and actin in H1299 cells transfected with control vector and vectors expressing wild-type and R175H mutant p53. (C) HCT116 p53+/+ and p53−/− cells were treated with 20 μM Etoposide, 10 nM Actinomycin D, and 0.2 μg/mL Doxorubicin for 12 and 24 h, and cell lysates were analyzed by western using PanK1, p53, p21 and actin antibodies. (D) U2OS cells were treated with 20 μM Etoposide and 10 nM Actinomycin D for 8 and 24 h, and mRNA levels of PanK1α and PanK1β were analyzed via RT-qPCR. (E) HCT116 p53+/+ and p53−/− cells were treated with 10 nM Actinomycin D for 12 and 24 h, and mRNA levels of PanK1α and PanK1β were analyzed via RT-qPCR.
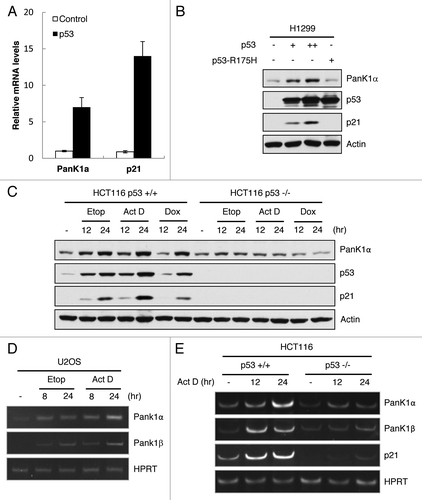
We next investigate whether p53 can regulate PanK1β expression. Our PanK1 antibody (Bethyl) is unable to detect endogenous PanK1β isoform in various cell lines tested, probably due to very low level expression of PanK1β. In order to examine the possibility that p53 also regulates PanK1β expression, RT-qPCR was performed in HCT116 and U2OS cells treated with DNA damaging agents. Interestingly, PanK1β mRNA level was induced upon DNA damage in both cell lines, similar to the kinetics seen for PanK1α, while p53-deficient HCT116 cells fail to upregulate PanK1β expression (). Together, our findings demonstrate that p53 upregulates the expression of both PanK1α and PanK1β in vitro.
PanK1 is not required for DNA damage-induced apoptosis
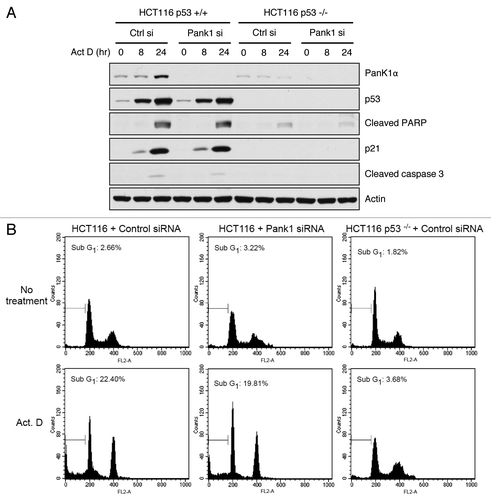
Since the major function of p53 is to act as a tumor suppressor that regulates cell fate under genotoxic stress, we wish to investigate whether PanK1 is a downstream modulator of p53-dependent cell cycle arrest or apoptosis. Depletion of PanK1 via siRNA in HCT116 cells containing WT-p53 did not have an effect on p21 induction or cleavage of proapoptotic effectors (caspase-3 and PARP) after Actinomycin D treatment (). Similarly, PanK1 knockdown in HCT116 cells did not have an effect on cell death after DNA damage, as measured by the sub-G1 population of apoptotic cells using FACS analysis (). Together, these results demonstrate that while both α- and β-isoforms of PanK1 are induced by p53, PanK1 does not modulate the effect of p53-dependent genotoxic stress response.
Figure 3. PanK1 is not required for DNA-damage-induced apoptosis. (A) HCT116 p53+/+ and p53−/− cells were transfected with control or PanK1 siRNA for 36 h and were subsequently treated with 10 nM Actinomycin D for 0, 8 or 24 h. Total cell extracts were assayed for PanK1, p53, p21, cleaved PARP, cleaved caspase 3 and actin. (B) HCT116 p53+/+ and p53−/− cells were transfected with control or PanK1 siRNA for 36 h and then treated with 10 nM Actinomycin D for 36 h. The cells were harvested, fixed with ice-cold methanol and analyzed by flow cytometry. Cells with sub-G1 DNA content were scored as apoptotic cells.
p53 regulates PanK1 expression under starvation and directly regulates PanK1 activity
The link between p53 and PanK1 suggests a role for p53 in CoA metabolism. As previously mentioned, regulation of CoA level in tissues is critical in response to various metabolic stresses in order to meet energy demands. Interestingly, p53 has been shown to modulate growth arrest and apoptosis under glucose starvation in vitro. Upon glucose withdrawal, p53 is activated by the energy-sensing AMPK, which leads to downstream expression of p53 targets.Citation13,Citation14 Thus, it is possible that p53 regulates the expression of PanK1 during starvation.
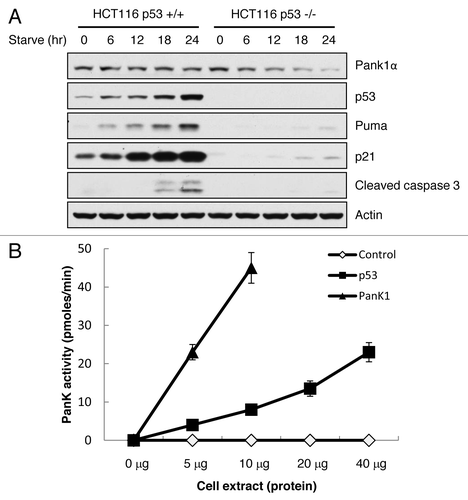
To test this hypothesis, we subjected isogenic HCT116 p53+/+ and p53−/− cells to glucose starvation. Upon glucose depletion, p53 levels are induced in HCT116 p53+/+ cells, and PanK1 expression remains unchanged. Surprisingly, PanK1 expression in HCT116 p53−/− cells decreased dramatically after glucose starvation in the absence of p53 upregulation (). To verify that p53 activity is indeed increased upon glucose starvation, we observed a steady increase in p53 downstream targets p21 and Puma as well as p53-induced apoptosis (cleaved caspase 3) in HCT116 p53+/+ cells, but not in p53−/− cells. Our data suggests that p53 is required to maintain PanK1 expression during energy deprivation. We then tested the PanK activity of cell lysates from H1299 cells that were transfected with either p53 or control expression vector. In the presence of p53 expression, PanK activity was significantly upregulated in those cells (). Together, these data indicate that p53 plays an important role in regulating metabolic pathways through transcriptional control of PANK1, independent of its canonical functions in apoptosis and cell growth arrest.
Figure 4. p53 regulates PanK1 expression under starvation and directly regulates PanK1 activity. (A) HCT116 p53+/+ and p53−/− cells were glucose-starved (DMEM + 10% FBS + no glucose) for the times indicated. Total cell extracts were assayed for PanK1, p53, Puma, p21, cleaved caspase 3 and actin. (B) H1299 cells were transfected with p53 or PanK1 (Pank1a)-expressing vectors, and total cell extracts were obtained to assay for PanK kinase activity in vitro.
A potential role of p53 in regulating gluconeogenesis in vivo
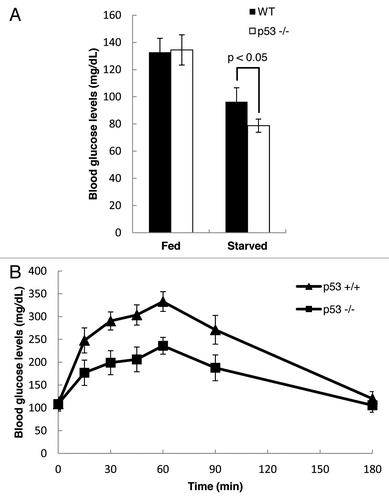
Previous studies showed that inactivation of Pank1 gene in mice leads to impairment of gluconeogenesis during starvation due to lower availability of free CoA. The liver carries out gluconeogenesis during fasting to provide energy for glucose-dependent tissues, such as red blood cells. There are two processes during gluconeogenesis that are CoA-dependent: (1) degradation of amino acid into TCA cycle intermediates; (2) activation of pyruvate carboxylase, the enzyme that catalyzes the first step of gluconeogenesis from pyruvate, by acetyl-CoA. Since Pank1 is responsible for CoA synthesis, we predicted that loss of p53-mediated regulation of Pank1 activity would lead to decreased availability of glucogenic substrates derived from these processes, resulting in a slower rate of glucose production. As expected, fasting blood glucose levels were significantly lower in the p53-null mice, while no differences were observed for blood glucose levels between the two genotypes under normal feeding (). Furthermore, a pyruvate challenge test performed after 16 h of fasting revealed a marked attenuation of gluconeogenic potential in the p53-null mice, as measured by their responding blood glucose levels after injection of sodium pyruvate (). Taken together, our findings suggest that p53-mediated Pank1 regulation has a significant impact on gluconeogenesis under fasting conditions.
Figure 5. p53 regulates gluconeogenesis in vivo. (A) Blood glucose levels were measured in WT and p53−/− mice that were either fed or starved for 2 d (WT fed, n = 4; WT starved, n = 4; p53−/− fed, n = 6; p53−/− starved, n = 6). (B) Pyruvate challenge test was performed using WT and p53−/− mice that were starved for 16 h (WT mice, n = 5; p53−/− mice, n = 7). Blood glucose levels were measured at different time points after intra-peritoneal injection of sodium pyruvate.
Discussion
Recent findings have demonstrated the importance of p53 in metabolic regulation. Here, we have provided evidence that linked p53 function to CoA metabolism through a novel p53 metabolic target PanK1, an enzyme that catalyzes the rate-limiting step of CoA synthesis. PanK1 was first identified as a potential p53 target from a ChIP-on-chip analysis, and subsequent luciferase reporter mutagenesis and gel shift assay revealed two p53 binding sites on the 5′-end of PANK1 exon 1α, indicating that PanK1 is likely a direct p53 target. Expressions of both α- and β-isoforms of PanK1 are shown to be induced by DNA damage in a p53-dependent manner, although the presence of PanK1 expression in p53-null cells suggests concomitant regulation by p53-independent pathways as well.
Many of the previously identified p53 metabolic targets play significant roles in modulating the effect of p53-dependent apoptosis or growth arrest. GAMT, while not sufficient to trigger cell death, is required to induce the full apoptotic response after p53 activation.Citation21 Similarly, it has been shown that TIGAR may partially rescue cells from p53-induced apoptosis, while TIGAR mutants exhibit impaired anti-apoptotic activity.Citation10 To our surprise, although PanK1 expression is upregulated under p53 activation in response to genotoxic stress, depletion of PanK1 by siRNA targeting did not affect p53-mediated cell death after DNA damage. Our findings suggest that p53 regulation on PanK1 expression may instead play a role in metabolic settings.
PanK1 is most highly expressed in the liver, in which it has been shown to carry out proper metabolic transformations during fasting that are mediated by adequate CoA synthesis; high CoA abundance is required for fatty acid and amino acid degradations that generate ketone bodies and glucose as alternative fuel sources under nutrient deprivation.Citation18,Citation20 Indeed, Pank1-knockout mice exhibit reduced fatty acid oxidation and gluconeogenesis during starvation due to lower availability of free CoA, which led to accumulation of long-chain acyl-CoA and triglycerides in the liver and lowered blood glucose levels.Citation20 Interestingly, gene expressions of all PanK isoforms are not elevated during fasting, implying that the existence of other factors, such as allosteric regulation, may be important in governing CoA levels.Citation19,Citation20 Nevertheless, in light of the p53 → PanK1 axis, we hypothesized that metabolic stress such as starvation may at least lead to maintenance of PANK1 gene expression through p53 activity. To our surprise, while PanK1 expression remained unchanged after glucose starvation in HCT116 p53+/+ cells, its p53−/− isogenic counterpart exhibited significant decrease in PanK1 protein levels, suggesting that p53 is required for the maintenance of PANK1 gene expression under energy deprivation. Consistent with this finding, we also observed a partial impairment in gluconeogenesis in fasted p53-null mice.
One interesting discovery in our study is that PanK1 expression appears to be p53-independent under conditions of excess nutrients. The transition from p53-independent to p53-dependent regulation of PanK1 after starvation implies that the unavailability of specific nutrients triggered a switch in the transcriptional machinery responsible for PanK1 expression. Previous works have shown that the activity of Sp1, a ubiquitously expressed housekeeping transcription factor that regulates the expression of thousands of genes implicated in a diverse array of cellular processes, is dependent upon the cellular nutritional and energy status. Fasting, glucose starvation and hypoxia can cause degradation of Sp1 or downregulation of the DNA binding activity of Sp1 via post-translational modifications such as O-linked glycosylation at serine/threonine residues.Citation22-Citation24 One possibility is that PanK1 expression is regulated by Sp1 during normal physiological conditions. However, in the face of energy deprivation, Sp1 activity diminishes to conserve nutrients, while p53 is activated in response to metabolic stress to maintain the expression of PanK1. Testing this hypothesis would allow us to gain better insight on how the metabolic response of p53 fits in the complex dynamics of energy homeostasis.
It is well-established that p53 responds to genotoxic stress in both normal and neoplastic cells. However, there is ever-increasing evidence that p53 also modulates energy balance in the adaptive response to metabolic stress. Our study here provides yet another novel insight on p53 nutrient stress response in a physiological setting. Unfortunately, our findings ultimately lead to an increasingly puzzling question of how p53 can differentially regulate a vast number of functionally diverse target genes in a context-specific manner. In other words, how is p53 programmed to respond differently under metabolic stress as opposed to genotoxic stress? Previous studies have shown that post-translational modifications of p53, particularly acetylation of lysine residues, play a crucial role in promoter-specific transcriptional activation of p53 targets. The K120 acetylation of p53 is required for transactivation of proapoptotic Puma gene, while K164 and C-terminal acetylations contribute to the activation of p21 growth arrest gene, but none is necessary for the induction of Mdm2 expression.Citation25,Citation26 Thus, it would be interesting to see if site-specific acetylation or other modifications of p53 is involved in orchestrating the p53 metabolic stress response through activation of PanK1 and/or other p53 metabolic targets.
Materials and Methods
Cell culture, transfection, siRNA-mediated knockdown and antibody
H1299 and U2OS cells were maintained in DMEM and HCT116 cells in McCoy’s 5A medium. All media were supplemented with 10% fetal bovine serum. Transfections with plasmid DNA and siRNA were performed using Lipofectamine2000 (Invitrogen) according to the manufacturer’s protocol. Ablation of PanK1 was performed by transfection of HCT116 cells with siRNA duplex targeting human PanK1 mRNA (5′ – GUGGAACGCUGGUUAAAUU – 3′). The rabbit polyclonal antibody specific for PanK1 protein was produced in collaboration with Bethyl Laboratories, Inc.
Chromatin Immunoprecipitation (ChIP)
Adherent cells were incubated in culture media containing 1% formaldehyde with gentle shaking for 10 min at room temperature, and crosslinking was stopped by addition of 2.5 M glycine to a final concentration of 0.125 M glycine. After two washes with cold PBS, cells were harvested in ice-cold lysis buffer [10 mM Tris-Cl (pH 8.0), 85 mM KCl, 0.5% NP-40, 5 mM EDTA, 0.25% Triton-X, and fresh proteinase inhibitor cocktail] and incubated on ice for 10 min. Nuclei were collected, suspended in cold RIPA buffer [10 mM Tris-Cl (pH 8.0), 150 mM NaCl, 0.1% SDS, 0.1% DOC, 1% Triton X-100, 5 mM EDTA, and fresh proteinase inhibitor cocktail] and sonicated to shear the genomic DNA to an average of 300 bp. Cleared extracts were blocked with protein A beads (Millipore), and p53 proteins were immunoprecipitated by FL-393 p53 antibody (Santa Cruz) followed by binding of antibodies to protein A beads. After eight washes by RIPA buffer with gentle rotation for 5 min each time, the proteins were eluted from the beads by 0.5 mL elution buffer [30 mM Tris-Cl (pH 8.0), 15 mM EDTA, 200 mM NaCl and 1% SDS]. The DNA samples were recovered by phenol extraction and ethanol precipitation after reversal of crosslinking. The purified DNA were then analyzed either by PCR within linear amplification range followed by agarose gel electrophoresis or by quantitative real-time PCR using Applied Biosystems 7500 Fast System. The following primers were used for PanK1 ChIP: forward primer 5′ – CGATGCCCATCTGGTTTTCACATCG – 3′ and reverse primer 5′ – GCCACCGACGAGTTTCAACA – 3′.
Luciferase activity assay
Promoter-containing fragments were amplified from human genomic DNA of H1299 cells and cloned into the pBVLuc luciferase reporter vector containing a minimal promoter. To test the potential p53 binding sites, fragment for pLucA containing the p53 BS was amplified using forward primer 5′ – GATGCCGCCTTCCCTTCTTA – 3′ and reverse primer 5′ – GAGGAAGCCGCGTTTGAAGT – 3′, while fragment for pLucB without the p53 BS was amplified using the same forward primer and reverse primer 5′ – CATCGCCATACAAAGCCCAA – 3′. Mutations of the various p53 binding sites were generated using site-directed mutagenesis with the following oligos: 5′ – CCCATCTCCTCTCTGTAATCCCAGAGAACTTGAT – 3′ and 5′ – ATCAAGTTCTCTGGGATTACAGAGAGGAGATGGG – 3′ for pLucA’1, 5′ – CCAGAGAATTTTATTCCCTATATTGAAACTCGTCGGTGGC – 3′ and 5′ – GCCACCGACGAGTTTCAATATAGGGAATAAAATTCTCTGG – 3′ for pLucA’23, and 5′ – GGCGGTGGCGGGTAGTACTGGGAATTCTCAGTGGCGGG – 3′ and 5′ – CCCGCCACTGAGAATTCCCAGTACTACCCGCCACCGCC – 3′ for pLucA’45. Transfection of H1299 cells were performed in 24-well plate using 0.2 μg luciferase reporter constructs, 0.05 μg pRL-tk Renilla construct and various amounts of p53-expressing vector. Luciferase activities were measured 24 h post-transfection using Dual-Luciferase Reporter Assay System (Promega). Firefly luciferase activities were normalized with Renilla luciferase activities to obtain the relative luciferase activity.
Gel mobility shift assay (EMSA)
Purified Flag-p53 proteins were obtained from transfected 293 cells. The 171-bp DNA probe containing the p53 binding sites were PCR-amplified from PANK1 promoter using primers 5′ – CTGTCTCTCCCAGACCCAT – 3′ and 5′ – AGTCTGGGAGGCGAGGAAG – 3′, labeled by T4 kinase (NEB, M0201S) and purified using the Bio-Spin 30 columns (Bio-Rad). The protein-DNA binding reactions (20 μl) contained 20 mM HEPES (pH 7.6), 80 mM NaCl, 0.1 mM EDTA, 12.5% glycerol, 2 mM MgCl2, 2 mM spermidine, 0.7 mM DTT, 200 ng/μL BSA, 20 ng/μL sheared sperm DNA, 10–20 fmol DNA probe, and 20 ng Flag-p53. In supershift assays, 200 ng pAb421 (Millipore) antibody was added to the reaction. Wild-type and mutant competition probes were PCR-amplified from pLucA and pLucA’2345, respectively, using the above primers.
RNA extraction and RT-qPCR
Total RNA was extracted using TRIzol Reagent (Life Technologies) according to the manufacturer’s protocol. cDNA was synthesized from total RNA using M-MulV Reverse Transcriptase kit (NEB). PCR analysis was performed using Applied Biosystems 7500 Fast System with the following primers: 5′ – CGCTGCGAAATACACCAATTTAACCAGCG – 3′ and 5′ – AAGAACAGGCCGCCATTCC – 3′ for human PanK1α, and 5′ – ACAGGACGCTGTGGGATGTAAA – 3′ and 5′ – CGCTGCGAAATACACCAATTTAACCAGCG – 3′ for human PanK1β.
PanK activity assay
Enzyme preparation and assays were performed as described previously.Citation27 The pantothenate kinase-specific activities in cell and tissue lysates were calculated as a function of protein concentration measured by Bradford method. Assays contained d-[1-14C]-pantothenate (45.5 μM; specific activity 55 mCi/mmol), ATP (2.5 mM, pH 7.0), MgCl2 (2.5 mM), Tris–HCl (0.1 M, pH 7.5) and increasing amounts of cell extract in a total volume of 40 μL. The mixture was incubated for 10 min at 37°C and the reaction was stopped by depositing a 5 μL aliquot onto a Whatmann DE81 ion-exchange filter disk that was washed with 1% acetic acid in 95% ethanol (25 ml/disk) three times to remove non-reacted pantothenate. 4’ – Phosphopantothenate was quantified by counting the dried disk in 3 mL scintillation solution.
Pyruvate challenge test and blood glucose measurements
For the pyruvate challenge test, mice were starved for 16 h and were then intraperitoneally injected with pyruvate dissolved in saline (2 mg/g of body mass). Blood glucose was measured using OneTouch Ultra Blue Test Strips (LifeScan Inc.).
Acknowledgments
This work was supported by the National Cancer Institute of the National Institutes of Health under Award 2P01 CA080058–12 and 5RO1 CA172023 to W. Gu. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. S.-J. Wang is supported by NIH cancer biology training grant T32-CA09503. The research in Y. Tang’s lab was supported by Albany Medical College Startup fund. We thank Suzanne Jackowski for valuable suggestions and reagents, and also thank Eric W. McIntush from Bethyl Laboratories, Inc. for developing the PanK1 antibody.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Lane DP. Cancer. p53, guardian of the genome. Nature 1992; 358:15 - 6; http://dx.doi.org/10.1038/358015a0; PMID: 1614522
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997; 88:323 - 31; http://dx.doi.org/10.1016/S0092-8674(00)81871-1; PMID: 9039259
- Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer 2002; 2:594 - 604; http://dx.doi.org/10.1038/nrc864; PMID: 12154352
- Maddocks OD, Vousden KH. Metabolic regulation by p53. J Mol Med (Berl) 2011; 89:237 - 45; http://dx.doi.org/10.1007/s00109-011-0735-5; PMID: 21340684
- Zilfou JT, Lowe SW. Tumor suppressive functions of p53. Cold Spring Harb Perspect Biol 2009; 1:a001883; http://dx.doi.org/10.1101/cshperspect.a001883; PMID: 20066118
- Chen JQ, Russo J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochim Biophys Acta 2012; 1826:370 - 84; PMID: 22750268
- Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, et al. p53 regulates mitochondrial respiration. Science 2006; 312:1650 - 3; http://dx.doi.org/10.1126/science.1126863; PMID: 16728594
- Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res 2004; 64:2627 - 33; http://dx.doi.org/10.1158/0008-5472.CAN-03-0846; PMID: 15059920
- Contractor T, Harris CR. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res 2012; 72:560 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-11-1215; PMID: 22123926
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006; 126:107 - 20; http://dx.doi.org/10.1016/j.cell.2006.05.036; PMID: 16839880
- Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A 2010; 107:7455 - 60; http://dx.doi.org/10.1073/pnas.1001006107; PMID: 20378837
- Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A 2010; 107:7461 - 6; http://dx.doi.org/10.1073/pnas.1002459107; PMID: 20351271
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 2005; 18:283 - 93; http://dx.doi.org/10.1016/j.molcel.2005.03.027; PMID: 15866171
- Okoshi R, Ozaki T, Yamamoto H, Ando K, Koida N, Ono S, et al. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J Biol Chem 2008; 283:3979 - 87; http://dx.doi.org/10.1074/jbc.M705232200; PMID: 18056705
- Scherz-Shouval R, Weidberg H, Gonen C, Wilder S, Elazar Z, Oren M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci U S A 2010; 107:18511 - 6; http://dx.doi.org/10.1073/pnas.1006124107; PMID: 20937856
- Halvorsen O, Skrede S. Regulation of the biosynthesis of CoA at the level of pantothenate kinase. Eur J Biochem 1982; 124:211 - 5; http://dx.doi.org/10.1111/j.1432-1033.1982.tb05927.x; PMID: 7084227
- Jackowski S, Rock CO. Regulation of coenzyme A biosynthesis. J Bacteriol 1981; 148:926 - 32; PMID: 6796563
- Robishaw JD, Neely JR. Coenzyme A metabolism. Am J Physiol 1985; 248:E1 - 9; PMID: 2981478
- Ramaswamy G, Karim MA, Murti KG, Jackowski S. PPARalpha controls the intracellular coenzyme A concentration via regulation of PANK1alpha gene expression. J Lipid Res 2004; 45:17 - 31; http://dx.doi.org/10.1194/jlr.M300279-JLR200; PMID: 14523052
- Leonardi R, Rehg JE, Rock CO, Jackowski S. Pantothenate kinase 1 is required to support the metabolic transition from the fed to the fasted state. PLoS One 2010; 5:e11107; http://dx.doi.org/10.1371/journal.pone.0011107; PMID: 20559429
- Ide T, Brown-Endres L, Chu K, Ongusaha PP, Ohtsuka T, El-Deiry WS, et al. GAMT, a p53-inducible modulator of apoptosis, is critical for the adaptive response to nutrient stress. Mol Cell 2009; 36:379 - 92; http://dx.doi.org/10.1016/j.molcel.2009.09.031; PMID: 19917247
- Ihara T, Tsujikawa T, Fujiyama Y, Bamba T. Alterations in the DNA binding activity of transcriptional factors activator protein-1, Sp1, and hepatocyte nuclear factor-1 in rat jejunum during starvation and refeeding. J Gastroenterol Hepatol 2003; 18:705 - 11; http://dx.doi.org/10.1046/j.1440-1746.2003.02924.x; PMID: 12753154
- Kang HT, Ju JW, Cho JW, Hwang ES. Down-regulation of Sp1 activity through modulation of O-glycosylation by treatment with a low glucose mimetic, 2-deoxyglucose. J Biol Chem 2003; 278:51223 - 31; http://dx.doi.org/10.1074/jbc.M307332200; PMID: 14532290
- Wei S, Chuang HC, Tsai WC, Yang HC, Ho SR, Paterson AJ, et al. Thiazolidinediones mimic glucose starvation in facilitating Sp1 degradation through the up-regulation of beta-transducin repeat-containing protein. Mol Pharmacol 2009; 76:47 - 57; http://dx.doi.org/10.1124/mol.109.055376; PMID: 19372209
- Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell 2006; 24:827 - 39; http://dx.doi.org/10.1016/j.molcel.2006.11.021; PMID: 17189186
- Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell 2008; 133:612 - 26; http://dx.doi.org/10.1016/j.cell.2008.03.025; PMID: 18485870
- Vallari DS, Jackowski S, Rock CO. Regulation of pantothenate kinase by coenzyme A and its thioesters. J Biol Chem 1987; 262:2468 - 71; PMID: 3029083