Abstract
Neuroblastoma is the most common solid tumor in childhood and represents 15% of all children’s cancer deaths. We have previously demonstrated that tripartite motif 16 (TRIM16), a member of the RING B-box coiled-coil (RBCC)/tripartite totif (TRIM) protein family, has significant effects on neuroblastoma proliferation and migration in vitro and tumorigenicity in vivo. However, the mechanism by which this putative tumor suppressor influences cell proliferation and tumorigenicity was undetermined. Here we show, for the first time, TRIM16’s striking pattern of expression and dynamic localization during cell cycle progression and neuroblastoma tumor development. In a tyrosine hydroxylase MYCN (TH-MYCN) neuroblastoma mouse model, immunohistochemical staining revealed strong nuclear TRIM16 expression in differentiating ganglia cells but not in the tumor-initiating cells. Furthermore in vitro studies clearly demonstrated that during G1 cell cycle phase, TRIM16 protein expression is upregulated and shifts to the nucleus of cells. TRIM16 also plays a role in cell cycle progression through changes in Cyclin D1 and p27 expression. Importantly, using TRIM16 deletion mutants, an uncharacterized protein domain of TRIM16 was found to be required for both TRIM16’s growth inhibitory effects and its nuclear localization. Taken together, our data suggest that TRIM16 acts as a novel regulator of both neuroblastoma G1/S progression and cell differentiation.
Introduction
There are over 70 tripartite motif (TRIM) proteins in the human genome. These proteins have evolutionary conserved domain structures and the role of TRIM proteins in cellular function can differ greatly from differentiation, transcription, cell cycle regulation, innate immunity and cell migration.Citation1 Several TRIM proteins have been implicated in cancer, offering novel targets for cancer therapies and prognostic markers.Citation2 TRIM16 is a ubiquitously expressed, predominately cytoplasmic protein, with mRNA expression levels highest in the embryonic brain and in adult testes.Citation3 We have previously demonstrated that overexpression of TRIM16 causes decreased proliferation in neuroblastoma and in lung, breast and skin cancer cell lines.Citation4-Citation7 Overexpression of TRIM16 is able to enhance retinoid-induced differentiation and causes downregulation of cell cycle factors E2F1 and pRB. Overexpression of TRIM16 also increases histone 3 acetylation and causes an upregulation of retinoid target genes. These findings indicate that TRIM16 plays an important role in the G1 phase of cell cycle, although the role of TRIM16 in cell cycle has not been directly studied.
The G1/S transition is the key step for cell cycle progression and is mediated by Cyclin Ds, CDK4/CDK6 (complexes operates at mid-G1) and cyclin E/CDK2 (operates at late G1).Citation8-Citation11 The Cip/Kip family (including p21, p27 and p57) can inhibit cell progression to S phase via inhibition of CDK kinases activities. Many TRIM proteins, such as the tumor suppressor promeylocytic leukemia protein (PML/TRIM19), are highly expressed during G1 phase and form PML bodies during G1 and partition during mitosis.Citation12,Citation13 Additionally, after retinoid treatment, TRIM16 protein co-localizes and homodimerizes with PML and forms nuclear bodies.Citation4,Citation14 These nuclear bodies are also formed after cellular stress.Citation15-Citation17 Moreover, p27 (also known as CDKN1B) and other factors involved in G1 have been found to be associated with these nuclear PML bodies.Citation18 In neuroblastoma, p27 has a key role in both cell cycle and is involved in retinoid-induced differentiation.
The decision of cells to initiate differentiation is commonly made in G1 phase of the cell cycle, and differentiation initiation requires cell cycle arrest.Citation8,Citation19 How the cell cycle machinery coordinates cell cycle arrest with differentiation activation is not fully understood in neuroblastoma. In vitro, TRIM16 overexpression induces or enhances keratinocyte and neuroblastoma differentiation, respectively.Citation6,Citation20 We have also shown that in human neuroblastoma tissues, TRIM16 protein is only expressed in the differentiated ganglion cell component of human tumors. However, it is yet to be determined if TRIM16’s role in neuroblastoma differentiation involves elements of G1 phase in the cell cycle. Also the expression and localization characteristics of TRIM16 through the phases of cell cycle have not been previously investigated. These are critical steps toward understanding TRIM16’s function in cancer and normal cells.
To further investigate TRIM16‘s role in neuroblastoma cell cycle regulation, we investigated the expression of TRIM16 in cells during cell cycle progression. Our results show that TRIM16 is most highly expressed during G1 phase of cell cycle, and that TRIM16 suppresses cell growth by prolonging the G1 phase of cell cycle. We have also demonstrated that nuclear localization of TRIM16 is associated with ganglia cell differentiation and inhibition of proliferation. Thus we have identified a novel functional domain of the TRIM16 protein, which is responsible for its growth-inhibitory effects and nuclear localization.
Results
TRIM16 protein has a nuclear localization in differentiating ganglia cells and is reduced in neuroblastoma tumor cells in vivo
Enforced overexpression of TRIM16 enhances neuronal differentiation in neuroblastoma cells,Citation6 suggesting that TRIM16 expression changes during neuroblast differentiation. To address this hypothesis, tissues from the histologically well-characterized TH-MYCN neuroblastoma mouse model were examined for TRIM16 protein expression.Citation21 In order to determine if TRIM16 is expressed dynamically during normal paravertebral ganglia development and neuroblastoma tumor formation, the TH-MYCN mice were culled at birth, and 7, 14 and 42 d of age, and processed for immunohistochemistry to examine the paravertebral ganglia regions from both wild-type and homozygous TH-MYCN transgenic mice. These time points sample a critical period of growth and differentiation in normal ganglia and also span tumor initiation and development in neuroblastoma tumors. In the wild-type (wt) animals, the newly differentiated ganglia at day 0–14 time points had strong, predominately nuclear TRIM16 expression (). In contrast, the mature and terminally differentiated ganglia cells at day 42 had low levels of diffuse (de-localized) TRIM16 staining. In the presence of two copies of transgenic MYCN, neuroblast cells in the hyperplastic regions of the ganglia go on to form mature tumors. These neuroblast cells of the ganglia were mostly negative for TRIM16 staining. In contrast, the more differentiated ganglia cells adjacent to the hyperplastic regions possessed strong nuclear TRIM16 staining (). The βIII-tubulin staining was used in this study as a marker of both ganglia and neuroblastoma tissue and confirmed that the tissues studied were paravertebral ganglia or neuroblastoma.
Figure 1. TRIM16 protein has nuclear localization in differentiating ganglia cells but is reduced in neuroblastoma tumors in vivo. Wild-type (wt) mice and homozygous TH-MYCN mice were culled at day 0, 7, 14, 42 time points. At day 42, only the transgenic mice have tumors. Parafilm-embedded paravertebral ganglia tissues were sectioned, and consecutive slides were used for immohistochemistry. Proteins of interest were stained by antibody conjugated to DAB (brown) stain and counterstained with hematoxylin (blue). β-III-tubulin (neuronal maker), cyclin E (G1/S marker), p27 (G0, G1 and neuronal differentiation marker) antibodies were used. The arrows show areas of hyperplasia/tumor initiating neuroblasts. Representative images are 40× captured with Aperio ScanScope XT.
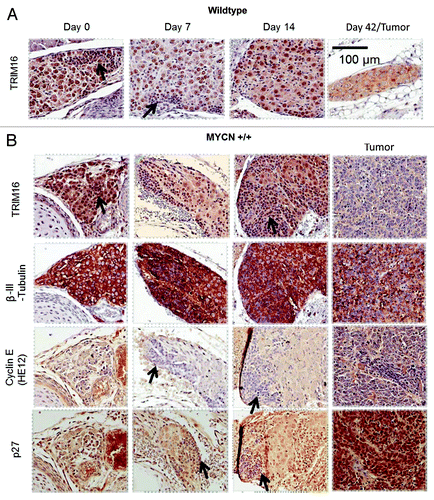
To gain greater insight into the cell cycle status of the cells involved in neuroblastoma development, the above tissues were stained with antibodies directed against components of G1 cell cycle phase. Both ganglia and hyperplastic cells (neuroblasts) were negative for cyclin E, showing that the ganglia cells were not in the late stages of G1. Cyclin E expression was observed in a minority of tumor cells, confirming that the antibody was working. Intense p27 staining was observed in the nucleus of the large differentiated ganglia cells. Strong p27 staining was not observed in the nucleus of the neuroblasts in the hyperplastic regions of the ganglia. This indicates the ganglia cells (and not the neuroblasts) were in G0, G1 or undergoing differentiation. Interestingly, strong ubiquitous p27 staining was also observed in the neuroblastoma tumors, an unexplained finding that is in accordance with immunohistochemical studies of human neuroblastomas.Citation22 Thus, during neuroblastoma development, the expression pattern of p27 and cyclin E indicated that the ganglia cells which expressed TRIM16 in the nucleus were arrested in early to mid-G1, which is consistent with early differentiation.
TRIM16 protein expression is cell cycle-dependent
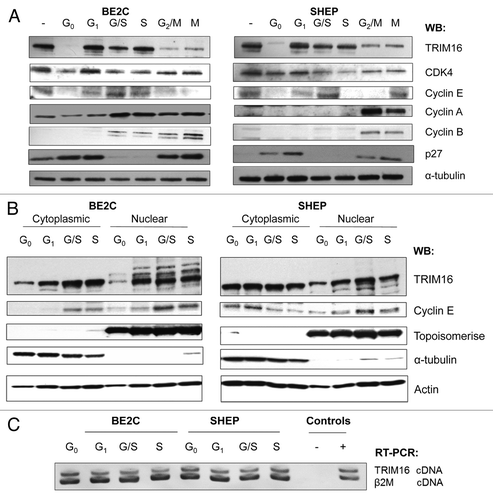
As TRIM16 expression correlated with differentiating cells, a process which requires the arrest of proliferation. We next investigated if TRIM16 is highly expressed during G0 and/or G1 cell cycle phases. To ascertain whether TRIM16 has a differential expression pattern through phases of the cell cycle, the BE2C and SHEP neuroblastoma cells were synchronized and analyzed by western blots. Treatment conditions were evaluated by using different markers of the cell cycle phases and analyzed in six different cell cycle phases: G0, G1 (CDK4), G1/S (Cyclin E), S, G2/M (Cyclin A) and mitosis (M, Cyclin B). Asynchronous cells were used as a control. Western blot analysis revealed that after synchronization to G0, via serum starvation, the level of TRIM16 protein expression decreased when compared with asynchronous cells (). After only 3 h of serum stimulation, the cells were entranced into G1 phase, and TRIM16 expression increased markedly when compared with G0 phase. Upon aphidicolin treatment, cells were arrested at the G1/S boundary, and the level of TRIM16 was increased in amount when compared with asychronous cells. After 3 h of aphidicolin treatment, the cells were released and came to S phase, and a high expression level of TRIM16 was maintained in these cells. The cells were then synchronized to G2/M or mitosis and showed a reduced level of TRIM16. Furthermore, the nuclear and cytoplasmic protein lysates were also analyzed by western blot; these results indicated that the high level of TRIM16 in G1 phase was more pronounced in the nucleus of the cells in both BE2C and SHEP (). RT-PCR showed that the induction of TRIM16 protein expression from G0 to G1 in the cell cycle was not due to an increase in TRIM16 mRNA levels (). To determine whether TRIM16’s dynamic expression association with the cell cycle was specific to neuroblastoma, the human embryonic fibroblast cell line HEK293, MDA-MB-231 (breast cancer cells) and CALU-6 (lung cancer cells) were utilized and subjected to synchronization. This analysis of whole-cell lysates yielded similar results to the neuroblastoma cells (Fig. S1).
Figure 2. TRIM16 protein expression is cell cycle dependent. (A) Western blots show TRIM16 expression at various phases of the cell cycle in two neuroblastoma cell lines. Two neuroblastoma cell lines (BE2C and SHEP) were used. Markers of the cell cycle phases: CDK4 for G1, Cyclin E for G1/S, Cyclin A G2/M and cyclin B for mitosis (M) were used. The α-tubulin antibody was used as a loading control. Representative panels are displayed of three replicate runs. (B) Cells were synchronized; the cytoplasmic and nuclear lysates were analyzed by western blot. Topoisomerase was used as a nuclear marker and α-tubulin as a cytoplasmic marker. (C) RT-PCR with TRIM16 primers and β 2-microglobulin (β2M) primers (template and loading control). cDNA samples were obtained from synchronized cells and subjected to competitive RT-PCR and gel electrophoresis. Representative gel image displayed.
TRIM16 localization shifts to the nucleus during the G1 phase of the cell cycle
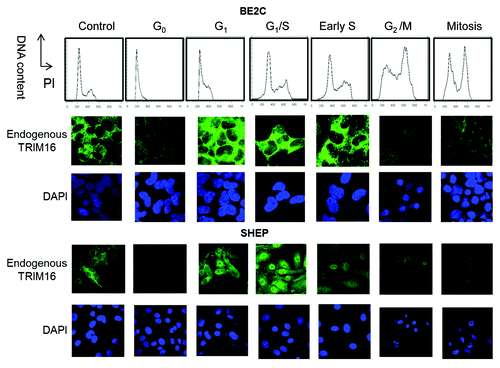
As TRIM16 nuclear protein increased during G1 of the cell cycle (in W analyses), it was important to ascertain if this could be confirmed by immunofluorescent studies. The localization of endogenous TRIM16 was assessed via immunofluorescent staining of endogenous TRIM16 and co-staining with DAPI nuclear/DNA marker (). In asychronous BE2C and SHEP neuroblastoma cell lines, TRIM16 was expressed predominately in the cytoplasm of the cells. At G0, TRIM16 expression decreased and was cytoplasmic; this was in stark contrast to cells that had entered the G1 phase. Cells in G1 had TRIM16 expression markedly increased in the nucleus, although the majority of TRIM16 remained cytoplasmic. Upon cell synchronization to the G1/S boundary, TRIM16 was increased in the nucleus compared with asynchronous cells. Strikingly, in SHEP cells, G1/S arrested cells achieved predominately nuclear TRIM16. S phase cells maintained high expression of TRIM16 but cells lost nuclear TRIM16. Cells synchronized to G2/M or mitosis did not display any involvement of TRIM16 with the DNA and had relatively low levels of expression. Therefore, during G1 cell cycle phase, TRIM16 protein is increased and also shifts to the nuclear compartment of the cell. CALU-6 (lung cancer) and HEK293 had a similar staining pattern to the neuroblastoma cells but, in addition, had induction of large nuclear bodies in the nucleus of G1 and G1/S synchronized cells (Fig. S2). Hence, the TRIM16 nuclear protein increases during G1 cell cycle phase is likely to be common yet compelling in cells where TRIM16 is expressed.
Figure 3. TRIM16 localization shifts to the nucleus during the G1 phase of the cell cycle. (A) Propidium iodide was used to optimize the synchronization of BE2C and SHEP cell lines by DNA content analysis. Immunofluorescent studies used confocal microscopy with the 100× objective on the Olympus FV1000. Endogenous TRIM16 is stained with Alexa Fluor 488 (green). DAPI (nuclear/DNA stain) has blue staining.
TRIM16 has a role in cell cycle progression
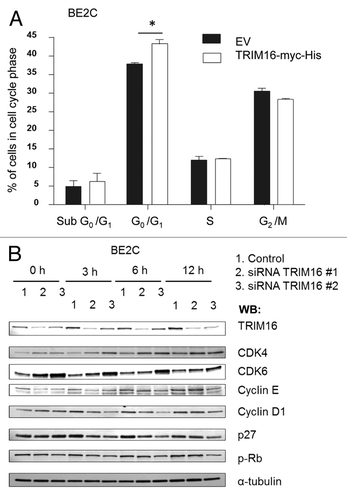
In order to establish whether TRIM16 has an active role in cell cycle progression, we analyzed how TRIM16 knockdown and overexpression affected the cell cycle. TRIM16 overexpression for 48 h resulted in an increased proportion of cells in G1, with over 44% of cells in G1 when TRIM16 was overexpressed, compared with the empty vector transfected cells with 39% of cells in G1 (). As synchronization of cells dramatically affects TRIM16 cell phenotype, and there was a marked induction at G1, demonstrated in and , it was likely that TRIM16 was important in synchronized cells for the progression into/through G1. This prompted investigation into whether TRIM16 was required for the transition from G0 to G1 in the cell cycle. BE2C cells were synchronized by serum starvation and simultaneously transfected with TRIM16-specific siRNAs. At the protein level, markers of the G1 cycle phase were used to analyze the serum-starved cells after the addition of serum. Two specific siRNA sequences were used to knocked-down TRIM16 protein levels at 36 h post transfection, and this knockdown was maintained during the subsequent time points (). The tumor suppressor p27 and cyclin D1 were significantly downregulated by the specific TRIM16 siRNAs. Critical mid-G1 factors, CDK4 and CDK6 showed a trend toward upregulation by TRIM16 knockdown. Rb and cyclin E showed no significant changes across the time points studied. This suggests that TRIM16 plays a role in modulation of G1 cell cycle components.
Figure 4. TRIM16 is involved in cell cycle progression. (A) BE2C neuroblastoma cells were transiently transfected with the TRIM16-myc-His plasmid or EV control, for 48 h before harvest. Propidium iodide was used to determine DNA content. *p < 0.05. (B) Western blot of TRIM16 knockdown by siRNA in the cells synchronized in G0 and serum released for time points up to 12 h, and probed with anti-CDK4, anti-CDK6, anti-cyclin E, anti-cyclin D1, anti-p27, anti-pRb and α-tubulin antibodies. BE2C neuroblastoma cells were used.
The “linker” domain of TRIM16 has nuclear localization and growth inhibition potential
As nuclear TRIM16 localization was likely to be important in the role of TRIM16 in both cell cycle progression and differentiation, GFP-fused deletion mutants were used to further study this characteristic in vitro. Confocal images were taken 36 h post-transfection of TRIM16 full-length and mutants in SHEP cells (). Empty vector (EV) and mutant 4 (M4) constructs display similar ubiquitous expression. Mutant 1 (M1) and mutant 2 (M2) both had cytoplasmic bodies and displayed some diffuse expression throughout the nucleus and cytoplasm. The expression level of mutant 1 protein was less than the other constructs. TRIM16 full-length and mutant 3 (M3) had a similar fluorescent pattern, with cells being either predominately cytoplasmic, or cytoplasmic and nuclear. Mutant 5 (M5) was cytoplasmic. A similar localization pattern was displayed in BE2C cells (Fig. S3)
Figure 5. The “linker” domain of TRIM16 has nuclear localization and growth inhibition potential. TRIM16 deletion domains were fused with GFP to aid observation of localization and transfection. (A) SHEP cells were transiently transfected with constructs and fixed for 36 h post transfection. Microscopy studies imaged exogenous GFP (green) directly by confocal microscopy at the 100× objective on the Olympus FV1000. DAPI was used as a nuclear stain (blue). Cyto stands for cytoplasmic, and Nuc stands for nuclear expression. (B) BE2C and SHEP cells were transiently transfected with domain mutant constructs. After 72 h transfections, the cells were analyzed via BrdU incorporation analysis.
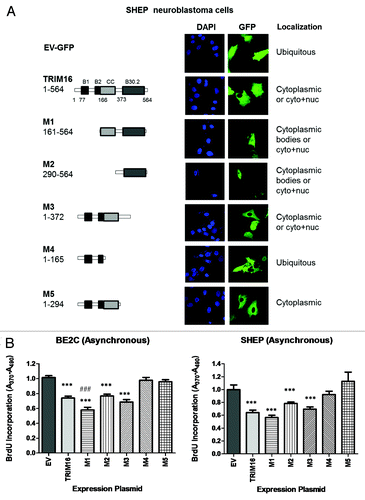
These domain mutants were also used to determine which domain causes the growth inhibition associated with the overexpression of full-length TRIM16. TRIM16 (wt), M1, M2, M3, constructs caused decreased proliferation in BE2C and SHEP cells, with M4 and M5 being comparable to the EV control (). Together these results implicate that a region between the TRIM16 amino acid (AA) 294 and AA373 (termed here as the “linker” domain) has both nuclear localization potential and proliferation inhibition potential. There is also possibly a region in AA165–294 (putative coiled-coil domain) which is responsible for nuclear exclusion/export of the TRIM16 protein.
Discussion
Even though previous studies have shown TRIM16 to have a role in cell proliferation and differentiation, these studies did not fully elucidate the mechanism by which the TRIM16 protein exerted these effects. Thus we began by thoroughly characterizing the level and localization of TRIM16’s protein through neuroblastoma differentiation and cell cycle progression. We found a significant and striking pattern of expression and localization for the TRIM16 protein, with highest nuclear protein expression during the G1 cell cycle phase. We have presented data here that gives a major insight into the function of this novel TRIM family member and has significant implications on the interpretation of previous research on TRIM16. Importantly, we have shown for the first time that TRIM16 expression and localization is influenced by cell cycle phase and its localization.
TRIM16, while being weakly expressed in G0 and mitosis, is most highly expressed during the G1 cell cycle phase. Furthermore, TRIM16’s localization becomes more nuclear during G1 and shifts back to the cytoplasm during S phase. This appears to be a common feature of TRIM16 in different type of cells, since other cell types also have the same pattern of expression as the neuroblastoma cell lines. In this work, we also show that TRIM16 transcription is not increased during the G0 to G1 phases. This result suggests that TRIM16’s role in G1 cell cycle phase may be through increased protein levels via decreased protein degradation of TRIM16. Interestingly, in one of the first publications on TRIM16 by Beer and colleagues,Citation20 studies showed a massive induction of TRIM16 protein (but not mRNA) in keratinocytes cells after serum starvation and activation with serum and other growth factors. Our result suggests that induction of TRIM16 in those keratinocytes cells was likely due to the cell cycle status of the cells studied, and that the biphasic induction of TRIM16 described by Beer et al. is more likely due to the cells re-entering the G1 cell cycle phase for a second time.
As TRIM16 overexpression promotes differentiation and decreased proliferation, cells where TRIM16 had been knocked-down would be expected to progress faster through G1 compared with control samples. However, the data presented does not indicate a faster progression through G1 when TRIM16 is suppressed in synchronized cells. Instead the data suggests a deregulation of G1 following knockdown of TRIM16. Cyclin D1 was downregulated by the TRIM16 knockdown. The time points used indicate a delay in this cell cycle regulator’s induction by at least 9 h upon TRIM16 depletion. Importantly, Cyclin D1 protein has been shown to enhance retinoid treatment response in breast cancer cells.Citation23 Cyclin D1 is also deregulated in many cancers.Citation24 The downregulation of cyclin D1 by TRIM16 could be through a feedback loop, in which TRIM16 knockdown increased E2F1 activity and thereby repressed cyclin D1 transcription.Citation6,Citation25 One interesting study has demonstrated that c-Myc has a similar effect on the cell cycle to that observed for TRIM16 knockdown.Citation26 When the c-Myc was overexpressed from G0-S phases, p27 and cyclin D1 were reduced, with CDK4 and CDK6 being induced. c-Myc is an oncogene, and the effect observed for TRIM16 knockdown may explain how TRIM16 acts as a tumor suppressor,Citation6 through influencing cell cycle components’ expression.
TRIM16 is dynamically expressed during normal parasympathetic ganglia development and comparatively suppressed in neuroblastoma cells. Evidence of this was gained by using the TH-MYCN mouse model of neuroblastoma.Citation21,Citation27 Notably, TRIM16 was highly expressed in the nuclei of the large differentiating ganglia cells of both wild-type and transgenic mice. This was in contrast to the tumor-initiating cells in the hyperplastic regions, where the majority of cells had reduced TRIM16 expression. Interestingly, in the highly proliferative neuroblastoma tumors, TRIM16 was expressed weakly in the cytoplasm. In the mature ganglia of the wild-type mice, TRIM16 was ubiquitously and moderately stained throughout the nucleus and cytoplasm of the ganglia cells and was greatly reduced compared with the high expression of TRIM16 observed in the ganglia at day 0. The decrease in TRIM16 expression in mature, non-dividing cells was reminiscent of TRIM16’s decrease in expression in G0 cell cycle phase in vitro. As the mature ganglia cells of the 6-week-old mice did not have predominately nuclear TRIM16, this suggests that nuclear TRIM16 expression is not associated with terminally differentiated cells.
As TRIM16 is downregulated at G0 in vitro, it probable that the nuclear TRIM16 expressing ganglia cells were in the G1 cell cycle phase or in the early stages of differentiation; this is also indicated by the high TRIM16 expression in the G1 phase and its comparisons with other cell cycle phase markers. Our results suggest that nuclear TRIM16 is, therefore, a marker of early differentiation or early G1 cell cycle phase in neuroblastoma cells. Our results also indicate that TRIM16 localization and expression is cell cycle-dependent in vivo. In the literature, TRIM16 is shown to display a similar expression phenomenon in differentiated keratinocytes as we observed in the differentiated neuroblasts.Citation20 TRIM16 was highly expressed during the early differentiation process of the skin, but not in the highly proliferative epithelium of wounded skin. The in vitro studies of this publication also demonstrated that overexpression of TRIM16 enhanced early differentiation in keratinocytes. Our own overexpression studiesCitation6 show that overexpression of TRIM16 enhances retinoid-induced differentiation in neuroblastoma cells, and that treatment by a differentiating agent can cause nuclear TRIM16 levels to increase.Citation4,Citation5 Taken together, the previous studies and the current study show that TRIM16 has a role in actively differentiating, rather than differentiated cells and it is important to note that neuronal differentiation (including via retinoid treatment) is known to occur via G1 cell cycle arrest. These data together allowed us to speculate that involvement of TRIM16 in differentiation was via interactions with G1 cell cycle phase machinery.
The “linker” domain of TRIM16 is required both for TRIM16 nuclear localization and its growth inhibitory effects in neuroblastoma. Confocal localization showed the “linker” region was required for TRIM16 protein to persist in the nucleus. Furthermore, the BrdU incorporation assay also demonstrated that overexpression of plasmids containing the “linker” region of TRIM16 produced a significant decrease in proliferating cells (like that seen for the wild-type protein). It is known that TRIM16 can reduce cell proliferation in several cancers in vitro,Citation4-Citation6 and our work indicates that the “linker” region is required in the tumor suppressor-like characteristics of TRIM16 in cancer cells.
It remains controversial if the “linker” domain is actually part of an undefined domain as presented in the Liu et al. paper,Citation3 or part of the coiled-coil domain (being a small third coiled-coil), as illustrated by Beer and colleagues.Citation20 A crystal structure of TRIM16 is required to resolve this controversy, and would also give insight into the surface amino acids of TRIM16. This would be important in determining whether putative localization sequences are located on the surface of the TRIM16 protein. Other TRIM proteins have been shown to possess localization signals. A nuclear export signal was found in a leucine-rich sequence in the coiled-coil domain of RFP (TRIM27).Citation28 Additionally, PML (TRIM19) has a putative bi-partite nuclear localization sequence (NLS).Citation29 Also, a nuclear receptor binding motif (NRBM)Citation3 is located in the “linker” region and may also affect the localization of TRIM16. This LxxLL motif, which appears in many retinoid receptors, is required for tissue differentiation signaling.Citation30 Importantly, nuclear export/import of TRIM16 may involve specific NLS and NES sequence sites on TRIM16, binding to shuttling proteins and the homodimerization of TRIM16 or heterodimerization of TRIM16 to other TRIM proteins.Citation14 More research needs to be undertaken to fully explore the exact sequences and molecules involved in the actions of the coiled-coil and “linker” domain in the localization of the TRIM16 protein.
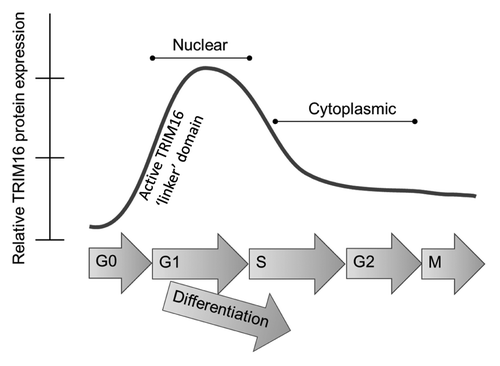
Results from this study have significantly contributed to the understanding and knowledge of TRIM16, and thus a new model of TRIM16 protein activity in neuroblastoma cells is proposed (). Although asynchronous cells have predominately cytoplasmic TRIM16, synchronization of cells to G1 shows TRIM16 localization is dynamic and is determined in part by cell cycle phase. TRIM16 localization is also dependent upon expression of both the “linker” and coiled-coil protein domains of TRIM16. Furthermore, nuclear TRIM16 is a novel marker of differentiating neuroblastoma cells and is dynamically expressed throughout ganglia differentiation. Nuclear TRIM16 is not only involved in differentiation, but is associated with proliferation inhibition, potentially acting via changes in Cyclin D1, CDK4/6 and p27. Pharmacological induction of nuclear TRIM16 presents itself as a potential target of differentiation and anti-proliferative treatment in neuroblastoma.
Figure 6. Schematic model of TRIM16 localization in neuroblastoma cells. Localization of TRIM16 is important for its growth inhibitory function. The localization of TRIM16 is influenced by cell differentiation, cell cycle phase and the activity of TRIM16’s protein domains. The linker domain of the TRIM16 protein is required for inhibition of cell cycle progression.
Materials and Methods
Cell culture and conditions
The human neuroblastoma cell lines used in these experiments were BE2C and SHEP cells. These cell lines were generously supplied by Dr J. Biedler (Memorial Sloan-Kettering Cancer Center, New York). The human embryonic kidney 293 cells (HEK 293), MDA-MB-231 and CALU-6 were purchased from the American Type Culture Collection. All cells were cultured at 37°C in 5% CO2 as an adherent monolayer in Dulbecco's modified Eagle's medium supplemented with L-glutamine and 10% fetal calf serum.
Cell cycle synchronization
Cells were synchronized at the G1/S boundary using 1 μg/mL Aphidicolin (AP) (Sigma) for 16 h incubation, and at the G2/M boundary using 0.5 μg/mL nocodazole (NZ) (Biomol) for 16 h. Synchronization in G0 was achieved by serum starvation to 0.1% fetal bovine serum for 36–48 h, and G1 cells were achieved by a 3 h in complete culture medium after serum starvation. S phase cells were at 3 h after AP release. Mitosis (M) phase cells were after 1 h of NZ release.
Flow cytometry
The cell cycle phase distribution of a cell population was determined by measuring cell DNA content by flow cytometry using propidium iodide (PI) (Sigma). The cells were trypsinized and resuspended at a concentration of 1 × 106 in complete medium, then pelleted by cold centrifugation at 500 standard gravity/g-force, rinsed with PBS and recollected as a cell pellet. The cells were then resuspended in 1 mL cold PBS and were fixed in 75% cold ethanol by adding 10 mL on top of the cell suspension and stored at 4°C, at least overnight. Prior to flow cytometric analysis, cells were pelleted and the entire supernatant carefully discarded. Cells were resuspended in 100 μL MilliQ H20, vortexed and heated at 90°C for 5 min. The samples were treated with RNase A (Sigma) for 30 min at room temperature and then stained with propidium iodide (50 μg/mL, final concentration) for 1 h at room temperature. The samples were analyzed using a BD Caliber Flow cytometer (BD). For cell cycle analysis, cells were divided into three subsets of cells that represented the G0 + G1 phase, S phase and G2 + M (as determined from comparison with untreated cell control gating).
Expression vectors
The pcDNA3.1(−).TRIM16.myc.His vector was created “in house” as previously described.Citation4,Citation5 The mutants are in the pCMV6-AC-GFP plasmid and were constructed by primer-specific PCR of full-length TRIM16 cDNA (NM_006470.3). Primers used to create GFP domain deletion vectors are displayed in the Table S1. The mutants used were M1 (AA161–564), M2 (AA290–564), M3 (AA1–372), M4 (AA1–165), M5 (AA1–294). The resulting mutants were transfected with Lipofectamine: DNA complexes in a 3:1 ratio for 8 h. Cell lysates were analyzed for presence of the transgene by western blot using TurboGFP antibody (Evrogen) according to the manufacturer’s instructions or via fluorescence and microscopy.
siRNA transfection
siRNAs were purchased commercially from Dharmacon. TRIM16 siRNA sequences: (1) PAGUAAUUCACCAUGCAGGUUU and (2) PUCUCCCUCCUGCAUUUGUGUU. Transfection was performed in the absence of serum, with cells being 50–70% confluent. Ten nM of siRNA and 4 μL Lipofectamine 2,000/mL was used in the final transfection solution, which was placed onto cells for 12 h (one well of a 6-well plate). Media was replaced, and cells were maintained in serum-free conditions for a further 24 h, before being fed complete culture media for time points, followed by cold PBS wash and cell lysis for protein extraction.
Immunofluorescence
Endogenous TRIM16 expression was determined on cells fixed in 4% paraformaldehyde and then with cold methanol. Two % BSA TBST was used to block non-specific antibody binding and dilute antibodies. 1/100 dilution TRIM16 rabbit antibody (Sigma), and 1/1,000 Alexafluor 488 anti-rabbit (Invitrogen) were used to detect TRIM16 protein. Appropriate isotype control antibodies were used (DAKO). Confocal images were captured using Olympus FV1000 confocal microscope and with 100× objective. GFP mutants’ expression were also visualized by confocal fluorescent microscope via the GFP tag on the transgene proteins. GFP vector expressing cells were treated with 4% paraformaldehyde in PBS, and after washing with PBS were permeabilized in 0.1% Triton X PBS for 5 min before mounting with vector shield media containing DAPI (Vector).
Western blotting and polymerase chain reaction (PCR)
Nuclear/cytoplasmic preparations were prepared with the NE-PER kit (Thermo Scientific) following manufacturer’s instructions. Whole-cell lysates were prepared in RIPA buffer. Twenty µg of lysates were loaded onto Criterion Gels TRIS-HCl 10% Polyacrylamide gels (Bio-Rad, NSW). Antibodies used were p27 (1/3000, BD), Cyclin E1(H12)/CDK4/CDK6/p-Rb (1/500, Cell Signaling), α-tubulin (1/5000, Sigma), Cyclin D1 (1/200, Calbiochem). RT-PCR of TRIM16 (EBBP) was performed as published in our previous studies.Citation4,Citation5
TH-MYCN mice and immunohistochemistry
TH-MYCN mice carry in the germline of the human MYCN cDNA under the control of the rat tyrosine hydroxylase (TH) promoter.Citation21,Citation27 A total of 38 mice resulting from the cross-breeding of hemizygous TH-MYCN mice were used in a blinded, histologic audit of tumor-prone paravertebral tissues over the developmental time period at day 0 (birth), day 7, 14, 42 (tumors only). TRIM16 antibody (Bethyl Laboratories) was used at 1:100 dilution; β-III-tubulin anti-rabbit antibody (Invitrogen) at 1:2,000. The sections were incubated with a biotinylated anti-rabbit secondary antibody (DAKO). The immune complexes were visualized by using liquid 3,3-diaminobenzidine (DAB) as a chromogen. Both Cyclin E1 (cell signaling) and p27 (BD) antibodies were at 1:100 with MOM kit (Dako) for mouse antibodies. Sections were counterstained with hematoxylin. Representative images are 40× captured with Aperio ScanScope XT.
Statistical analysis
Averaged replicates of three independent experiments were used in molecular and tissue culture studies. Results were statistically analyzed using the two-tailed, unpaired Student’s t-test. Results are expressed as mean values with 95% confidence intervals. Error bars represents standard error. p < 0.05 was considered statistically significant.
| Abbreviations: | ||
| TRIM16 | = | tripartite motif 16 |
| RBCC | = | the RING B-box coiled-coil |
| TH-MYCN | = | tyrosine hydroxylase MYCN |
| PML | = | promeylocytic leukemia protein |
| AP | = | aphidicolin |
| NZ | = | nocodazole |
| PI | = | propidium iodide |
| PCR | = | polymerase chain reaction |
Additional material
Download Zip (769.1 KB)Acknowledgments
We thank Dr Sela Pouha who assisted with the flow cytometer work. We are also grateful to Dr Eric Sekyere, Ms Margo Van Bekkum and Ms Joanna Keating, who aided and advised with the TH-MYCN mouse model. This research was supported by National Health and Medical Research Council (NHMRC) Biomedical Scholarship, Programme Grants from the NHMRC Australia, Cancer Institute NSW and Cancer Council NSW. The Children’s Cancer Institute Australia for Medical Research is affiliated with the University of NSW and Sydney Children’s Hospital.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Ethical Approval
The present study was approved by the Animal Care and Ethics Committee of the University of New South Wales and was conducted under the Animal Research Act 1985 (New South Wales, Australia) and the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/23825
References
- Meroni G, Diez-Roux G. TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. Bioessays 2005; 27:1147 - 57; http://dx.doi.org/10.1002/bies.20304; PMID: 16237670
- Hatakeyama S. TRIM proteins and cancer. Nat Rev Cancer 2011; 11:792 - 804; http://dx.doi.org/10.1038/nrc3139; PMID: 21979307
- Liu H-LC, Golder-Novoselsky E, Seto MH, Webster L, McClary J, Zajchowski DA. The novel estrogen-responsive B-box protein (EBBP) gene is tamoxifen-regulated in cells expressing an estrogen receptor DNA-binding domain mutant. Mol Endocrinol 1998; 12:1733 - 48; http://dx.doi.org/10.1210/me.12.11.1733; PMID: 9817599
- Cheung BB, Bell J, Raif A, Bohlken A, Yan J, Roediger B, et al. The estrogen-responsive B box protein is a novel regulator of the retinoid signal. J Biol Chem 2006; 281:18246 - 56; http://dx.doi.org/10.1074/jbc.M600879200; PMID: 16636064
- Raif A, Marshall GM, Bell JL, Koach J, Tan O, D’andreti C, et al. The estrogen-responsive B box protein (EBBP) restores retinoid sensitivity in retinoid-resistant cancer cells via effects on histone acetylation. Cancer Lett 2009; 277:82 - 90; http://dx.doi.org/10.1016/j.canlet.2008.11.030; PMID: 19147277
- Marshall GM, Bell JL, Koach J, Tan O, Kim P, Malyukova A, et al. TRIM16 acts as a tumour suppressor by inhibitory effects on cytoplasmic vimentin and nuclear E2F1 in neuroblastoma cells. Oncogene 2010; 29:6172 - 83; http://dx.doi.org/10.1038/onc.2010.340; PMID: 20729920
- Cheung BB, Koach J, Tan O, Kim P, Bell JL, D’andreti C, et al. The retinoid signalling molecule, TRIM16, is repressed during squamous cell carcinoma skin carcinogenesis in vivo and reduces skin cancer cell migration in vitro. J Pathol 2012; 226:451 - 62; http://dx.doi.org/10.1002/path.2986; PMID: 22009481
- Galderisi U, Jori FP, Giordano A. Cell cycle regulation and neural differentiation. Oncogene 2003; 22:5208 - 19; http://dx.doi.org/10.1038/sj.onc.1206558; PMID: 12910258
- Watanabe Y, Watanabe T, Kitagawa M, Taya Y, Nakayama K-i, Motoyama N. pRb phosphorylation is regulated differentially by cyclin-dependent kinase (Cdk) 2 and Cdk4 in retinoic acid-induced neuronal differentiation of P19 cells. Brain Res 1999; 842:342 - 50; http://dx.doi.org/10.1016/S0006-8993(99)01844-2; PMID: 10526130
- Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999; 98:859 - 69; http://dx.doi.org/10.1016/S0092-8674(00)81519-6; PMID: 10499802
- Sherr CJ. G1 phase progression: cycling on cue. Cell 1994; 79:551 - 5; http://dx.doi.org/10.1016/0092-8674(94)90540-1; PMID: 7954821
- Chan JYH, Li L, Fan YH, Mu ZM, Zhang WW, Chang KS. Cell-cycle regulation of DNA damage-induced expression of the suppressor gene PML. Biochem Biophys Res Commun 1997; 240:640 - 6; http://dx.doi.org/10.1006/bbrc.1997.7692; PMID: 9398618
- Everett RD, Lomonte P, Sternsdorf T, van Driel R, Orr A. Cell cycle regulation of PML modification and ND10 composition. J Cell Sci 1999; 112:4581 - 8; PMID: 10574707
- Bell JL, Malyukova A, Holien JK, Koach J, Parker MW, Kavallaris M, et al. TRIM16 acts as an E3 ubiquitin ligase and can heterodimerize with other TRIM family members. PLoS ONE 2012; 7:e37470; http://dx.doi.org/10.1371/journal.pone.0037470; PMID: 22629402
- Kurki S, Latonen L, Laiho M. Cellular stress and DNA damage invoke temporally distinct Mdm2, p53 and PML complexes and damage-specific nuclear relocalization. J Cell Sci 2003; 116:3917 - 25; http://dx.doi.org/10.1242/jcs.00714; PMID: 12915590
- Dellaire G, Bazett-Jones DP, Dellaire G, Bazett-Jones DP. PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. Bioessays 2004; 26:963 - 77; http://dx.doi.org/10.1002/bies.20089; PMID: 15351967
- Bernardi R, Papa A, Pandolfi PP. Regulation of apoptosis by PML and the PML-NBs. Oncogene 2008; 27:6299 - 312; http://dx.doi.org/10.1038/onc.2008.305; PMID: 18931695
- Quignon F, De Bels F, Koken M, Feunteun J, Ameisen JC, de Thé H. PML induces a novel caspase-independent death process. Nat Genet 1998; 20:259 - 65; http://dx.doi.org/10.1038/3068; PMID: 9806544
- Borriello A, Pietra VD, Criscuolo M, Oliva A, Tonini GP, Iolascon A, et al. p27Kip1 accumulation is associated with retinoic-induced neuroblastoma differentiation: evidence of a decreased proteasome-dependent degradation. Oncogene 2000; 19:51 - 60; http://dx.doi.org/10.1038/sj.onc.1203231; PMID: 10644979
- Beer HD, Munding C, Dubois N, Mamie C, Hohl D, Werner S. The estrogen-responsive B box protein: a novel regulator of keratinocyte differentiation. J Biol Chem 2002; 277:20740 - 9; http://dx.doi.org/10.1074/jbc.M111233200; PMID: 11919186
- Hansford LM, Thomas WD, Keating JM, Burkhart CA, Peaston AE, Norris MD, et al. Mechanisms of embryonal tumor initiation: distinct roles for MycN expression and MYCN amplification. Proc Natl Acad Sci USA 2004; 101:12664 - 9; http://dx.doi.org/10.1073/pnas.0401083101; PMID: 15314226
- Bergmann E, Wanzel M, Weber A, Shin I, Christiansen H, Eilers M. Expression of P27(KIP1) is prognostic and independent of MYCN amplification in human neuroblastoma. Int J Cancer 2001; 95:176 - 83; http://dx.doi.org/10.1002/1097-0215(20010520)95:3<176::AID-IJC1030>3.0.CO;2-Z; PMID: 11307151
- Niu MY, Ménard M, Reed JC, Krajewski S, Pratt MAC. Ectopic expression of cyclin D1 amplifies a retinoic acid-induced mitochondrial death pathway in breast cancer cells. Oncogene 2001; 20:3506 - 18; http://dx.doi.org/10.1038/sj.onc.1204453; PMID: 11429697
- Gillett C, Fantl V, Smith R, Fisher C, Bartek J, Dickson C, et al. Amplification and overexpression of cyclin D1 in breast cancer detected by immunohistochemical staining. Cancer Res 1994; 54:1812 - 7; PMID: 8137296
- Watanabe G, Albanese C, Lee RJ, Reutens A, Vairo G, Henglein B, et al. Inhibition of cyclin D1 kinase activity is associated with E2F-mediated inhibition of cyclin D1 promoter activity through E2F and Sp1. Mol Cell Biol 1998; 18:3212 - 22; PMID: 9584162
- Mateyak MK, Obaya AJ, Sedivy JM. c-Myc regulates cyclin D-Cdk4 and -Cdk6 activity but affects cell cycle progression at multiple independent points. Mol Cell Biol 1999; 19:4672 - 83; PMID: 10373516
- Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J 1997; 16:2985 - 95; http://dx.doi.org/10.1093/emboj/16.11.2985; PMID: 9214616
- Cao T, Borden KL, Freemont PS, Etkin LD. Involvement of the rfp tripartite motif in protein-protein interactions and subcellular distribution. J Cell Sci 1997; 110:1563 - 71; PMID: 9247190
- Duprez E, Saurin AJ, Desterro JM, Lallemand-Breitenbach V, Howe K, Boddy MN, et al. SUMO-1 modification of the acute promyelocytic leukaemia protein PML: implications for nuclear localisation. J Cell Sci 1999; 112:381 - 93; PMID: 9885291
- Loinder K, Söderström M. Functional analyses of an LXXLL motif in nuclear receptor corepressor (N-CoR). J Steroid Biochem Mol Biol 2004; 91:191 - 6; http://dx.doi.org/10.1016/j.jsbmb.2004.04.006; PMID: 15336696