Abstract
Previously, we have identified the RUNX1 gene as hypomethylated and overexpressed in post-chemotherapy (CT) primary cultures derived from epithelial ovarian cancer (EOC) patients, when compared with primary cultures derived from matched primary (prior to CT) tumors. Here we show that RUNX1 displays a trend of hypomethylation, although not significant, in omental metastases compared with primary EOC tumors. Surprisingly, RUNX1 displayed significantly higher expression not only in metastatic tissue, but also in high-grade primary tumors and even in low malignant potential tumors. The RUNX1 expression levels were almost identical in primary tumors and omental metastases, suggesting that RUNX1 hypomethylation might have a limited impact on its overexpression in advanced (metastatic) stage of the disease.
Knockdown of the RUNX1 expression in EOC cells led to sharp decrease of cell proliferation and induced G1 cell cycle arrest. Moreover, RUNX1 suppression significantly inhibited EOC cell migration and invasion. Gene expression profiling and consecutive network and pathway analyses confirmed these findings, as numerous genes and pathways known previously to be implicated in ovarian tumorigenesis, including EOC tumor invasion and metastasis, were found to be downregulated upon RUNX1 suppression, while a number of pro-apoptotic genes and some EOC tumor suppressor genes were induced.
Taken together, our data are indicative for a strong oncogenic potential of the RUNX1 gene in EOC progression and suggest that RUNX1 might be a novel EOC therapeutic target. Further studies are needed to more completely elucidate the functional implications of RUNX1 and other members of the RUNX gene family in ovarian tumorigenesis.
Introduction
Epithelial ovarian cancer (EOC) accounts for 4% of all cancers in women and is the leading cause of death from gynecologic malignancies.Citation1 Despite treatment improvements, long-term survival rates for patients with advanced disease remain disappointing.Citation2 The molecular basis of EOC initiation and progression is still poorly understood,Citation3 as experimental models of ovarian cancer lack disease specificity.Citation4 To establish novel therapeutic and diagnostic strategies against this deadly disease, it is essential to understand its molecular pathology.
Disruption of normal gene regulation is important for carcinogenesis resulting in loss or gain of genetic function. Recently, the importance of epigenetic perturbation of gene regulation in cancer,Citation5 including EOC,Citation6 has begun to be more fully appreciated. The most studied epigenetic alteration is DNA methylation, the addition of a methyl moiety to the cytosine-5 position within the context of a CpG dinucleotide, mediated by DNA methyltransferases.Citation5 DNA methylation patterns are reset early in the embryogenesis and reestablished early during development. After that, they are thought to be relatively stable. In cancer, the physiological regulation of DNA methylation is disrupted, leading to drastic changes of the distribution pattern of 5-methylcytosine. The heavy methylation found in the bulk of chromatin is reduced, while the normally unmethylated CpG islands located in the promoter and first exon of genes become hypermethylated. Promoter hypermethylation often leads to inactivation of different tumor-suppressing genes and is associated with many important pathways involved in cancer, such as DNA repair, cell cycle regulation, apoptosis, carcinogen metabolism, hormonal response and cell adherence.Citation7 Aberrant DNA methylation is also involved in the development of resistance to chemotherapy (CT).Citation8 The role of DNA hypomethylation in carcinogenesis is less studied. Recent studies have demonstrated that global decrease in the level of DNA methylation is related to hypomethylation of repeated sequences, increase in genetic instability as well as re-activation of proto-oncogenes and pro-metastasis genes.Citation9
Similar to other malignancies, aberrant DNA methylation, including global hypomethylation of heterochromatin and local CpG island methylation, occurs in EOC and contributes to ovarian tumorigenesis and mechanisms of chemoresistance.Citation6 Applying a more global array-based technology, several studies have demonstrated that DNA methylation changes in ovarian cancer are cumulative with disease progression and CT resistance.Citation10-Citation12 Using a similar approach (methylated DNA immunoprecipitation coupled to CpG island tiling arrays), we have recently shown that DNA hypermethylation occurs in less invasive/early stages of ovarian tumorigenesis, while advanced disease was associated with DNA hypomethylation of a number of oncogenes implicated in cancer progression, invasion/metastasis and probably chemoresistance.Citation13 This observation was further confirmed when comparing the DNA methylation profiles of primary cell cultures derived from matched tumor samples obtained prior to, and following, CT treatment from two serous EOC patients with advanced disease. The runt-related transcription factor 1 (RUNX1) was among the genes identified to be notably hypomethylated and overexpressed in the post-CT primary cultures.
The RUNX1 gene belongs to the RUNX gene family, which encodes transcription factors (including RUNX2 and RUNX3) that bind DNA as components of the core-binding factor (CBF) complex in partnership with the CBFβ cofactor.Citation14 This complex activates and represses transcription of key regulators of growth, survival and differentiation pathways.Citation15-Citation17 RUNX1 is essential for definitive hematopoiesis, megakaryocyte maturation, T- and B-cell lineages and neuronal development.Citation18 The prosurvival activity of RUNX1 is mediated by transcriptional regulation of enzymes involved in sphingolipid metabolism, which may reduce intracellular long-chain ceramides with elevation of extracellular sphingosine 1 phosphate.Citation19 RUNX1 also suppresses the onset of apoptosis in response to exogenous tumor necrosis factor α by opposing activation of c-Jun-NH2-kinase and p38MAPK, keys mediators of ceramides-induced death.Citation19
The importance of RUNX1 in hematopoiesis and its tumor suppressor function in leukemia are well established,Citation20 although RUNX1 gene amplifications and gain-of-RUNX1 function mutations have been postulated to have leukemogenic effects.Citation21,Citation22 Similarly, recent studies in solid tumors present contrasting roles of RUNX1 as either a tumor suppressor or oncogene (reviewed in ref. Citation20). The implication of RUNX1 in EOC tumorigenesis is currently unknown, although it was shown that in conjunction with some matrix metalloproteinases (MMP-2 and -9), RUNX1 could contribute to the invasive stage of endometrial and ovarian endometrioid carcinomas.Citation23
This prompted us to further investigate if RUNX1 expression is epigenetically modulated (due to DNA hypomethylation) in advanced EOC, and whether the RUNX1 gene is functionally implicated in EOC tumorigenesis, including disease progression and response to treatment.
Results
RUNX1 gene exhibits specific hypomethylation in serous EOC omental metastases compared with primary serous EOC tumors
Previously, we have identified the RUNX1 gene as hypomethylated and overexpressed in post-CT primary cell cultures, derived from two serous EOC patients, when compared with matched primary cultures, obtained prior to CT. RUNX1 has two promoters driving the expression of three isoforms: the distal promoter P1 and the proximal promoter P2.Citation20 P1 controls the longest isoform RUNX1c. RUNX1a is the shortest, and RUNX1b the most expressed form, and the proximal P2 promoter drives expression of both of them (Fig. S1). Here, we have further validated the RUNX1 methylation status in primary tumors and omental metastases. BSP analysis was performed targeting a 419 bp DNA fragment of the proximal promoter (P2) region of RUNX1 gene, stretching between nt −2,475 to −2,056 upstream of its first exon and containing 13 putative CpG methylation targets (; see also Fig. S1 for RUNX1 gene structure in relation to these CpG sites). As seen in , the BSP analysis displayed a trend of hypomethylation of some of these targets in metastatic tissues, compared with primary EOC tumors, although the difference in methylation was not significant. No differences were observed when comparing RUNX1 methylation status between primary EOC tumors and normal ovarian tissue samples (data not shown).
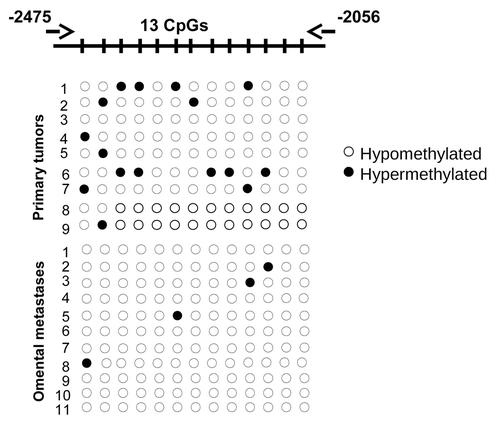
Figure 1. BSP analysis of the methylation status of RUNX1 in grade 3 primary serous EOC tumors compared with omental metastases. Filled circles represent methylated CpGs, and open circles represent unmethylated CpGs. CpG plot of the analyzed region is also presented (CpGs are displayed with vertical marks). The indicated positions on the CpG plot represent the number of nucleotides stretching upstream of the first exon of the RUNX1 gene.
Analysis of RUNX1 expression in serous EOC tumors by immunohistochemistry (IHC)
We further evaluated RUNX1 protein expression by IHC in serous EOC tumors and ovarian normal tissue samples, using tissue microarrays (TMAs). Our TMAs included triplicate cores of 117 serous EOC tumors, including 13 low-malignant potential (LMP) tumors, 52 high-grade tumors and 52 omental metastases. Thirteen normal ovarian tissue samples were also included as controls. shows the major clinical characteristics of patients for whom extensive follow-up clinical data (up to 5 y) were available. The age ranged from 41 to 83 y (median: 64 y). High-grade tumors were mainly grade 3 (99%) and stage III (80%). The majority of patients (87%) received a combination of platinum and taxol. The median baseline CA125 was around 800 U/ml. Forty percent of the patients had a progression or a recurrence within the first 6 mo of follow-up; for 35.4% of the patients, the progression-free survival (PFS) interval was in the range of 7 to 24 mo, and 21.2% of the patients displayed PFS values higher than 25 mo ().
Table 1. Patients’ characteristics
Surprisingly, RUNX1 displayed significantly higher expression not only in metastatic tissue, but also in LMP and high-grade primary tumors when compared with normal tissue, as the expression levels were almost identical between the primary tumors and omental metastases (). These findings indicate that the hypomethylation of some of the CpG targets might have a limited, if any, impact on its expression in advanced (metastatic) stage of the disease. Kaplan-Meier survival curves based on RUNX1 expression analyses in cohort of 52 high-grade serous ovarian adenocarcinoma patients displayed no association with PFS (data not shown).
Figure 2. Analysis of RUNX1 expression in serous EOC tumors by IHC. (A) Representative IHC images of RUNX1 protein expression in normal ovarian tissues, LMP tumors, high-grade tumors and omental metastases. (B) Box-plot presentation of RUNX1 protein expression levels in normal ovarian tissues, LMP tumors, high-grade tumors and omental metastases.
Phenotype analysis of RUNX1 suppression in EOC cells: Possible implications in EOC cell proliferation, cell cycle control, migration and invasion
Next, we decided to verify if short hairpin RNA (shRNA)-mediated RUNX1 gene knockdown could produce any cancer-related phenotypic changes in EOC cells. We tested several EOC cell lines for endogenous RUNX1 expression by sqRT-PCR and western analysis (data not shown). Among these, the SKOV3 cell line displayed strong RUNX1 expression and was further used to generate stably transfected shRNA-RUNX1 clones. Clone selection for further analyses was based on sqRT-PCR and western blot validation of the RUNX1 gene/protein expression in selected clones compared with empty vector-transfected clones. Among the clones analyzed, the shRNA-RUNX1 knockdown clone sh-cl4 displayed a significant decrease of RUNX1 expression levels compared with the mock-transfected control () and was selected for further analyses.
Figure 3. Analysis of RUNX1 expression in SKOV3 cells. (A) Semi-quantitative duplex RT-PCR analysis of RUNX1 mRNA expression levels in the shRNA-RUNX1 clone sh-cl4, compared with the mock-transfected clone ctrl3. The 18S rRNA gene was used as internal standard. (B) Western blot analysis of RUNX1 protein expression in the shRNA-RUNX1 clone sh-cl4, compared with the mock-transfected clone ctrl3. β-actin was used as a loading control.
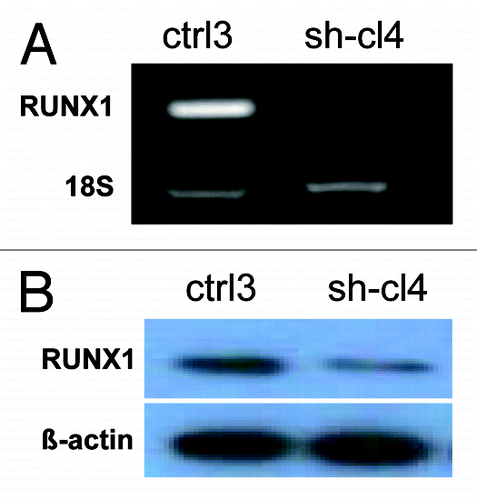
We investigated the impact of RUNX1 gene suppression on SKOV3 cell proliferation, cell cycle control, migration, invasion and sensitivity to cisplatin and paclitaxel (drugs, conventionally used for first-line EOC CT). The RUNX1 gene knockdown led to sharp decrease of the number of viable adherent cells (represented by cell index) compared with control cells (). This observation was further supported by the colony formation assay showing that the numbers of clones formed by cells with stably reduced RUNX1 expression were significantly lower than that of control cells (). Taken together, our observations strongly indicate an influence of RUNX1 transcripts on EOC cell proliferation and further on their propensity to form colonies. Moreover, when compared with control clones, the shRNA-RUNX1 clone exhibited a significant accumulation of cells in the G1 phase, with a corresponding reduction of cells in the S and G2/M phases at 0, 3, 6, 9 and 24 h, after removing hydroxyurea (). These data indicate that RUNX1 depletion induces G1 arrest and, thus, explain the drastic reduction in the proliferation rates of these cells observed earlier.
Figure 4. ShRNA-mediated knockdown of the RUNX1 expression in SKOV3 cells: (A) effect on cell proliferation; (C) effect on cell cycle control. (B) Representative images of colony forming assays following RUNX1 knockdown.
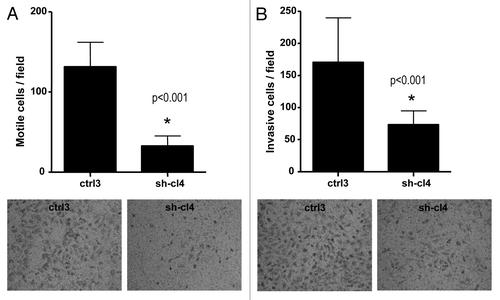
Additionally, RUNX1 suppression significantly inhibited both migration and invasion of SKOV3 cells. As shown in , the numbers of SKOV3 cells that passed through the filter using the sh-cl4 clone were remarkably less than that in the negative control (ctrl3) clone, which is indicative of a role for RUNX1 in the regulation of invasion and migration in EOC.
Figure 5. Effect of RUNX1 knockdown on SKOV3 cell migration and invasion. (A) Migration was assessed using Boyden chamber assay. Cells from the shRNA-RUNX clone (sh-cl4) and the control clone (ctrl3) were seeded into the upper chambers in 0.1% FBS containing medium at a density of 2.5 × 104 per well, and 600 μl of 1% FBS-containing medium was placed in the lower chamber as a chemoattractant. After 24 h at 37°C in 5% CO2, the cells were fixed with cold methanol and stained with blue trypan solution. Migrated cells on the underside of the filter were photographed and counted by phase contrast microscopy. (B) Cell invasion was assayed in a similar way, as the upper chambers were coated with Matrigel. Here, NIH3T3 conditioned medium was added in the lower chamber as a chemoattractant (see “Materials and Methods” for details). All experiments were performed in triplicate. For each experiment, cell number was calculated as the total count from 10 random fields per filter (at magnification ×40). Differences between shRNA-RUNX1-transfected and vehicle-transfected SKOV3 cells were determined by a Student’s t-test, where p < 0.05 was considered significant.
Finally, RUNX1 suppression had no significant impact on SKOV3 cisplatin and paclitaxel sensitivity (data not shown).
Molecular mechanisms of RUNX1 action in EOC cells
To better understand the molecular mechanisms of RUNX1 action in EOC cells, we employed the Agilent Whole Human Genome microarrays, containing ~44,000 genes to identify global gene expression changes upon RUNX1 suppression in SKOV3 cells. We compared the gene expression of the previously selected clones shRNA- RUNX1 (sh-cl4) against the corresponding control clone (ctrl3). All microarray experiments were performed in duplicates, as two hybridizations were performed for the RUNX1-suppressing cell clone against the corresponding control, using a fluorescent dye reversal (dye-swap) technique. For each comparison, a subset of differentially expressed genes was selected, displaying at least 2-fold difference in both duplicate microarray experiments. Using these selection criteria, we found 334 genes to be upregulated and 607 genes to be downregulated in SKOV3 cells following RUNX1 knockdown, as the RUNX1 gene displayed 3.48-fold suppression in the shRNA-RUNX1 (sh-cl4) clone, compared with the corresponding control (Table S1). In order to investigate if some the differentially expressed genes represent RUNX1 direct target genes, we searched for human RUNX1 binding sites using the oPOSSUM software (www.cisreg.ca/cgi-bin/oPOSSUM/opossum).Citation24 We found that almost half of both upregulated genes (140/334) and downregulated genes (282/607) contained at least one conserved RUNX1 binding site in their regulatory regions (see Table S1 for details). shows a list of selected functionally related groups of genes that were differentially expressed (≥ 2-fold) in SKOV3 cells upon RUNX1 knockdown. As seen from , genes with previously shown implication in mechanisms of cell growth and proliferation, cell adhesion, regulation of transcription, metabolism and transport were predominantly suppressed, while RUNX1 knockdown was associated with the induction of apoptosis-related genes. Comparable numbers of genes functionally related to cell cycle control and signal transduction were equally up- and downregulated following RUNX1 suppression. Table S1 shows the complete list of the differentially expressed genes (≥ 2-fold) following RUNX1 knockdown in SKOV3 cells.
Table 2. Selected differentially expressed gene groups in SKOV3 cells upon RUNX1 knock-down
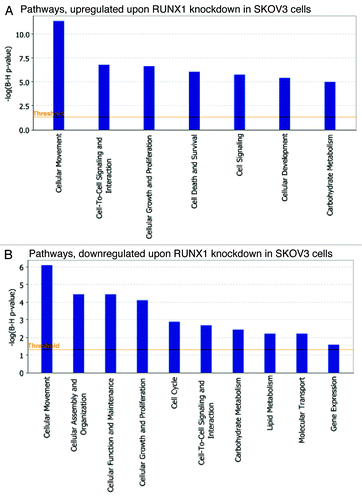
Pathway and network analyses, generated through the use of the IPA software confirmed the major functionally related gene groups, found to be differentially expressed in the shRNA-RUNX1 clone. As seen from , similar pathways were both induced and suppressed upon RUNX1 knockdown: these included pathways functionally related to cellular movement, cellular growth and proliferation, cell-to-cell signaling and interaction and carbohydrate metabolism. Pathways implicated in cell death and survival, cell signaling and cellular development were upregulated (), while those linked to cellular assembly and organization, cellular function and maintenance, cell cycle, lipid metabolism, molecular transport and gene expression were mostly suppressed ().
Figure 6. Functional analysis for a data set of differentially expressed genes (≥ 2-fold) following RUNX1 suppression in SKOV3 cells. (A) Functional analysis of upregulated genes. (B) Functional analysis of downregulated genes. Top functions that meet a p-value cutoff of 0.05 are displayed.
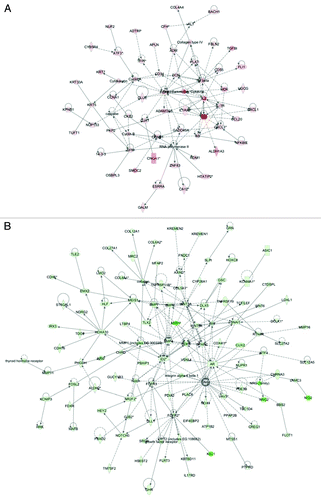
Common networks obtained upon merging the five top-scoring networks recognized some important nodes that are specifically up- or downregulated upon RUNX1 suppression in SKOV3 cells (). Thus, major nodes and associated interaction partners that were upregulated upon RUNX1 knockdown (displayed in ) comprised genes implicated in signal transduction (ADM, DCN, FGF2, SFN, TGF β), cell adhesion (CD34, CXCL3), cell-to-cell signaling (GJA1), cell cycle control (CCNA1, CCNB1, GADD45A), gene expression (ATF3; members of the RNA polymerase II network), immune response (IL1 network, including IL1A) and cytoskeleton formation (KRT5, cytokeratin). Gene nodes that were downregulated upon RUNX1 knockdown in SKOV3 cells are presented in ; these were mostly involved in signal transduction (AR, BMP4, BMP6, BMP7, FGFR2, FGFR3, Notch, NOTCH3, WNT3A, WNT5A, members of the AKT network), regulation of transcription (DLX5, HOXA10, ID2, LEF1, MEIS1, SNAI1), cell growth and/or maintenance (collagen, SPARC), metabolism (MMP1, MMP7) and cell adhesion (CDH1). Interestingly, the majority of the up- and downregulated gene nodes listed above (including ADM, DCN, FGF2, TGF β, CD34, GADD45A, ATF3, IL1A, BMP4, BMP6, BMP7, FGFR3, WNT5A, DLX5, ID2, LEF1, MEIS1 and SPARC) represent RUNX1 direct target genes (Table S1).
Figure 7. Network analysis of dynamic gene expression in SKOV3 cells based on the 2-fold common gene expression list obtained following shRNA-mediated RUNX1 knockdown. (A) Upregulated networks. (B) Downregulated networks. The five top-scoring networks for each cell line were merged and are displayed graphically as nodes (genes/gene products) and edges (the biological relationships between the nodes). Intensity of the node color indicates the degree of up- (red) or downregulation (green). Nodes are displayed using various shapes that represent the functional class of the gene product (square, cytokine, vertical oval, transmembrane receptor, rectangle, nuclear receptor, diamond, enzyme, rhomboid, transporter, hexagon, translation factor, horizontal oval, transcription factor, circle, other). Edges are displayed with various labels that describe the nature of relationship between the nodes: ____, binding only; →, acts on. The length of an edge reflects the evidence supporting that node-to-node relationship, in that edges supported by article from literature are shorter. Dotted edges represent indirect interaction.
Validation of microarray findings with semi-quantitative RT-PCR (sqRT-PCR)
To validate microarray results, we arbitrarily selected nine differentially expressed genes and quantified their expression by sqRT-PCR in SKOV3 cells following shRNA-RUNX1 knockdown compared with control (vehicle-transfected) SKOV3 cells. summarizes the gene expression measurements of all validated genes. We found that both methods (microarray analysis and sqRT-PCR) detected similar patterns for the up- and downregulated genes selected for validation.
Table 3. Semi-quantitative RT-PCR validation of microarray data
Discussion
As previously demonstrated, the RUNX1 protein can activate or repress target gene expression depending on whether it interacts with a co-activator or co-repressor, and, as a consequence, it has displayed both oncogenic and tumor-suppressive functions in different cancer types.Citation20 Thus, RUNX1 overexpression displays context-specific effects, inducing growth arrest or senescence in most primary cell types, but promoting proliferation and/or survival in cells expressing collaborating oncogenes or mutated tumor suppressor pathways.Citation25-Citation28 The dysfunction of RUNX1 is strongly correlated with hematological disorders. Indeed RUNX1, also known as AML1 (acute myeloid leukemia-1), has long been recognized as an important translocation breakpoint in human leukemias, with the TEL-AML1 t (12;21) fusion accounting for 20% of acute lymphoblastic leukemia (ALL) cases and the AML1-ETO t (8;21) fusion accounting for 12% of AMLs.Citation17 RUNX1 is also frequently mutated in AML and myelodisplastic syndrome,Citation25,Citation29,Citation30 suggesting a tumor suppressor role of this gene in these malignancies. However, oncogenic functions for RUNX1 are also demonstrated, since RUNX1 amplification is associated with some cases of childhood acute ALL, as well as with Down syndrome-related acute megakaryoblastic leukemia,Citation31 and RUNX1 positively modulates myeloid leukemogenesis in animal models.Citation32 Similarly, there is a potential RUNX1 tumor suppressor role in breast, intestinal and esophagus cancer;Citation33-Citation36 however, RUNX1 overexpression has been associated with development of invasive/metastatic endometrial carcinoma,Citation37,Citation38 and a recent study was indicative for the role of RUNX1 in driving cancer stem cell proliferation and promoting carcinogenesis in epithelial tissues.Citation39 These data warrant further studies to elucidate the role of RUNX1 in formation and/or progression of human cancers.
Our previous findings, based on analyses in primary cultures, derived from matched tumor samples obtained prior to and following CT treatment from two serous EOC patients, were suggestive for RUNX1 overexpression in advanced (metastatic) EOC, which might be due to epigenetic mechanisms associated with DNA hypomethylation of its putative promoter region.Citation13 Here, we have shown that the stretch of DNA sequence, located between nt −2,475 to −2,056 upstream of the first exon of RUNX1 isoforms 1a and 1b displays rather limited and non-significant hypomethylation in EOC omental metastases compared with primary serous EOC tumors, which probably has no impact on RUNX1 expression. Moreover, our IHC analyses were indicative for equally strong RUNX1 protein overexpression both in grade 3 serous EOC tumors and metastatic tissues. However, a recent in silico study based on the Cancer Genome Atlas (TCGA) data and focused on the clinical relevance of epigenetic modifications of Notch superfamily genes in high-grade serous EOC identified a significant inverse relationship between RUNX1 methylation status and its mRNA expression levels.Citation40 Moreover, patients with a lower RUNX1 methylation level and higher RUNX1 expression level had significantly poorer overall survival compared with patients with a higher RUNX1 methylation level and lower RUNX1 expression level. Thus, we cannot fully exclude that epigenetic alterations of RUNX1 expression could influence its role in ovarian tumorigenesis. Interestingly, RUNX1 knockdown was associated with downregulation of members of the Notch pathway, including NOTCH3 (see below and ).
We have found a significantly higher RUNX1 expression not only in metastatic tissue, but also in high-grade primary tumors and even in LMP tumors, compared with normal ovarian tissue. Normal ovarian tissues controls consistently displayed low RUNX1 expression; minimal expression was also detected in other human adult tissues (data not shown). The above findings persuaded us to investigate the functional implication of RUNX1 in mechanisms of EOC tumorigenesis. Our functional analyses are strongly indicative of evident oncogenic capacity of RUNX1 in serous EOC, including its potential role in EOC cell proliferation, cell cycle control and cell migration/invasion ( and ). Thus, our data confirm recent findings that RUNX1is required for tumor and cancer cell growth and/or invasion/metastasis, as found for skin, head and neck, prostate, colon and rectal and endometrioid cancers.Citation37,Citation39,Citation41,Citation42
To better elucidate the molecular mechanisms and biological pathways implicated in RUNX1-mediated action in EOC cells, we used a complementary gene expression profiling using the DNA microarray technology to monitor cellular changes in gene expression and discover the molecular targets upon RUNX1 suppression in EOC cells. To our knowledge, the present work represents the first effort to define global changes in gene expression upon modulation of RUNX1 gene expression in epithelial cancer cells. The gene expression data and consecutive IPA network and pathway analyses were quite confirmatory of the data obtained by the RUNX1 functional assays. Indeed, microarray data sustained RUNX1 correlation with EOC cell proliferation (including cell cycle control), migration and invasion, since RUNX1 knockdown resulted in reduced of genes associated with cell proliferation, cell migration and cell invasion and cell cycle control, while inducing some pro-apoptotic genes ( and ).
IPA network analysis was indicative for some important gene nodes linked to RUNX1 suppression in EOC cells, as most of these substantiate and/or complement the functional data obtained. Thus, RUNX1 knockdown resulted in upregulation of gene nodes known to be implicated in apoptosis induction (ATF3, CCNA1, CCNB1, GADD45A, IL1A, TGF β)Citation43-Citation50 or suppressing cell cycle progression and survival (CD55, SFN),Citation51-Citation54 as well as gene nodes linked to suppression of cell invasion and/or metastasis (ADM, CXCL3, DCN, FGF2, GJA1)Citation55-Citation59 or increasing cell adhesion (CD34)Citation60 (). Among these, CCNA1, DCN, GADD45A and SFN have been previously recognized as potential EOC tumor suppressor genes,Citation61-Citation64 while increased expression of ADM and ATF in EOC tumors was associated with positive disease outcomeCitation65 and decreased tumor invasion.Citation66 The CCNB1 was found to be downregulated in advanced EOC,Citation67 and GJA1 inhibition was shown to promote EOC drug resistance.Citation68 The TGFβ gene and related pathway displays a dual role in EOC, both promoting and inhibiting tumorigenesis.Citation69
In parallel, upon RUNX1 knockdown, we have observed a predominant and strong downregulation of major gene nodes (including AR, BMP4, BMP6, BMP7, CDH1, DLX5, FGFR2, FGFR3, HOXA10, ID2, LEF1, MEIS1, MMP1, MMP7, members of the Notch pathway including NOTCH3, SNAI1, SPARC, WNT3A, WNT5A) with proven functional implication in EOC cellular growth and proliferation, migration, invasion and metastasis (). All these gene nodes are also known to be implicated in EOC tumorigenesis (BMP4, BMP6, BMP7, CDH1, LEF1, MEIS1, NOTCH3, WNT3A, WNT5A),Citation70-Citation74 including disease progression (AR, FGFR2, FGFR3, DLX5, HOXA10)Citation75-Citation78 and EOC tumor invasion/metastasis (ID2, MPP1, MPP7, SNAI1, SPARK)Citation79-Citation82 (). Our data confirm previous findings for the possible implication of RUNX1 in metalloproteinase-mediated invasion and metastasis in gynecological cancers.Citation23 Additionally, RUNX1 suppression was associated with downregulation of numerous genes from the AKT pathway (), representing one of the major oncogenic pathways implicated in EOC etiology.Citation83 Thus, our findings support the concept of oncogenic functionality of RUNX1 in EOC carcinogenesis. Given that all three RUNX proteins recognize common DNA sequence motifs, it would be important to investigate the role of the other members of the RUNX gene family in EOC cancer formation and progression. Similarly to RUNX1, the RUNX2 gene was also identified as hypomethylated and overexpressed in post-CT EOC primary cultures compared with matched primary cultures derived prior to CT,Citation13 and its role in EOC progression is currently under investigation. Moreover, RUNX2 was recently associated with advanced EOC progression and poor disease prognosis.Citation84 As for the RUNX3 gene, the current literature data are rather contradictory, since RUNX3 was initially recognized as an EOC tumor suppressor gene that is downregulated in EOC tumors due to DNA hypermethylation;Citation85 however, novel findings are indicative of RUNX3 overexpression and oncogenic function in ovarian carcinoma.Citation86
In conclusion, we have shown that the RUNX1 transcription factor is significantly overexpressed in serous EOC tumors, including LMP tumors, compared with normal ovarian tissue. BSP validation of the RUNX1 methylation status in primary EOC tumors and omental metastasis was mostly indicative for limited or no implication of epigenetics mechanisms (DNA hypomethylation) in RUNX1 overexpression in metastatic tissues. Consecutive functional analyses of RUNX1 in EOC cells pointed toward its association with EOC cell proliferation (including cell cycle control), migration and invasion. Gene expression profiling and consecutive network and pathway analyses confirmed these findings, as numerous genes and pathways known previously to be implicated in ovarian tumorigenesis, including EOC tumor invasion and metastasis, were found to be suppressed upon RUNX1 knockdown, while a number of pro-apoptotic genes and some EOC tumor suppressor genes were found to be induced. Hence, RUNX1 is possibly required for EOC tumor and cancer cell growth and invasion and could represent a potential EOC therapeutic target. Further in vitro and in vivo studies, including also the other RUNX genes, are warranted to more completely elucidate the functional implications of the RUNX transcription factors in ovarian tumorigenesis.
Materials and Methods
Patients and tissue specimens
Snap frozen and formalin-fixed paraffin-embedded (FFPE) tissues of 117 serous EOC tumors were obtained at the Hotel-Dieu de Quebec Hospital. These included 13 borderline, or low-malignant potential (LMP) tumors, 52 high-grade adenocarcinomas and 52 omental metastases. None of the patients received chemotherapy before surgery (see for detailed clinicopathological characteristics). All tumors were histologically classified according to the criteria defined by the World Health Organization.Citation87 The CT treatment was completed for all patients, and the response to treatment was known. Disease progression was evaluated following the guidelines of the Gynecology Cancer Intergroup.Citation87 Progression-free survival (PFS) was defined as the time from surgery to the first observation of disease progression, recurrence or death. Thirteen normal ovarian samples were derived from women subjected to hysterectomy with oophorectomy due to non-ovarian pathologies. The study was approved by the Clinical Research Ethics Committee of the Hotel-Dieu de Quebec Hospital, and all patients signed an informed consent for voluntary participation.
Cell cultures
The EOC cell lines OVCAR3, SKOV3 and C13 were purchased from American Tissue Type Collection; OV-90, OV2008, TOV-112 and TOV-21 cell lines were a kind gift from Dr Anne-Marie Mes-Masson (Montreal University), while A2780s and A2780cp cell lines were a kind gift from Dr Benjamin Tsang (Ottawa University). The cell lines were passaged in different culture media supplemented with 10% fetal bovine serum, as described previously.Citation88
Bisulfite sequencing PCR (BSP) analysis
BSP analysis was performed, as previously described.Citation89 Briefly, genomic DNAs from primary and metastatic EOC tumor specimens were isolated using the Qiagen DNeasy Blood and Tissue Kit. Bisulfite modification of genomic DNAs was done using the Methyl Detector kit (Active Motif). For BSP, a 419-bp fragment was amplified using primer pairs specific for bisulfite-modified sequences but not harboring CpGs, located at nt −2,475 (AATTTGATTTTTTTTTGGGAGA) to nt-2,056 (TCCACTTTCTAACTCTATCCCTAAA) upstream of the RUNX1 transcription start (ATG) codon. BSP primer selection was performed using the Methyl Primer Express Software v1.0 (Applied Biosystems). PCR was done for 35 cycles (94°C, 30 sec; 60°C, 50 sec; 72°C, 1 min). PCR products were sent for dideoxy-sequencing analysis at the Genomics Analysis Platform at Laval University (www.bioinfo.ulaval.ca/seq/en/).
Tissue microarrays (TMAs) construction and IHC
TMAs were constructed as previously described.Citation90 Briefly, one representative block of each ovarian tumor and normal ovarian tissue was selected for the preparation of the tissue arrays. Three 0.6 mm cores of tumor were taken from each tumor block and placed, 0.4 mm apart, on a recipient paraffin block using a commercial tissue arrayer (Beecher Instruments). The cores were randomly placed on one of two recipient blocks to avoid IHC evaluation biases. Four-micron-thick sections were cut for the hematoxylin-eosin (HE) staining and IHC analyses.
IHC was performed, as previously described.Citation89,Citation90 Briefly, 4-µm tissue sections were deparaffinized and then heated in an autoclave for 12 min to retrieve the antigenicity before blocking with endogenous peroxidase. Following treatment with 3% H2O2 for 10 min to quench the endogenous peroxidise activity, sections were incubated with anti-RUNX1 antibody (1:100 dilution) (Abnova Corporation) at room temperature for 2 h. Sections were then incubated with a biotinylated secondary antibody (Dako) and then exposed to a streptavidin complex (Dako). Complete reaction was revealed by 3,3′-diaminobenzidine, and slides were counterstained with hematoxylin. RUNX1 protein expression was assessed by semiquantitative scoring of the intensity of staining and recorded as absent (0), weak (1+), moderate (2+) or strong (3+). The relationship between RUNX1 expression in serous ovarian carcinomas and normal ovarian tissues was evaluated by the Wilcoxon two-sample test. A significant association was considered when p-value was below 0.05. A Kaplan Meier curve and the log-rank test were performed based on PFS values to test the effect of the intensity of RUNX1 (3, 2 vs. 0, 1) on disease progression.
Short hairpin RNA (shRNA)-mediated RUNX1 knockdown in SKOV3 cells
We used the pSilencer 4.1 puro vector (Ambion) to construct a plasmid that exogenously expresses RUNX1-specific shRNA (RUNX1 target sequence: AAATGCTACCGCAGCCATGAA), as described.Citation91 SKOV3 cells were stably transfected with the shRNA-RUNX1 plasmid using the ExGen 500 transfection reagent (Fermentas Canada Inc.) and according to the manufacturer’s instructions. Cells were consecutively grown for 2 weeks in selection medium containing 5 μg/mL puromycin (Wisent) to isolate stable clones. Cells were also mock-transfected with the pSilencer 4.1 puro vector, and stably transfected clones were isolated as controls. Stable clones with inhibited RUNX1 expression were evaluated and validated by western blot and semi-quantitative RT-PCR.
Western blot analysis
Western blot analysis was performed as previously described.Citation89 Briefly, protein lysates were prepared by resuspending cell pellets in Laemmli sample buffer containing 5% β-mercaptoethanol. Protein lysates were separated by 6–12% Tris-glycine gel electrophoresis and transferred onto a polyvinylidene difluoride membrane using a semidry apparatus (Bio-Rad Laboratories). The membrane was blocked with 5% nonfat dry milk in TBST (20 mmol/L TRIS-HCl, 0.5 M NaCl and 0.1% Tween 20), incubated with the anti-RUNX1 mouse monoclonal antibody (1:200) (Santa Cruz Biotechnology) and anti-β-actin antibody (1:5,000) (Santa Cruz Biotechnology) at 4°C overnight. After 3× 15 min washes with TBST (20 mmol/L TRIS-HCl, 0.5 M NaCl and 0.1% Tween 20) at room temperature, the membrane was incubated with horseradish peroxidase-conjugated secondary antibody and detected with ECL solution (Thermo Fisher Scientific).
Cell proliferation assay using impedance measurement with the xCELLigence system
Cell proliferation (cell index) was checked by the xCELLigence Real-Time Cell Analyzer (RTCA) instrument according to the instructions of the supplier (Roche Applied Science and ACEA Biosciences). The xCELLigence system consists of four main components: the RTCA analyzer, the RTCA DP station, the RTCA computer with integrated software and disposable E-plate 16. Cells were seeded in triplicate at 2 × 104 cells/well in the E-Plate 16, a specialized 16-well plate used with the RTCA instrument. Each of the 16 wells on the E-Plate 16 contains an integral sensor electrode array, so that cells inside each well can be monitored and assayed. Cell growth was monitored for 20 h.
Colony formation assay
Colony formation assay was performed, as previously described.Citation89 Briefly, SKOV3 cells were seeded at 500 cells per 60-mm culture dish. After 14 d, the dishes were washed twice in PBS, fixed with cold methanol, stained with Coomassie Blue (Sigma-Aldrich) for 5 min, washed with water and air-dried. The number of colonies was determined by imaging with a Multimage™ Cabinet (Alpha Innotech Corporation) and using AlphaEase Fc software.
Cell migration and invasion assays
Cell migration assays were performed in a modified Boyden-chamber assay using a Transwell two-chamber insert (6.5 mm diameter) separated by a polycarbonate filter of 5-μm pores (Costar). shRNA-RUNX1 transfected, control (vehicle-transfected) and intact SKOV3 cells were seeded into the upper chambers in 0.1% FBS containing medium at a density of 2.5 × 104 per well, and 600 μl of 1% FBS containing medium was placed in the lower chamber as a chemoattractant. After 24 h at 37°C in 5% CO2, the cells were fixed with cold methanol and stained with trypan blue solution. Cells on the upper surface of the filter were removed with cotton buds. Migrated cells on the underside of the filter were photographed and counted by phase contrast microscopy, by selecting 10 random fields per filter (at magnification ×40). The experiments were performed in triplicate. Cell invasion was assayed in a similar way, as the 5-μm pore polycarbonate filters were coated with 40 µl of Matrigel™ at a concentration of 0.5 mg/ml (BD Biosciences). Here, 600 μl of NIH3T3 conditioned medium was added in the lower chamber as a chemoattractant. Differences between shRNA-RUNX1-transfected, vehicle-transfected and intact SKOV3 cells were determined by a Student’s t-test, where p < 0.05 was considered significant.
Flow cytometry
For flow cytometer analysis, 7.5 × 104 SKOV3 cells were treated with 20 mM hydroxyurea (Sigma) for synchronization at the G1/S boundary. After 16 h of incubation, cells were washed once with PBS and resuspended in 1 ml of complete media (time 0). Then, cells were harvested by trypsinization at 0, 3, 6, 9 and 24 h, washed three times with PBS and fixed with ice-cold 95% ethanol overnight. Cells were washed with PBS (3×) and incubated with propidium iodide (50 μg/ml) (Sigma) in the dark at room temperature for 30 min. Flow cytometric analysis was performed on a Beckman Coulter EPICS XL-MCL analyzer. The cell cycle phase distribution was calculated from the resultant DNA using the cell QuesPro software.
MTT (cytotoxicity) assay
The MTT cell proliferation assay (Sigma) was used to measure the cell growth inhibition effects of cisplatin and paclitaxel in SKOV3 cell clones suppressing RUNX1, as previously described.Citation89 Briefly, cell suspensions (at 2 × 104 cells/ml) were transferred to 96-well plates in triplicates and incubated for 3 d with different drugs’ concentrations (ranging between 1 nM and 100 µM). Then, 38 µl of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT, 5 mg/ml) was added to each well 4 h before the end of the incubation. After centrifugation and removing the supernatant, 200 µL of dimethyl sulphoxide (DMSO) were added to resolve the crystals, and the optical density was measured by microplate reader at 595 nm.
Gene expression profiling and data analysis
Gene expression analysis was performed as previously described.Citation88 Briefly, total RNA was extracted from the shRNA-RUNX1 knockdown clone (sh-cl4) and the corresponding control (mock-transfected) SKOV3 clone (ctrl3). The quality of the RNA samples was examined by capillary electrophoresis using the Agilent 2100 Bioanalyzer (Agilent). Fluorescently labeled cRNA targets were generated from 0.5 µg of total RNA from each corresponding SKOV3 cell clone, using the Fluorescent Linear Amplification Kit (Agilent) and 10.0 mM Cyanine 3- or 5-labeled CTP (PerkinElmer), and following user’s manual. Cyanine labeled cRNA from the clone suppressing RUNX1 (sh-cl4) was mixed with the same amount of reverse-color cyanine-labeled cRNA from the corresponding control (ctrl3) clone and hybridized on the Agilent Whole Human Genome microarrays, containing 44,000 genes. Array hybridization, washing, scanning, data extraction and analyses were performed as previously described.Citation88 Network analysis of the microarray data was completed using the Ingenuity Pathway Analysis (IPA) software (www.ingenuity.com).
Semi-quantitative duplex RT-PCR (sqRT-PCR)
Analysis of RUNX1 gene expression in stably transfected RUNX1 knockdown clones (shRNA-RUNX1) and the corresponding mock-transfected SKOV3 clones was performed by sqRT-PCR as previously described.Citation88 The 18S rRNA gene was used as an internal standard. Comparative signal intensity was evaluated using the ImajeJ software (http://rsb.info.nih.gov/ij/). Primers were designed for these loci with the sequences freely available from the Entrez Nucleotide database and the Primer3 algorithm for primer design (www.genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi).
| Abbreviations: | ||
| EOC | = | epithelial ovarian cancer |
| IHC | = | immunohistochemistry |
| CT | = | chemotherapy |
| LMP | = | low malignant potential |
| PFS | = | progression-free survival |
| IPA | = | Ingenuity Pathway Analysis |
| TMA | = | tissue microarray |
| sh-RNA | = | short hairpin RNA |
| BSP | = | bisulfite sequencing PCR |
| sqRT-PCR | = | semi-quantitative reverse transcriptase-PCR |
Additional material
Download Zip (291 KB)Acknowledgments
This study was sustained by a grant to D.B. from the Cancer Research Society of Canada. Clinical specimens were provided by the Banque de tissus et de données of the Réseau de recherche sur le cancer of the Fonds de recherche du Québec-Santé, which is affiliated with the Canadian Tumor Repository Network.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/23963
References
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin 2010; 60:277 - 300; http://dx.doi.org/10.3322/caac.20073; PMID: 20610543
- Marchetti C, Pisano C, Facchini G, Bruni GS, Magazzino FP, Losito S, et al. First-line treatment of advanced ovarian cancer: current research and perspectives. Expert Rev Anticancer Ther 2010; 10:47 - 60; http://dx.doi.org/10.1586/era.09.167; PMID: 20014885
- Ricci F, Bernasconi S, Perego P, Ganzinelli M, Russo G, Bono F, et al. Ovarian carcinoma tumor-initiating cells have a mesenchymal phenotype. Cell Cycle 2012; 11:1966 - 76; http://dx.doi.org/10.4161/cc.20308; PMID: 22544328
- Cho KR. Murine models of ovarian cancer for preclinical testing of targeted therapeutics: has their time arrived?. Cell Cycle 2012; 11:430 - 1; http://dx.doi.org/10.4161/cc.11.3.19275; PMID: 22262181
- Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128:683 - 92; http://dx.doi.org/10.1016/j.cell.2007.01.029; PMID: 17320506
- Balch C, Fang F, Matei DE, Huang TH, Nephew KP. Minireview: epigenetic changes in ovarian cancer. Endocrinology 2009; 150:4003 - 11; http://dx.doi.org/10.1210/en.2009-0404; PMID: 19574400
- Momparler RL. Cancer epigenetics. Oncogene 2003; 22:6479 - 83; http://dx.doi.org/10.1038/sj.onc.1206774; PMID: 14528271
- Maier S, Dahlstroem C, Haefliger C, Plum A, Piepenbrock C. Identifying DNA methylation biomarkers of cancer drug response. Am J Pharmacogenomics 2005; 5:223 - 32; http://dx.doi.org/10.2165/00129785-200505040-00003; PMID: 16078859
- Szyf M, Pakneshan P, Rabbani SA. DNA demethylation and cancer: therapeutic implications. Cancer Lett 2004; 211:133 - 43; http://dx.doi.org/10.1016/j.canlet.2004.04.009; PMID: 15219937
- Bauerschlag DO, Ammerpohl O, Bräutigam K, Schem C, Lin Q, Weigel MT, et al. Progression-free survival in ovarian cancer is reflected in epigenetic DNA methylation profiles. Oncology 2011; 80:12 - 20; http://dx.doi.org/10.1159/000327746; PMID: 21577013
- Watts GS, Futscher BW, Holtan N, Degeest K, Domann FE, Rose SL. DNA methylation changes in ovarian cancer are cumulative with disease progression and identify tumor stage. BMC Med Genomics 2008; 1:47; http://dx.doi.org/10.1186/1755-8794-1-47; PMID: 18826610
- Li M, Balch C, Montgomery JS, Jeong M, Chung JH, Yan P, et al. Integrated analysis of DNA methylation and gene expression reveals specific signaling pathways associated with platinum resistance in ovarian cancer. BMC Med Genomics 2009; 2:34; http://dx.doi.org/10.1186/1755-8794-2-34; PMID: 19505326
- Keita M, Wang Z-Q, Pelletier J-F, Bachvarova M, Plante M, Gregoire J, et al. Global methylation profiling in serous ovarian cancer is indicative for distinct aberrant DNA methylation signatures associated with tumor aggressiveness and disease progression. Gynecol Oncol 2013; 128:356 - 63; PMID: 23219462
- Huang X, Peng JW, Speck NA, Bushweller JH. Solution structure of core binding factor beta and map of the CBF alpha binding site. Nat Struct Biol 1999; 6:624 - 7; http://dx.doi.org/10.1038/10670; PMID: 10404216
- Wang CQ, Jacob B, Nah GS, Osato M. Runx family genes, niche, and stem cell quiescence. Blood Cells Mol Dis 2010; 44:275 - 86; http://dx.doi.org/10.1016/j.bcmd.2010.01.006; PMID: 20144877
- Wong WF, Kohu K, Chiba T, Sato T, Satake M. Interplay of transcription factors in T-cell differentiation and function: the role of Runx. Immunology 2011; 132:157 - 64; http://dx.doi.org/10.1111/j.1365-2567.2010.03381.x; PMID: 21091910
- Chuang LS, Lai SK, Murata-Hori M, Yamada A, Li HY, Gunaratne J, et al. RUNX3 interactome reveals novel centrosomal targeting of RUNX family of transcription factors. Cell Cycle 2012; 11:1938 - 47; http://dx.doi.org/10.4161/cc.20278; PMID: 22544322
- Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 1996; 84:321 - 30; http://dx.doi.org/10.1016/S0092-8674(00)80986-1; PMID: 8565077
- Kilbey A, Terry A, Jenkins A, Borland G, Zhang Q, Wakelam MJ, et al. Runx regulation of sphingolipid metabolism and survival signaling. Cancer Res 2010; 70:5860 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-10-0726; PMID: 20587518
- Scheitz CJ, Tumbar T. New insights into the role of Runx1 in epithelial stem cell biology and pathology. J Cell Biochem 2012; http://dx.doi.org/10.1002/jcb.24453; PMID: 23150456
- Wotton S, Stewart M, Blyth K, Vaillant F, Kilbey A, Neil JC, et al. Proviral insertion indicates a dominant oncogenic role for Runx1/AML-1 in T-cell lymphoma. Cancer Res 2002; 62:7181 - 5; PMID: 12499254
- Robinson HM, Broadfield ZJ, Cheung KL, Harewood L, Harris RL, Jalali GR, et al. Amplification of AML1 in acute lymphoblastic leukemia is associated with a poor outcome. Leukemia: official journal of the Leukemia Society of America. Leukemia Research Fund, UK 2003; 17:2249 - 50; http://dx.doi.org/10.1038/sj.leu.2403140
- Planagumà J, Liljeström M, Alameda F, Bützow R, Virtanen I, Reventós J, et al. Matrix metalloproteinase-2 and matrix metalloproteinase-9 codistribute with transcription factors RUNX1/AML1 and ETV5/ERM at the invasive front of endometrial and ovarian carcinoma. Hum Pathol 2011; 42:57 - 67; http://dx.doi.org/10.1016/j.humpath.2010.01.025; PMID: 20970160
- Ho Sui SJ, Mortimer JR, Arenillas DJ, Brumm J, Walsh CJ, Kennedy BP, et al. oPOSSUM: identification of over-represented transcription factor binding sites in co-expressed genes. Nucleic Acids Res 2005; 33:3154 - 64; http://dx.doi.org/10.1093/nar/gki624; PMID: 15933209
- Blyth K, Cameron ER, Neil JC. The RUNX genes: gain or loss of function in cancer. Nat Rev Cancer 2005; 5:376 - 87; http://dx.doi.org/10.1038/nrc1607; PMID: 15864279
- Wotton SF, Blyth K, Kilbey A, Jenkins A, Terry A, Bernardin-Fried F, et al. RUNX1 transformation of primary embryonic fibroblasts is revealed in the absence of p53. Oncogene 2004; 23:5476 - 86; http://dx.doi.org/10.1038/sj.onc.1207729; PMID: 15133495
- Wolyniec K, Wotton S, Kilbey A, Jenkins A, Terry A, Peters G, et al. RUNX1 and its fusion oncoprotein derivative, RUNX1-ETO, induce senescence-like growth arrest independently of replicative stress. Oncogene 2009; 28:2502 - 12; http://dx.doi.org/10.1038/onc.2009.101; PMID: 19448675
- Linggi B, Müller-Tidow C, van de Locht L, Hu M, Nip J, Serve H, et al. The t(8;21) fusion protein, AML1 ETO, specifically represses the transcription of the p14(ARF) tumor suppressor in acute myeloid leukemia. Nat Med 2002; 8:743 - 50; http://dx.doi.org/10.1038/nm726; PMID: 12091906
- Mikhail FM, Sinha KK, Saunthararajah Y, Nucifora G. Normal and transforming functions of RUNX1: a perspective. J Cell Physiol 2006; 207:582 - 93; http://dx.doi.org/10.1002/jcp.20538; PMID: 16250015
- Speck NA, Gilliland DG. Core-binding factors in haematopoiesis and leukaemia. Nat Rev Cancer 2002; 2:502 - 13; http://dx.doi.org/10.1038/nrc840; PMID: 12094236
- Roumier C, Fenaux P, Lafage M, Imbert M, Eclache V, Preudhomme C. New mechanisms of AML1 gene alteration in hematological malignancies. Leukemia: official journal of the Leukemia Society of America. Leukemia Research Fund, UK 2003; 17:9 - 16; http://dx.doi.org/10.1038/sj.leu.2402766
- Yanagida M, Osato M, Yamashita N, Liqun H, Jacob B, Wu F, et al. Increased dosage of Runx1/AML1 acts as a positive modulator of myeloid leukemogenesis in BXH2 mice. Oncogene 2005; 24:4477 - 85; http://dx.doi.org/10.1038/sj.onc.1208675; PMID: 15856017
- Janes KA. RUNX1 and its understudied role in breast cancer. Cell Cycle 2011; 10:3461 - 5; http://dx.doi.org/10.4161/cc.10.20.18029; PMID: 22024923
- Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 2012; 486:353 - 60; PMID: 22722193
- Fijneman RJ, Anderson RA, Richards E, Liu J, Tijssen M, Meijer GA, et al. Runx1 is a tumor suppressor gene in the mouse gastrointestinal tract. Cancer Sci 2012; 103:593 - 9; http://dx.doi.org/10.1111/j.1349-7006.2011.02189.x; PMID: 22171576
- Dulak AM, Schumacher SE, van Lieshout J, Imamura Y, Fox C, Shim B, et al. Gastrointestinal adenocarcinomas of the esophagus, stomach, and colon exhibit distinct patterns of genome instability and oncogenesis. Cancer Res 2012; 72:4383 - 93; http://dx.doi.org/10.1158/0008-5472.CAN-11-3893; PMID: 22751462
- Planagumà J, Díaz-Fuertes M, Gil-Moreno A, Abal M, Monge M, García A, et al. A differential gene expression profile reveals overexpression of RUNX1/AML1 in invasive endometrioid carcinoma. Cancer Res 2004; 64:8846 - 53; http://dx.doi.org/10.1158/0008-5472.CAN-04-2066; PMID: 15604243
- Doll A, Gonzalez M, Abal M, Llaurado M, Rigau M, Colas E, et al. An orthotopic endometrial cancer mouse model demonstrates a role for RUNX1 in distant metastasis. Int J Cancer 2009; 125:257 - 63; http://dx.doi.org/10.1002/ijc.24330; PMID: 19384951
- Scheitz CJ, Lee TS, McDermitt DJ, Tumbar T. Defining a tissue stem cell-driven Runx1/Stat3 signalling axis in epithelial cancer. EMBO J 2012; 31:4124 - 39; http://dx.doi.org/10.1038/emboj.2012.270; PMID: 23034403
- Ivan C, Hu W, Bottsford-Miller J, Zand B, Dalton HJ, Liu T, et al. Epigenetic analysis of the Notch superfamily in high-grade serous ovarian cancer. Gynecol Oncol 2013; 128:506 - 11; http://dx.doi.org/10.1016/j.ygyno.2012.11.029; PMID: 23200915
- Yeh HY, Cheng SW, Lin YC, Yeh CY, Lin SF, Soo VW. Identifying significant genetic regulatory networks in the prostate cancer from microarray data based on transcription factor analysis and conditional independency. BMC Med Genomics 2009; 2:70; http://dx.doi.org/10.1186/1755-8794-2-70; PMID: 20025723
- Slattery ML, Lundgreen A, Herrick JS, Caan BJ, Potter JD, Wolff RK. Associations between genetic variation in RUNX1, RUNX2, RUNX3, MAPK1 and eIF4E and riskof colon and rectal cancer: additional support for a TGF-β-signaling pathway. Carcinogenesis 2011; 32:318 - 26; http://dx.doi.org/10.1093/carcin/bgq245; PMID: 21088106
- Xu K, Zhou Y, Qiu W, Liu X, Xia M, Liu L, et al. Activating transcription factor 3 (ATF3) promotes sublytic C5b-9-induced glomerular mesangial cells apoptosis through up-regulation of Gadd45α and KLF6 gene expression. Immunobiology 2011; 216:871 - 81; http://dx.doi.org/10.1016/j.imbio.2011.02.005; PMID: 21396734
- Ehrhardt H, Schrembs D, Moritz C, Wachter F, Haldar S, Graubner U, et al. Optimized anti-tumor effects of anthracyclines plus Vinca alkaloids using a novel, mechanism-based application schedule. Blood 2011; 118:6123 - 31; http://dx.doi.org/10.1182/blood-2010-02-269811; PMID: 21926351
- Porter LA, Singh G, Lee JM. Abundance of cyclin B1 regulates gamma-radiation-induced apoptosis. Blood 2000; 95:2645 - 50; PMID: 10753846
- Zerbini LF, Wang Y, Czibere A, Correa RG, Cho JY, Ijiri K, et al. NF-kappa B-mediated repression of growth arrest- and DNA-damage-inducible proteins 45alpha and gamma is essential for cancer cell survival. Proc Natl Acad Sci USA 2004; 101:13618 - 23; http://dx.doi.org/10.1073/pnas.0402069101; PMID: 15353598
- Pollock AS, Turck J, Lovett DH. The prodomain of interleukin 1alpha interacts with elements of the RNA processing apparatus and induces apoptosis in malignant cells. FASEB J 2003; 17:203 - 13; http://dx.doi.org/10.1096/fj.02-0602com; PMID: 12554699
- Kim JE, Kim SJ, Jeong HW, Lee BH, Choi JY, Park RW, et al. RGD peptides released from beta ig-h3, a TGF-beta-induced cell-adhesive molecule, mediate apoptosis. Oncogene 2003; 22:2045 - 53; http://dx.doi.org/10.1038/sj.onc.1206269; PMID: 12673209
- Yan C, Boyd DD. ATF3 regulates the stability of p53: a link to cancer. Cell Cycle 2006; 5:926 - 9; http://dx.doi.org/10.4161/cc.5.9.2714; PMID: 16628010
- Glinsky GV. Genomic models of metastatic cancer: functional analysis of death-from-cancer signature genes reveals aneuploid, anoikis-resistant, metastasis-enabling phenotype with altered cell cycle control and activated Polycomb Group (PcG) protein chromatin silencing pathway. Cell Cycle 2006; 5:1208 - 16; http://dx.doi.org/10.4161/cc.5.11.2796; PMID: 16760651
- Hamstra DA, Pagé M, Maybaum J, Rehemtulla A. Expression of endogenously activated secreted or cell surface carboxypeptidase A sensitizes tumor cells to methotrexate-alpha-peptide prodrugs. Cancer Res 2000; 60:657 - 65; PMID: 10676650
- Laronga C, Yang HY, Neal C, Lee MH. Association of the cyclin-dependent kinases and 14-3-3 sigma negatively regulates cell cycle progression. J Biol Chem 2000; 275:23106 - 12; http://dx.doi.org/10.1074/jbc.M905616199; PMID: 10767298
- Dhar S, Squire JA, Hande MP, Wellinger RJ, Pandita TK. Inactivation of 14-3-3sigma influences telomere behavior and ionizing radiation-induced chromosomal instability. Mol Cell Biol 2000; 20:7764 - 72; http://dx.doi.org/10.1128/MCB.20.20.7764-7772.2000; PMID: 11003671
- Meng S, Arbit T, Veeriah S, Mellinghoff IK, Fang F, Vivanco I, et al. 14-3-3sigma and p21 synergize to determine DNA damage response following Chk2 inhibition. Cell Cycle 2009; 8:2238 - 46; http://dx.doi.org/10.4161/cc.8.14.8998; PMID: 19502805
- Fukai N, Shichiri M, Ozawa N, Matsushita M, Hirata Y. Coexpression of calcitonin receptor-like receptor and receptor activity-modifying protein 2 or 3 mediates the antimigratory effect of adrenomedullin. Endocrinology 2003; 144:447 - 53; http://dx.doi.org/10.1210/en.2002-220463; PMID: 12538603
- Aggarwal BB, Sung B. Pharmacological basis for the role of curcumin in chronic diseases: an age-old spice with modern targets. Trends Pharmacol Sci 2009; 30:85 - 94; http://dx.doi.org/10.1016/j.tips.2008.11.002; PMID: 19110321
- Iozzo RV, Buraschi S, Genua M, Xu SQ, Solomides CC, Peiper SC, et al. Decorin antagonizes IGF receptor I (IGF-IR) function by interfering with IGF-IR activity and attenuating downstream signaling. J Biol Chem 2011; 286:34712 - 21; http://dx.doi.org/10.1074/jbc.M111.262766; PMID: 21840990
- Korah RM, Sysounthone V, Golowa Y, Wieder R. Basic fibroblast growth factor confers a less malignant phenotype in MDA-MB-231 human breast cancer cells. Cancer Res 2000; 60:733 - 40; PMID: 10676661
- Plante I, Stewart MK, Barr K, Allan AL, Laird DW. Cx43 suppresses mammary tumor metastasis to the lung in a Cx43 mutant mouse model of human disease. Oncogene 2011; 30:1681 - 92; http://dx.doi.org/10.1038/onc.2010.551; PMID: 21151177
- Krause DS, Fackler MJ, Civin CI, May WS. CD34: structure, biology, and clinical utility. Blood 1996; 87:1 - 13; PMID: 8547630
- Rivera A, Mavila A, Bayless KJ, Davis GE, Maxwell SA. Cyclin A1 is a p53-induced gene that mediates apoptosis, G2/M arrest, and mitotic catastrophe in renal, ovarian, and lung carcinoma cells. Cell Mol Life Sci 2006; 63:1425 - 39; http://dx.doi.org/10.1007/s00018-006-5521-5; PMID: 16799873
- Nash MA, Deavers MT, Freedman RS. The expression of decorin in human ovarian tumors. Clin Cancer Res 2002; 8:1754 - 60; PMID: 12060613
- Hollander MC, Kovalsky O, Salvador JM, Kim KE, Patterson AD, Haines DC, et al. Dimethylbenzanthracene carcinogenesis in Gadd45a-null mice is associated with decreased DNA repair and increased mutation frequency. Cancer Res 2001; 61:2487 - 91; PMID: 11289119
- Akahira J, Sugihashi Y, Suzuki T, Ito K, Niikura H, Moriya T, et al. Decreased expression of 14-3-3 sigma is associated with advanced disease in human epithelial ovarian cancer: its correlation with aberrant DNA methylation. Clin Cancer Res 2004; 10:2687 - 93; http://dx.doi.org/10.1158/1078-0432.CCR-03-0510; PMID: 15102672
- Baranello C, Mariani M, Andreoli M, Fanelli M, Martinelli E, Ferrandina G, et al. Adrenomedullin in ovarian cancer: foe in vitro and friend in vivo?. PLoS ONE 2012; 7:e40678; http://dx.doi.org/10.1371/journal.pone.0040678; PMID: 22859951
- Syed V, Mukherjee K, Lyons-Weiler J, Lau KM, Mashima T, Tsuruo T, et al. Identification of ATF-3, caveolin-1, DLC-1, and NM23-H2 as putative antitumorigenic, progesterone-regulated genes for ovarian cancer cells by gene profiling. Oncogene 2005; 24:1774 - 87; http://dx.doi.org/10.1038/sj.onc.1207991; PMID: 15674352
- Lee YH, Heo JH, Kim TH, Kang H, Kim G, Kim J, et al. Significance of cell cycle regulatory proteins as malignant and prognostic biomarkers in ovarian epithelial tumors. Int J Gynecol Pathol 2011; 30:205 - 17; http://dx.doi.org/10.1097/PGP.0b013e3182063e71; PMID: 21464733
- Li J, Wood WH 3rd, Becker KG, Weeraratna AT, Morin PJ. Gene expression response to cisplatin treatment in drug-sensitive and drug-resistant ovarian cancer cells. Oncogene 2007; 26:2860 - 72; http://dx.doi.org/10.1038/sj.onc.1210086; PMID: 17072341
- Chou JL, Chen LY, Lai HC, Chan MW. TGF-β: friend or foe? The role of TGF-β/SMAD signaling in epigenetic silencing of ovarian cancer and its implication in epigenetic therapy. Expert Opin Ther Targets 2010; 14:1213 - 23; http://dx.doi.org/10.1517/14728222.2010.525353; PMID: 20925461
- McLean K, Gong Y, Choi Y, Deng N, Yang K, Bai S, et al. Human ovarian carcinoma–associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J Clin Invest 2011; 121:3206 - 19; http://dx.doi.org/10.1172/JCI45273; PMID: 21737876
- Symowicz J, Adley BP, Gleason KJ, Johnson JJ, Ghosh S, Fishman DA, et al. Engagement of collagen-binding integrins promotes matrix metalloproteinase-9-dependent E-cadherin ectodomain shedding in ovarian carcinoma cells. Cancer Res 2007; 67:2030 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-06-2808; PMID: 17332331
- Rask K, Nilsson A, Brännström M, Carlsson P, Hellberg P, Janson PO, et al. Wnt-signalling pathway in ovarian epithelial tumours: increased expression of beta-catenin and GSK3beta. Br J Cancer 2003; 89:1298 - 304; http://dx.doi.org/10.1038/sj.bjc.6601265; PMID: 14520463
- Crijns AP, de Graeff P, Geerts D, Ten Hoor KA, Hollema H, van der Sluis T, et al. MEIS and PBX homeobox proteins in ovarian cancer. Eur J Cancer 2007; 43:2495 - 505; http://dx.doi.org/10.1016/j.ejca.2007.08.025; PMID: 17949970
- Lu KH, Patterson AP, Wang L, Marquez RT, Atkinson EN, Baggerly KA, et al. Selection of potential markers for epithelial ovarian cancer with gene expression arrays and recursive descent partition analysis. Clin Cancer Res 2004; 10:3291 - 300; http://dx.doi.org/10.1158/1078-0432.CCR-03-0409; PMID: 15161682
- Elattar A, Warburton KG, Mukhopadhyay A, Freer RM, Shaheen F, Cross P, et al. Androgen receptor expression is a biological marker for androgen sensitivity in high grade serous epithelial ovarian cancer. Gynecol Oncol 2012; 124:142 - 7; http://dx.doi.org/10.1016/j.ygyno.2011.09.004; PMID: 22001143
- Zecchini S, Bombardelli L, Decio A, Bianchi M, Mazzarol G, Sanguineti F, et al. The adhesion molecule NCAM promotes ovarian cancer progression via FGFR signalling. EMBO Mol Med 2011; 3:480 - 94; http://dx.doi.org/10.1002/emmm.201100152; PMID: 21739604
- Tan Y, Cheung M, Pei J, Menges CW, Godwin AK, Testa JR. Upregulation of DLX5 promotes ovarian cancer cell proliferation by enhancing IRS-2-AKT signaling. Cancer Res 2010; 70:9197 - 206; http://dx.doi.org/10.1158/0008-5472.CAN-10-1568; PMID: 21045156
- Tanwar PS, Kaneko-Tarui T, Lee HJ, Zhang L, Teixeira JM. PTEN loss and HOXA10 expression are associated with ovarian endometrioid adenocarcinoma differentiation and progression. Carcinogenesis 2012; 33:2351 - 61; http://dx.doi.org/10.1093/carcin/bgs405; PMID: 22962306
- Meng Y, Gu C, Wu Z, Zhao Y, Si Y, Fu X, et al. Id2 promotes the invasive growth of MCF-7 and SKOV-3 cells by a novel mechanism independent of dimerization to basic helix-loop-helix factors. BMC Cancer 2009; 9:75; http://dx.doi.org/10.1186/1471-2407-9-75; PMID: 19257909
- Karam A, Dorigo O. MMPs in ovarian cancer as therapeutic targets. Anticancer Agents Med Chem 2012; 12:764 - 72; http://dx.doi.org/10.2174/187152012802650174; PMID: 22292752
- Lu ZY, Dong R, Li D, Li WB, Xu FQ, Geng Y, et al. SNAI1 overexpression induces stemness and promotes ovarian cancer cell invasion and metastasis. Oncol Rep 2012; 27:1587 - 91; PMID: 22344746
- Chen J, Wang M, Xi B, Xue J, He D, Zhang J, et al. SPARC is a key regulator of proliferation, apoptosis and invasion in human ovarian cancer. PLoS ONE 2012; 7:e42413; http://dx.doi.org/10.1371/journal.pone.0042413; PMID: 22879971
- Nicosia SV, Bai W, Cheng JQ, Coppola D, Kruk PA. Oncogenic pathways implicated in ovarian epithelial cancer. Hematol Oncol Clin North Am 2003; 17:927 - 43; http://dx.doi.org/10.1016/S0889-8588(03)00056-X; PMID: 12959183
- Li W, Xu S, Lin S, Zhao W. Overexpression of runt-related transcription factor-2 is associated with advanced tumor progression and poor prognosis in epithelial ovarian cancer. J Biomed Biotechnol 2012; 2012:456534; http://dx.doi.org/10.1155/2012/456534; PMID: 23093845
- Zhang S, Wei L, Zhang A, Zhang L, Yu H. RUNX3 gene methylation in epithelial ovarian cancer tissues and ovarian cancer cell lines. OMICS 2009; 13:307 - 11; http://dx.doi.org/10.1089/omi.2009.0030; PMID: 19645591
- Lee CW, Chuang LS, Kimura S, Lai SK, Ong CW, Yan B, et al. RUNX3 functions as an oncogene in ovarian cancer. Gynecol Oncol 2011; 122:410 - 7; http://dx.doi.org/10.1016/j.ygyno.2011.04.044; PMID: 21612813
- Vergote I, Rustin GJ, Eisenhauer EA, Kristensen GB, Pujade-Lauraine E, Parmar MK, et al. Re: new guidelines to evaluate the response to treatment in solid tumors [ovarian cancer]. Gynecologic Cancer Intergroup. J Natl Cancer Inst 2000; 92:1534 - 5; http://dx.doi.org/10.1093/jnci/92.18.1534; PMID: 10995813
- L’Espérance S, Bachvarova M, Tetu B, Mes-Masson AM, Bachvarov D. Global gene expression analysis of early response to chemotherapy treatment in ovarian cancer spheroids. BMC Genomics 2008; 9:99; http://dx.doi.org/10.1186/1471-2164-9-99; PMID: 18302766
- Mercier PL, Bachvarova M, Plante M, Gregoire J, Renaud MC, Ghani K, et al. Characterization of DOK1, a candidate tumor suppressor gene, in epithelial ovarian cancer. Mol Oncol 2011; 5:438 - 53; http://dx.doi.org/10.1016/j.molonc.2011.07.003; PMID: 21856257
- Tetu B, Popa I, Bairati I, L'Esperance S, Bachvarova M, Plante M, et al. Immunohistochemical analysis of possible chemoresistance markers identified by micro-arrays on serous ovarian carcinomas. Modern pathology: an official journal of the United States and Canadian Academy of Pathology. Inc 2008; 21:1002 - 10
- Lai SR, Andrews LG, Tollefsbol TO. RNA interference using a plasmid construct expressing short-hairpin RNA. Methods Mol Biol 2007; 405:31 - 7; http://dx.doi.org/10.1007/978-1-60327-070-0_4; PMID: 18369815