Abstract
Aurora kinase B is a critical component of the chromosomal passenger complex, which is involved in the regulation of microtubule-kinetochore attachments and cytokinesis. By using conditional knockout cells and chemical inhibition, we show here that inactivation of Aurora B results in delayed G1/S transition and premature mitotic exit. Aurora B deficiency results in delayed DNA replication in cultured fibroblasts as well as liver cells after hepatectomy. This is accompanied by increased transcription of the cell cycle inhibitor p21Cip1. Lack of Aurora B does not prevent mitotic entry but results in a premature exit from prometaphase in the presence of increased p21Cip1-Cdk1 inactive complexes. Aurora B-null cells display reduced degradation of cyclin B1, suggesting the presence of phenomenon known as adaptation to the mitotic checkpoint, previously described in yeast. Elimination of p21Cip1 rescues Cdk1 activity and prevents premature mitotic exit in Aurora B-deficient cells. These results suggest that Aurora B represses p21Cip1, preventing delayed DNA replication, Cdk inhibition and premature mitotic exit. The upregulation of p21Cip1 observed after inhibition of Aurora B may have important implications in cell cycle progression, tetraploidy, senescence or cancer therapy.
Introduction
Progression through the cell cycle requires the controlled activation of different families of kinases that regulate diverse cellular processes required for cell division.Citation1 Early work in Drosophila led to the identification of aurora mutants, which carry a loss-of-function mutation in a serine/threonine kinase essential for centrosome separation and the formation of bipolar spindles.Citation2 A single Aurora protein exists in budding (increase-inploidy 1; Ipl1) or fission (Ark1) yeast, whereas two family members, Aurora A and Aurora B are present in worms, flies and frogs. Three different Aurora family members, known as Aurora A, B and C, exist in mammals.Citation3-Citation5 These kinases contain a conserved catalytic domain and N-terminal domains that vary in sequence and in length. Aurora B and C are close paralogs that probably arose from a relatively recent common ancestor, and they show certain functional overlap.Citation6-Citation8
Aurora B is the enzymatic activity of the chromosome passenger complex (CPC), which localizes to the kinetochores from prophase to metaphase and to the central spindle and midbody in cytokinesis.Citation4,Citation9,Citation10 Other mammalian CPC proteins include the inner centromere protein incenp, survivin and borealin (also known as DasraB), which controls the targeting, enzymatic activity and stability of Aurora B.Citation9 The CPC is crucial for the destabilization of aberrant microtubule-to-kinetochore attachments and the spindle assembly checkpoint (SAC)-dependent delay in mitotic progression until these defects are corrected.Citation4,Citation5,Citation10-Citation12 Substrate phosphorylation depends on the distance of the substrate from Aurora B at the inner centromere, thus indicating that recruitment of the CPC to the kinetochore prevents the stabilization of improper attachments and activates the SAC to delay the metaphase to anaphase transition.Citation13 Aurora B therefore plays a critical role in generating unattached kinetochores, thus triggering a SAC-mediated arrest. During cytokinesis, Aurora B localizes to the midbody remnant, where its local inactivation is crucial for completion of abscission.Citation14,Citation15
Whether Aurora B plays additional roles in interphase has not been addressed in detail. A role for Aurora B in the G1/S transition has been described in lymphocytes, in which this kinase can form complexes with mTOR and may modulate differentiation by regulating specific epigenetic marks.Citation16,Citation17 More recent data suggest that Aurora B directly phosphorylates p53 and results in decreased induction of target genes.Citation18,Citation19 Using Aurora B conditional knockout cells and chemical inhibition, we show here that lack of Aurora B results in decreased G1/S transition in vitro and in vivo. In addition, Aurora B inactivation results in decreased Cdk1 activity and premature mitotic exit. These defects are accompanied by transcriptional upregulation of the cell cycle inhibitor p21Cip1. Elimination of p21Cip1 rescues the premature mitotic exit in the absence of Aurora B, suggesting that this kinase contributes to full Cdk1 activity by repressing the expression of this cell cycle inhibitor.
Results
Aurora B is required for timely entry into S-phase
We made use of Aurora B conditional knockout cellsCitation8 to specifically ablate Aurora B in quiescent cells (G0) and test the effect of its absence during the cell cycle. The Aurora B-encoding gene (Aurkb) was specifically deleted in primary Aurkb(lox/lox) mouse embryonic fibroblasts (MEFs) by expressing the Cre recombinase in confluent- and serum-arrested cells (). Expression of Cre results in an efficient recombination between loxP sites and deletion of AurkB exons 2–6, as we have reported previously.Citation8 Serum was added 2 d later, and entry into S-phase was monitored by DNA content () and incorporation of the nucleotide analog BrdU (). Lack of Aurora B resulted in a significant decreased in the number of cells that entered into S-phase 12–18 h after the addition of serum, a time when the number of S-phase cells peaks in controls cells (). Importantly, the number of Aurkb(Δ/Δ) cells that incorporated BrdU was still growing after 24 h, whereas control cells had already bypassed S-phase (). In addition, the number of cells that entered mitosis in these assays was similar in Aurora B-null and control cultures (see below), suggesting that lack of Aurora-B results in a delay in DNA replication rather than in a complete arrest in the G1/S transition. Similar results were found after chemical inhibition of Aurora B using ZM447439 (commonly abbreviated as ZM1; ), indicating that these effects were not an artifactual consequence of the genetic ablation of the Aurora B-encoding gene.
Figure 1. Aurora B is required for the G1/S transition in mammalian cells. (A) Schematic representation of the protocol followed for acute deletion of Aurora B in quiescent cells. Primary Aurkb(lox/lox) MEFs were seeded at 80% confluence and allowed to form a completely confluent culture during 2 d. These cells were then cultured in 0.1% FBS and transduced with adenoviruses expressing GFP (AdGFP) or the Cre recombinase (AdCre). Two days after the infection, cells were seeded in new plates at low confluence in the presence of 10% FBS to induce entry into the cell cycle. The Aurora B/C inhibitor ZM447439 (ZM1) was also added at this point to non-infected cells. (B) Progression into S-phase at different time points after addition of serum. Cells were analyzed by propidium iodide staining showing a dramatic delay in S-phase entry upon the genetic ablation or chemical inactivation of Aurora B. Representative FACS profiles of these samples at time serum was added (t = 0) or 18 h after mitogenic stimulation. n.s., not significant; ***, p < 0.001. (C) Incorporation of BrdU was also tested by treating these cultures with a short (20 min) pulse of BrdU before harvesting the cells. BrdU incorporation was analyzed by immunofluorescence with specific antibodies (green; arrows show positive cells). DAPI (DNA) is shown in red. ***, p < 0.001. (D) Immunodetection of phosphorylated p70S6K at Thr389, or phosphorylated pRb at Ser780 2, 10 or 18 h after stimulation with serum. The expression of p21Cip1 was increased at all time-points in Aurkb(Δ/Δ) cells. Immunodetection of Akt or β-actin was used as a loading control. (E) Transcriptional induction of p21Cip1 at different time points after serum addition. Cre recombinase was added 48 h before serum addition as represented in . p21Cip1 mRNA levels in Cre-transduced cells were normalized vs. those of Flp-treated cells in each time point. *, p < 0.05; **, p < 0.01; ***, p < 0.001. (F) Analysis of p21Cip1 protein levels in NIH3T3 cells after stimulation with serum and treatment with doxorubicin (Doxo), ZM1, Cre or combination of these treatments in wild-type Aurkb(+/+) cells. As a control, Doxo-treated cells were transduced with a virus expressing short-hairpin RNAs against the p21Cip1 transcript (shp21Cip1; right lanes). Aurora B is used as a control that is normally induced during the cell cycle.
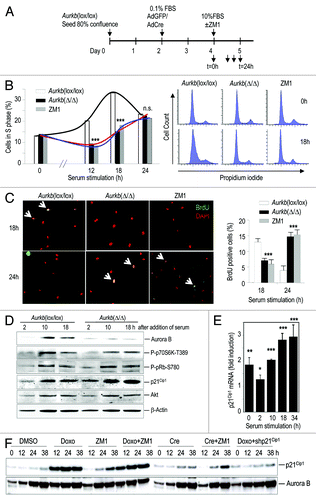
Aurora B has been proposed to modulate several routes that could affect G1/S progression such as mTOR signaling,Citation16 direct phosphorylation of the retinoblastoma protein (pRb)Citation20 and, more recently, p53 signaling.Citation18,Citation19 As represented in , genetic ablation of Aurora B did not result in significant differences in the phosphorylation of p70S6K or pRb but dramatically increased the levels of the p53 target p21Cip1 (). This increase is at least partially due to transcriptional induction, and it was obvious even before the addition of serum (, t = 0 corresponds to 48 h in the presence of Cre; see ). The p53-p21Cip1 pathway is known to be induced by DNA damage, and Cre recombinase could trigger some DNA damage-like response. However, treatment with Cre recombinase did not alter entry into S-phase either in Aurkb(+/+) cells or in many other MEF cultured tested in our laboratory (data not shown), and ZM1 is sufficient to delay S-phase entry in the absence of Cre (). We also compared the effect of doxorubicin (a DNA damage inducer), the Aurora B inhibitor ZM1 and Cre recombinase in the induction of p21Cip1 in Aurkb(+/+) cells. As depicted in , treatment with Cre provokes some induction of p21Cip1 in these wild-type MEFs. However, this induction is less efficient that the one provoked by ZM1, and the effect of the combination of Cre and ZM1 is not significantly different from the effect induced by ZM1 alone (). Since Aurora B was ablated in G0, these results suggest a role for Aurora B in preventing p21Cip1 induction and a p21Cip1-dependent delay in G1 progression.
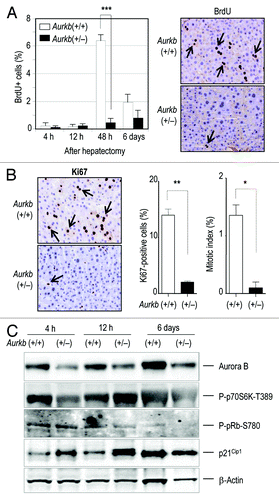
We further tested the defective entry into S-phase in vivo by inducing cell proliferation in quiescent hepatocypes. Since complete ablation of Aurora B results in embryonic lethality,Citation8 we used heterozygous mice in these assays. Aurkb(+/−) and Aurkb(+/+) were subjected to 2/3 hepatectomy, and the entry into the cell cycle was analyzed by BrdU incorporation, Ki67 staining or mitotic index. Control mice displayed a significant increase in the levels of BrdU incorporation 48 h after hepatectomy, whereas this proliferative response was severely impaired in Aurkb(+/−) mice (). Similar data were obtained when monitoring Ki67 or mitotic index 48 h after hepatectomy (). This deficient entry into S-phase in vivo was also accompanied by a significant upregulation of p21Cip1 as soon as 4 h after hepatectomy, whereas the levels of p70S6K phosphorylation did not follow a consistent pattern (). pRb phosphorylation was slightly reduced at later time points (12 h and 6 d) in agreement with reduced proliferation in these hepatocytes. A similar delay in proliferation in the skin was observed during wound healing in Aurkb(+/−) mice (Fig. S1), suggesting a defective acute proliferative response in the presence of reduced levels of Aurora B in vivo.
Figure 2. Defective entry into S-phase in Aurora B-deficient hepatocytes. (A) BrdU incorporation in vivo after 2/3 partial hepatectomy in Aurkb(+/+) and Aurkb(+/−) mice. The percentage of BrdU-positive cells (as detected by immunohistochemistry) is shown 4, 12 and 48 h as well as 6 d after hepatectomy. Incorporation of BrdU (arrows) is highly reduced by 48 h in Aurora B-mutant mice. 2–3 mice of each genotype were used per time point. ***, p < 0.001. (B) Representative images of proliferation markers (Ki67 level) 48 h after hepatectomy. Quantification of Ki67 or mitotic figures (arrows) in these samples also indicates a significant reduction in cell cycle entry and progression in Aurkb(+/−)(n = 2) vs. control (n = 3) mice. *; p < 0.05; **, p < 0.01. (C) Immunodetection of phospho-p70S6K, phospo-pRb and p21Cip1 levels in Aurkb(+/+) and Aurkb(+/−) liver protein lysates at different time points after partial hepatectomy. Immunodetection of β-actin was used as a loading control.
Premature mitotic exit in Aurora B-null cells
We then analyzed mitotic entry and exit in the absence of Aurora B. Confluent MEFs were infected with adenoviruses expressing either GFP (AdGFP) or the Cre recombinase (AdCre) in the presence of low serum (0.1% FBS) and seeded at low confluency 48 h later (t = 0) in the presence of 10% FBS to allow cells entering into a new cell cycle (). Lack of Aurora B resulted in decreased phosphorylation of histone H3 and abnormal mitotic figures, including monopolar or multipolar spindles (). These mutant cells exited from mitosis in the absence of chromosome segregation, generating tetraploid cells in agreement with previous data using RNA interference or small-molecule kinase inhibitors.Citation21-Citation23 These tetraploid cells frequently displayed multinuclei or micronuclei as well as ring-shaped nuclei,Citation8,Citation22 possibly as a consequence of chromatin decondensation and mitotic exit from monopolar spindles ().
Figure 3. Premature mitotic exit in Aurora B-deficient fibroblasts. (A) Schematic representation of the assay performed to study mitosis in Aurora B-null cells. (B) Examination of these cultures by immunofluorescence at 48 h after cell cycle entry [day 6 in (A)] indicates abnormal mitotic figures in the absence of Aurora B with reduced phosphorylation of histone H3 (P-H3, green signal; see also ref. Citation8) and multipolar spindles (left panel) identified by α-tubulin (red), γ-tubulin (white) and DAPI (blue). Monopolar spindles are shown in the right panels and these cells frequently exit from mitosis generating ring-shape nuclei. α-tubulin, red; DAPI, blue. Scale bars, 10 μm. (C) Wild-type or Aurora B-null cells expressing mCherry-H2B were monitored by time-lapse microscopy for 24 h [days 5–6 in (A)]. The percentage of cells that entered mitosis was similar in control and Aurora B-null cells. (D) Representation of progression throughout the different phases of mitosis in these cells. Every raw corresponds to a single cell, and cells were aligned at the time they entered mitosis. The ring in the last cell indicates the formation of ring-shaped nuclei. The duration of mitosis (DOM) in these cultures is shown in the plot. ***, p < 0.001. (E) Representative images of mCherry-H2B (red)-expressing cells showing a normal division in control cells and defective mitotic exit in the absence of chromosome segregation or cytokinesis in Aurora B-null cultures. About 15% of these mutant cells exited mitosis forming ring-shaped tetraploid nuclei. Numbers indicate the time (min) since each cell entered into mitosis.
![Figure 3. Premature mitotic exit in Aurora B-deficient fibroblasts. (A) Schematic representation of the assay performed to study mitosis in Aurora B-null cells. (B) Examination of these cultures by immunofluorescence at 48 h after cell cycle entry [day 6 in (A)] indicates abnormal mitotic figures in the absence of Aurora B with reduced phosphorylation of histone H3 (P-H3, green signal; see also ref. Citation8) and multipolar spindles (left panel) identified by α-tubulin (red), γ-tubulin (white) and DAPI (blue). Monopolar spindles are shown in the right panels and these cells frequently exit from mitosis generating ring-shape nuclei. α-tubulin, red; DAPI, blue. Scale bars, 10 μm. (C) Wild-type or Aurora B-null cells expressing mCherry-H2B were monitored by time-lapse microscopy for 24 h [days 5–6 in (A)]. The percentage of cells that entered mitosis was similar in control and Aurora B-null cells. (D) Representation of progression throughout the different phases of mitosis in these cells. Every raw corresponds to a single cell, and cells were aligned at the time they entered mitosis. The ring in the last cell indicates the formation of ring-shaped nuclei. The duration of mitosis (DOM) in these cultures is shown in the plot. ***, p < 0.001. (E) Representative images of mCherry-H2B (red)-expressing cells showing a normal division in control cells and defective mitotic exit in the absence of chromosome segregation or cytokinesis in Aurora B-null cultures. About 15% of these mutant cells exited mitosis forming ring-shaped tetraploid nuclei. Numbers indicate the time (min) since each cell entered into mitosis.](/cms/asset/a5ce31bb-5f4b-40be-8137-4b5eee819ffc/kccy_a_10924004_f0003.gif)
We then monitored mitotic entry and progression in these cultures by videomicroscopy. AdGFP- or AdCre-infected cells were stimulated with serum and recorded from 24–48 h after cell cycle re-entry (). The percentage of cells that entered mitosis during this period was similar in both cultures (), indicating that Aurora B-null cells did not display major problems in mitotic entry. Aurkb (lox/lox) control cells spent 49.5 ± 2.7 min in mitosis (n = 20; as determined by cell morphology and DNA condensation) in these cultures. Aurkb (Δ/Δ) cells, on the other hand, displayed similar progression in the early phases of mitosis but exited from mitosis (defined by chromosome decondensation and re-formation of the nucleus) without forming a proper metaphase plate (). Interestingly, most of these mutant cells prematurely exited from mitosis in 40.75 ± 8.4 min (n = 20; ). A low percentage (15%) of Aurkb (Δ/Δ) cells displayed slightly longer duration of mitosis (about 60 min) and usually contained collapsed spindles and exited from mitosis with typical ring-like nuclei (). These results indicate that Aurora B-null cells do not display major defects in mitotic entry but are not able to maintain the mitotic state and prematurely exit mitosis from prometaphase-like structures.
Increased p21Cip1 and defective Cdk1 activity in Aurora B-null cells
To get some molecular insights into the altered regulation of mitosis in Aurora B-null cells, MEFs were synchronized with aphidicolin and released in the presence or absence of nocodazole or taxol. Wild-type cells displayed phosphorylation of Cdk substrates 18–24 h after the aphidicolin release suggesting the presence of mitotic cells in these cultures. Aurkb (Δ/Δ) cells displayed decreased phosphorylation of Cdk substrates as well as decreased signal of MPM2, an antibody that recognizes a mixture of antigens mostly composed of Cdk substrates (). Interestingly, the levels of cyclin B1, securin and Tpx2 were increased in DMSO-treated Aurkb (Δ/Δ) cells despite the low presence of Cdk-dependent phosphorylation. In the presence of microtubule poisons, wild-type cells arrested in mitosis and displayed high levels of Cdk substrates that decreased with time due to mitotic slippage and continuous degradation of cyclin B1 and securin. As already reported,Citation8 Aurora B-knockout cells did not arrest in the presence of taxol and displayed a reduced arrest in the presence of nocodazole as monitored by the phosphorylation of Cdk substrates. Unexpectedly, these cells displayed high levels of cyclin B1 and securin (). Whereas most wild-type interphasic cells displayed no cyclin B1 signal in their nucleus, Aurora B-null cultures displayed frequent interphasic figures with high levels of cyclin B1 signal. These cells are typical Aurora B-null, post-mitotic cells, as detected by the presence of multiple nuclei, micronuclei or multiple microtubule-organizing centers (; Fig. S2). These abnormal cells with high cyclin B1 levels were also present after mitotic exit in the presence of taxol or nocodazole (Fig. S3). Whereas p21Cip1 frequently displayed a diffuse distribution in these fibroblasts, it was mostly nuclear in Aurkb (Δ/Δ) cells with high nuclear staining of cyclin B1 (Fig. S4). Whether these two molecules are forming a complex that is retained in the nucleus of these abnormal, Aurora B-null, tetraploid cells is unclear at this moment.
Figure 4. Aberrant Cdk1 activity and cyclin B1 degradation in Aurora B-null cultures. (A) Aurkb(lox/lox) cells were transduced with AdGFP- or AdCre- [to generate Aurkb(Δ/Δ) cells as indicated in ] expressing viruses and arrested in S-phase with aphidicolin, released in the presence of DMSO, nocodazole (Noc) or taxol and harvested for protein lysates at the indicated time points. The protein levels of Aurora B, cyclin B1, securin and Tpx2 and the phosphorylation of MPM2 antigens or Cdk substrates were analyzed by immunoblot with specific antibodies. (B) Immunofluorescence to detect cyclin B1 (CycB1, green) or α-tubulin (α-tub, red) in Aurkb(lox/lox) or Aurkb(Δ/Δ) cultures 48 h after serum addition as represented in . Cells with several nuclei or micronuclei (arrow) as well as cells with multiple centrosomes (arrowheads) frequently accumulate nuclear cyclin B1 in the absence of Aurora B. DAPI, blue. Scale bars, 10 μm. (C) Induction of p21Cip1 and formation of p21Cip1-Cdk1 complexes with reduced Cdk1 activity in Aurora B-null cells. The protein levels of p21Cip1 in the extract or after immunoprecipitation of Cdk1 are shown by specific antibodies. The levels of Cdk1, cyclin B1 or phosphorylated (Tyr15) Cdk1 are also detected after immunoprecipitation of Cdk1. The ability of these complexes to phosphorylate histone H1 (P-H1) is also shown. Time points indicate hours after stimulation with serum in the absence or presence of nocodazole (Noc).
![Figure 4. Aberrant Cdk1 activity and cyclin B1 degradation in Aurora B-null cultures. (A) Aurkb(lox/lox) cells were transduced with AdGFP- or AdCre- [to generate Aurkb(Δ/Δ) cells as indicated in Fig. 3] expressing viruses and arrested in S-phase with aphidicolin, released in the presence of DMSO, nocodazole (Noc) or taxol and harvested for protein lysates at the indicated time points. The protein levels of Aurora B, cyclin B1, securin and Tpx2 and the phosphorylation of MPM2 antigens or Cdk substrates were analyzed by immunoblot with specific antibodies. (B) Immunofluorescence to detect cyclin B1 (CycB1, green) or α-tubulin (α-tub, red) in Aurkb(lox/lox) or Aurkb(Δ/Δ) cultures 48 h after serum addition as represented in Figure 3A. Cells with several nuclei or micronuclei (arrow) as well as cells with multiple centrosomes (arrowheads) frequently accumulate nuclear cyclin B1 in the absence of Aurora B. DAPI, blue. Scale bars, 10 μm. (C) Induction of p21Cip1 and formation of p21Cip1-Cdk1 complexes with reduced Cdk1 activity in Aurora B-null cells. The protein levels of p21Cip1 in the extract or after immunoprecipitation of Cdk1 are shown by specific antibodies. The levels of Cdk1, cyclin B1 or phosphorylated (Tyr15) Cdk1 are also detected after immunoprecipitation of Cdk1. The ability of these complexes to phosphorylate histone H1 (P-H1) is also shown. Time points indicate hours after stimulation with serum in the absence or presence of nocodazole (Noc).](/cms/asset/59738b9c-ef21-4d94-a96e-06bf53193ed6/kccy_a_10924004_f0004.gif)
Mitotic exit is a consequence of the activation of Cdc20-bound anaphase-promoting complex (APC/C) and the subsequent targeting of cyclin B1 an securin with ubiquitin for proteasome-dependent degradation.Citation24 Degradation of cyclin B1 results in the inactivation of Cdk1, activation of PP1A-B55α,δ complexes and mitotic exit.Citation24,Citation25 Since Aurora B-null cells exited from mitosis prematurely and displayed increased levels of cyclin B1 and securin, we next tested whether Cdk1 could be inactivated by alternative mechanisms in these cells. We therefore analyzed Cdk1 complexes and their activity in Aurora B-null and control cultures several time points after cell cycle entry. As indicated in , both wild-type and Aurora B-null cells were able to activate Cdk1 at low levels 12 h after stimulation with serum and more strongly at 34 h, when a large percentage of the population was in mitosis. However, Aurora B-deficient cells accumulated high levels of p21Cip1, and, as a consequence, the pool of Cdk1 bound to p21Cip1 increased (). No major differences were found in the amount of cyclin B1 bound to Cdk1, as it is well established that Cip/Kip inhibitors form trimeric complexes with Cdks and cyclins.Citation26 However, Cdk1 activity was clearly reduced in these extracts. Since no significant increase was found in the inhibitory phosphorylation of Cdk1, the reduced Cdk1 activity is likely a consequence of the increased binding of the Cdk inhibitor p21Cip1 to Cdk1 ().
Premature mitotic exit in the absence of Aurora B is p21Cip1-dependent
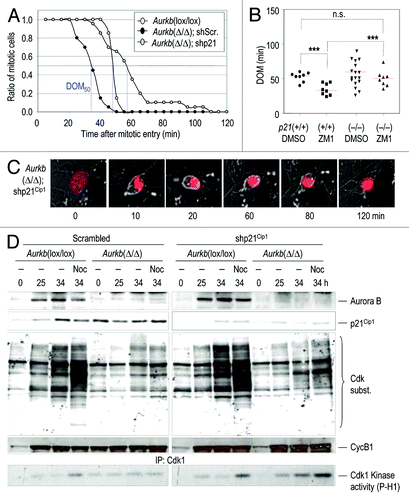
To test the functional relevance of p21Cip1 in the premature exit in Aurora B-null cells, we made use of short hairpin interfering RNAs (shRNAs) specific for the Cdkn1a (encoding p21Cip1) transcript. Aurkb (lox/lox) were infected with AdGFP or AdCre, as well as with vectors expressing shRNAs against p21Cip1 (shp21) or scrambled shRNAs (shScr), then stimulated with serum and monitored by videomicroscopy. Wild-type cells displayed a DOM50 (time after mitotic entry in which half of the population exits from mitosis) of 50 ± 3 min, whereas, in agreement with our previous results, Aurora B-null cells displayed a DOM50 of 35 ± 5 min. Interestingly, knockdown of p21Cip1 rescued the early mitotic exit in the absence of Aurora B, and these cells showed an increased DOM50 of 58 ± 8 min (). This rescue in the DOM did not affect other cellular defects in the absence of Aurora B, and these cells were in a prometaphase-like state with condensed chromosomes until they decondensed DNA and exited mitosis as tetraploid cells. We also tested whether lack of p21Cip1 rescued premature exit by using p21Cip1-knockout MEFs. Wild-type p21(+/+) MEFs prematurely exited from mitosis (DOM = 32 ± 6 min) when treated with the Aurora B inhibitor ZM1. However, p21(−/−) MEFs displayed a normal duration of mitosis (50 ± 5 min) when treated with the same doses of the Aurora B inhibitor (). The reason for the wide distribution in the DOM in untreated p21(−/−) MEFs is currently unknown, but it may be related to deregulation of Cdk1 in the absence of this cell cycle inhibitor. A few of these p21Cip1-knockdown or -knockout cells remained in mitosis for longer periods (80–100 min; ) suggesting increased Cdk1 activity and delayed mitotic exit in the absence of p21Cip1. At the molecular level, knockdown of p21Cip1 resulted in increased presence of Cdk-dependent mitotic phospho-epitopes at 25 or 34 h after cell cycle entry or in the presence of nocodazole. These mitotic markers were accompanied by increased Cdk1 kinase activity upon p21Cip1 downregulation (). All together, these results suggest that induction of p21Cip1 in the absence of Aurora B prevents the maintenance of high levels of Cdk1 activity and makes cells susceptible to rapid exit from mitosis due to partial Cdk1 inactivation.
Figure 5. Downregulation of p21Cip1 rescues premature mitotic exit in Aurora B-null cells. (A) Aurkb(Δ/Δ) cultures were transduced with retroviruses expressing short hairpin RNAs against the p21Cip1 transcript (shp21) or scrambled (shScr) sequences. Aurkb(lox/lox) cells were used as a control. In the graph, cells were aligned at mitotic entry and the plot represents the ratio of cells that remained in mitosis at different time points. Blue lines represent DOM50, i.e., the time after mitotic entry in which half of the cell population has exited mitosis. (B) Duration of mitosis (DOM) in p21Cip1(p21)(+/+) and p21(−/−) cells in the presence of DMSO or the Aurora B inhibitor ZM1. n.s., not significant; ***, p < 0.001. (C) Representative time-lapse images from Aurkb(Δ/Δ); shp21 cells in the assay described in (A). Expression of mCherry-H2B is in red. (D) Immunodetection of Aurora B, p21Cip1 and cyclin B1 (CycB1) in cells transduced with shp21 or shScr-expressing retroviruses. Activity of Cdk1 is shown by phosphorylation of its substrates and direct kinase assay on histone H1 after immunoprecipitation with Cdk1-specific antibodies. Time points indicate hours after stimulation with serum in the absence or presence of nocodazole (Noc).
Discussion
A role for Aurora B in interphase
The induction of Aurora B during G2 and M-phase and the requirements for this kinase in microtubule-kinetochore attachment and biorientation of chromosomes have been widely described in the last few years.Citation4,Citation5 Aurora B is targeted for degradation by APC/C-Cdh1 during mitotic exit, yet this kinase is known to be expressed at the basal level throughout the cell cycle.Citation27 Genetic ablation of the Aurora B-encoding gene in Aurkb(Δ/Δ) cells results in a significant delay (> 6 h in MEFs and > 1 d in liver cells; and ) in the entry into S-phase after stimulation of quiescent cells. As a reference, the defect in S-phase entry is significantly stronger than that observed in primary MEFs deficient in Cdk4, Cdk6 or both Cdk4/6 (ref. Citation28), two major kinases regulating G1/S transition in mammals. Similarly, S-phase entry in Aurkb(+/−) mice after partial hepatectomy is dramatically impaired, whereas double Cdk4;Cdk2-null mice display a quite normal regeneration of the liver in vivo.Citation29 Several evidences have recently suggested a role for Aurora B in G1. Aurora B and Survivin are known to form a complex with mTOR in T lymphocytes, and they promote the phosphorylation of mTOR targets such as p70S6K (ref. Citation16). mTOR actually co-localizes with the CPC at the central spindle midzone and the midbody regions during cytokinesis.Citation30 On the other hand, Survivin has been reported to bind directly to Cdk4 and to trigger phosphorylation of the retinoblastoma protein (pRb),Citation31 and Aurora B is thought to directly phosphorylate pRb at Ser780 during a endoreduplication checkpoint.Citation20 However, our data suggest that p70S6K or pRb are not critical targets of Aurora B, at least in fibroblasts or liver cells.
Recent reports have demonstrated that Aurora B can directly phosphorylate p53, suppressing the transcriptional activity and function of this factor.Citation18,Citation19,Citation32 Aurora B may be also targeted for degradation by the SCF-Fbw7 complex, which, in turn, is regulated by p53, suggesting regulatory feedback loops between these molecules.Citation33,Citation34 Similar networks have been described for Aurora A and p53.Citation34-Citation39 We reported previously that p53 is induced in Aurora B-null embryosCitation8 in agreement with the proposal that Aurora B-dependent phosphorylation of p53 may result in the increased degradation of this transcription factor.Citation19 Inactivation of Aurora B also results in increased p53 activity and the subsequent induction of p21Cip1 in several different cell types.Citation18,Citation19 Yet, the functional consequences of these findings in the regulation of the cell cycle were not well understood. Here, we have shown that p21Cip1 is significantly induced in Aurora B-null cells, and these cultures display a significant delay in the entry into S-phase. We have not addressed whether this induction is or not p53-dependent, as it is known that the G1 arrest induced by survivin inactivation, despite the induction of p53, is not rescued by concomitant ablation of this factor.Citation40,Citation41 Since genetic ablation of Aurora B is induced in quiescent/G0 cells, these defects are not the indirect consequence of mitotic aberrations but indicate a specific requirement for Aurora B in the control of p21Cip1 levels during G1.
Aurora B modulates Cdk1 activity in mitosis
Elimination of Aurora B by RNA interference or specific inhibitors results in defective chromosome congression, accompanied by lagging chromosomes and extensive chromatin bridging at anaphase, finally leading to polyploidy.Citation4,Citation21,Citation22 Aurora B is also required for proper cytokinesis, and it is a central regulator abscission checkpoint that protects cells against tetraploidization.Citation15,Citation42 Aurora B is essential for SAC activation in response to taxol, but its requirement for SAC activation in the absence of microtubules is still a matter of debate given the partial effects found in multiple assays and the limited efficiency and specificity of chemical inhibitors.Citation43,Citation44 Aurora B-null cells display aberrant BubR1 localization and a partial SAC-dependent arrest in the presence of high concentration of nocodazole, supporting the notion that Aurora B is directly involved, but not essential, in the SAC (ref. Citation8; Fig. S3).
In this manuscript we describe a new function of Aurora B in sustaining mitotic progression. In the absence of Aurora B, Cdk1 activity is diminished as a consequence of the induction of the Cdk inhibitor p21Cip1 and the presence of an increased pool of p21Cip1-Cdk1 complexes. Aurora B-null cells exit prematurely from mitosis, suggesting that the presence of p21Cip1 may impair the balance between Cdk1 and PP2A activities in favor of the phosphatase. In fact, since Cdk1 activity is reduced, cells may eventually exit from mitosis without a complete degradation of cyclin B1, a possibility that is further supported by the presence of high levels of cyclin B1 in Aurora B-deficient cells.
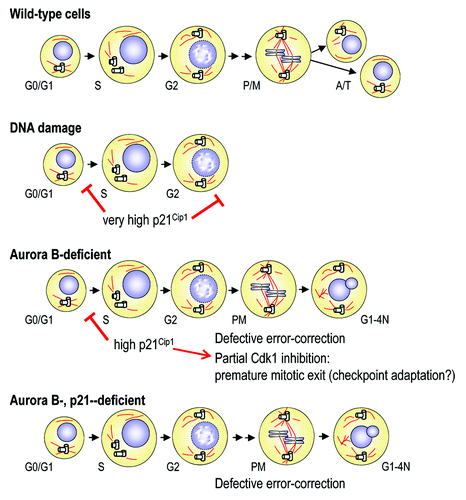
Exiting mitosis without cyclin B1 degradation is known as “adaptation” to the SAC. Yeast cells can adapt to the checkpoint through inactivation of the mitotic kinase Cdk1 mediated by inhibitory phosphorylation or expression of Cdk1 inhibitors. In vertebrates, adaptation to the SAC has not been described so far. In fact, it is well established that cells exit from mitosis in the absence of complete bipolar attachment of kinetochores by mitotic slippage, i.e., partial but continuous activation of APC/C-Cdc20 and degradation of cyclin B1 even in the presence of these aberrations.Citation45,Citation46 The presence of reduced degradation of cyclin B (and securin) in Aurora B-null cells suggests the presence of certain levels of mitotic checkpoint adaptation in these primary fibroblasts. It is important to note that this adaptation may be difficult to see in immortal or tumoral cells in which p21Cip1 is not significantly expressed. The punctuated pattern of cyclin B1 accumulation is reminiscent of what has previously been observed with Tpx2 (nuclear accumulation in interphase) as a consequence of defective APC/C-Cdh1 activation after mitotic slippage,Citation45 and therefore suggests defective degradation of cyclin B1 during mitotic exit in Aurora B-null cells. Overexpression of p21Cip1 has been previously shown to result in nuclear sequestration of inactive p21Cip1-Cdk1-Cyclin B1 complexes.Citation47,Citation48 Upon DNA damage, p21Cip1 induction results in strong G1 inhibition and G2 arrest due to these inactive complexes. Aurora B-null cells display strong nuclear accumulation of cyclin B1 in non-mitotic cells. Although these cells could be thought to correspond to G2 cells we think this is not the case since: (1) We have no evidence of G2 arrest in the absence of Aurora B (); (2) These cells are frequently binucleated or contain multiple microtubule-organizing centers (; Fig. S2), indicating that they are a result of abnormal mitosisCitation8; and (3) Accumulation of cyclin is also seen after mitotic exit in the presence of taxol or nocodazole (Fig. S3). All these data suggest that whereas very high levels of p21Cip1, as the ones obtained after DNA damage (doxorubicine; ), result in G2 arrest (data not shown and refs. Citation47 and Citation48), intermediate high levels of p21Cip1 allow mitotic entry but result in premature exit due to weak Cdk1 activity (). Further research will be necessary to formally prove the relevance of this possible adaptation to the SAC in mammals. Importantly, downregulation of p21Cip1 rescues full Cdk1 activity and the premature exit from mitosis, suggesting the relevance of the Aurora B-p21Cip1 interaction described here in the maintenance of the mitotic state. As upregulation of p21Cip1 occurs in G0/G1, this additional role of Aurora B in the control of mitosis is actually a consequence of the interphase function of this kinase in suppressing the levels of the cell cycle inhibitor. It is important to note that lack of p21Cip1 may also prolong the duration of mitosis in normal cells (; no statistical differences in the mean but variances were statistically different p < 0.0001), suggesting that this cell cycle inhibitor may play important roles in the control of Cdk1 activity during mitosis.
Figure 6. Relationships between Aurora B and p21Cip1 during the cell cycle. DNA damage agents induce very high levels of p21Cip1 resulting in G1 and G2 arrest, and prevent entry into mitosis. Lack of Aurora B results in significant upregulation of p21Cip1 and the subsequent delay in G1/S transition. In these cells, the intermediate high levels of p21Cip1 are insufficient to prevent mitotic entry, but Aurora B-null cells display defective Cdk1 activation and premature exit. Concomitant ablation of p21Cip1 rescues Cdk1 activity and the duration of mitosis, although these cells exit from the cell cycle as tetraploid cells due to defective error-correction of improper microtubule-to-kinetochore attachments and lack of chromosome segregation.
Tetraploidy, senescence and cancer therapy
Premature mitotic exit (perhaps by adaptation to the SAC) provides an efficient mechanism for ultimately preventing mitosis upon inappropriate conditions. The most obvious scenario is represented by cellular or oncogene-induced senescence, which is usually characterized by p53-dependent or independent upregulation of p21Cip1. The excess in p21Cip1 may result in premature exit from mitosis without chromosome segregation due to defective Cdk1 activity. In fact, senescence is commonly characterized by the presence of large cells with large nuclei that are frequently tetraploid.Citation49 The mitotic machinery is commonly downregulated during the process of cellular senescence, and the mechanism described here may therefore participate in the morphological changes that accompany senescence, i.e., lack of cell division and tetraploidy.
Aurora kinases have recently received much attention due to their possible involvement in tumor development and their therapeutic value.Citation50,Citation51 All three Aurora kinases are overexpressed in different types of cancers.Citation50,Citation52 Whereas initial efforts were focused to Aurora A, recent data suggest that Aurora B is a relevant cancer target.Citation53,Citation54 The critical roles of this kinase in the regulation of G1/S during interphase, or in the prevention of premature mitotic exit described in this manuscript may therefore have relevant implications for cancer therapy. For instance, Aurkb(+/−) mice display reduced formation of tumors upon specific treatments,Citation8 and this may be explained by increased induction of p21Cip1, as we have shown here in the liver of these heterozygous mutants. Further understanding of the clinical effect of Aurora B inhibition in p53- or p21Cip1-positive or -deficient tumors will be necessary to optimize the use of Aurora B inhibitors in the clinic.Citation50,Citation55-Citation57
Materials and Methods
Cell culture
Mouse embryo fibroblasts from Aurora B conditional knockoutCitation8 or p21Cip1-null miceCitation58 were obtained from E14.5 embryos and cultured using routine protocols.Citation59 All cultures were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 2 mM glutamine, 1% penicillin/streptomycin and 10% fetal bovine serum (FBS) or donor calf serum (CS). For acute deletion of Aurora B in cultured cells we followed the schemes represented in and . The adenoviruses expressing GFP or the Cre recombinase (Ad5 CMV-Cre) were obtained from the University of Iowa. Infection was performed during 2 d in a cell culture synchronized in G0 by serum deprivation and/or confluence. The lentiviral vectors expressing short hairpin RNAs against p21Cip1 were used as reported previously.Citation60 ZM447439 (abbreviated as ZM1, Tocris Bioscience) and doxorubicin (Sigma) were used at 20 μM and 1 μM, respectively. Nocodozole (Sigma) was used at 3.5 μM to completely prevent the formation of microtubules,Citation61 and taxol (Sigma) was used at 500 nM. To measure entry into S-phase, cells were pulsed for 30 min with 10 μM BrdU and stained using an anti-BrdU antibody (PharMingen). DNA content was analyzed by flow cytometry (FACS; Becton-Dickinson). The duration of mitosis was scored by videomicroscopy using Deltavision apparatus as indicated previously.Citation8
Hepatectomy and wound healing
All protocols in live animals were performed following the current European and Spanish Guidelines for Humane End Points for Animals used in biomedical research. All animals were maintained in a mixed 129/Sv (25%) × CD1 (25%) × C57BL/6J (50%) background. Skin woundsCitation62 and 2/3 partial hepatectomyCitation63 were essentially performed as reported previously. After hepatectomy, mice were injected with 50 mg/kg BrdU 2 h before mice were sacrificed for samples. For histological observation, dissected organs were fixed in 10%-buffered formalin (Sigma) and embedded in paraffin wax. Sections of 3- or 5-μm thickness were stained with hematoxylin and eosin (H&E). Additional immunohistochemical examination of the tissues and pathologies analyzed were performed using specific antibodies against BrdU (GE Healthcare), or Ki67 (Master Diagnostica).
mRNA quantification
Total RNA from cultured cells was isolated using the RNeasy mini kit (Qiagen) and quantitative reverse transcription-PCR (qRT-PCR) was performed using the SuperScript III Platinum SYBR Green One-Step qRT-PCR kit with ROX system (Invitrogen). GAPDH was used for normalization of mRNA levels.
Immunofluorescence and protein analysis
For immunofluorescence, MEFs were fixed with 4% PFA and permeabilized with 0.15% Triton X. Cells were then blocked with 3% BSA and incubated with MPM2 antibodies (Millipore, 05-368) or primary antibodies against the following antigens: α-tubulin (Sigma, T5168 and T9026), γ-tubulin (Sigma, T6557), Aurora B (Abcam, ab2254), BrdU (GE Healthcare; RPN202), Cyclin B1 (Millipore, MAB3684), p21Cip1 (Santa Cruz Biotechnology, sc-397) and phospho-histone H3 Ser10 (P-H3; Millipore, 05-636) for 2–4 h at 37°C. The matching secondary antibodies, with different Alexa dies (488, 594, 647), are from Molecular Probes (Invitrogen). Images were obtained using a confocal ultra-spectral microscope (Leica TCS-SP5-AOBS-UV).
For immunodetection in protein lysates, cells were washed twice with ice-cold PBS and lysed in RIPA lysis buffer (37 mM NaCl, 0.5% NP-40, 0.1% SDS, 1% TX-100, 20 mM TRIS-HCl pH 7.4, 2 mM EDTA, 10% glicerol 1 mM PMSF) supplemented with protease and phosphatase inhibitory cocktails (Sigma). After 30 min on ice, samples were cleared by centrifugation. Proteins were separated on SDS-PAGE, transferred to nitrocellulose membranes (BioRad), probed using specific antibody and detected using fluorescent donkey (Rockland) or goat (Invitrogen) anti-rabbit or anti-mouse secondary antibodies followed detection using the Odyssey Infrared Imaging System (Li-Cor Biosciences). After transfer of the protein lysates, we probed nitrocellulose membranes with MPM2 antibodies (Millipore, 05-368) or primary antibodies against Aurora B (Abcam, ab2254), p21Cip1 (Santa Cruz Biotechnology, sc-397), phospho-p70S6K (T389, Cell Signaling), phospho-pRb (S780, Cell Signaling), Akt (Cell Signaling, 9272), phospho-Cdk substrates (Cell Signaling, 2324), securin (Abcam, ab3305), Tpx2 (Lifespan Biosciences, LS-B146), Cdk1 (Santa Cruz Biotechnology, sc-54), Phospho-Tyr15-Cdk1 (Santa Cruz Biotechnology, sc-7989) or β-Actin (Sigma, A4700). Secondary antibodies were coupled to Alexa 680 and 800 dies (Invitrogen) for immunodetection. Cdk1 kinase assays were performed essentially as described previously.Citation59
| Abbreviations: | ||
| APC/C | = | anaphase-promoting comple |
| CPC | = | chromosomal passenger complex |
| SAC | = | spindle assembly checkpoint |
Additional material
Download Zip (1.3 MB)Acknowledgments
M.T. and G.F.M. were supported by the Foundation La Caixa and the Comunidad de Madrid. This work was supported by grants from the Ministerio de Economía y Competitividad (MINECO; SAF2012-38215), Fundación Ramón Areces, the OncoCycle Programme (S2010/BMD-2470) from the Comunidad de Madrid, the OncoBIO Consolider-Ingenio 2010 Programme (CSD2007-00017) from the MINECO and the European Union Seventh Framework Programme (MitoSys project; HEALTH-F5-2010-241548).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
M.T. performed most of the mitotic assays, whereas G.F.M. analyzed G1/S progression. I.P.C. helped in the analysis of the data. C.H. performed the hepatectomy assay. M.M. designed and supervised the project and wrote the manuscript.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/24004
Related Research Data
References
- Malumbres M. Physiological relevance of cell cycle kinases. Physiol Rev 2011; 91:973 - 1007; http://dx.doi.org/10.1152/physrev.00025.2010; PMID: 21742793
- Glover DM, Leibowitz MH, McLean DA, Parry H. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell 1995; 81:95 - 105; http://dx.doi.org/10.1016/0092-8674(95)90374-7; PMID: 7720077
- Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol 2001; 2:21 - 32; http://dx.doi.org/10.1038/35048096; PMID: 11413462
- Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol 2003; 4:842 - 54; http://dx.doi.org/10.1038/nrm1245; PMID: 14625535
- Lens SM, Voest EE, Medema RH. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev Cancer 2010; 10:825 - 41; http://dx.doi.org/10.1038/nrc2964; PMID: 21102634
- Brown JR, Koretke KK, Birkeland ML, Sanseau P, Patrick DR. Evolutionary relationships of Aurora kinases: implications for model organism studies and the development of anti-cancer drugs. BMC Evol Biol 2004; 4:39; http://dx.doi.org/10.1186/1471-2148-4-39; PMID: 15476560
- Slattery SD, Moore RV, Brinkley BR, Hall RM. Aurora-C and Aurora-B share phosphorylation and regulation of CENP-A and Borealin during mitosis. Cell Cycle 2008; 7:787 - 95; http://dx.doi.org/10.4161/cc.7.6.5563; PMID: 18239465
- Fernández-Miranda G, Trakala M, Martín J, Escobar B, González A, Ghyselinck NB, et al. Genetic disruption of aurora B uncovers an essential role for aurora C during early mammalian development. Development 2011; 138:2661 - 72; http://dx.doi.org/10.1242/dev.066381; PMID: 21613325
- Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol 2007; 8:798 - 812; http://dx.doi.org/10.1038/nrm2257; PMID: 17848966
- Carmena M, Wheelock M, Funabiki H, Earnshaw WC. The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol 2012; 13:789 - 803; http://dx.doi.org/10.1038/nrm3474; PMID: 23175282
- Lens SM, Medema RH. The survivin/Aurora B complex: its role in coordinating tension and attachment. Cell Cycle 2003; 2:507 - 10; http://dx.doi.org/10.4161/cc.2.6.559; PMID: 14504461
- Cimini D. Detection and correction of merotelic kinetochore orientation by Aurora B and its partners. Cell Cycle 2007; 6:1558 - 64; http://dx.doi.org/10.4161/cc.6.13.4452; PMID: 17603301
- Liu D, Vader G, Vromans MJ, Lampson MA, Lens SM. Sensing chromosome bi-orientation by spatial separation of aurora B kinase from kinetochore substrates. Science 2009; 323:1350 - 3; http://dx.doi.org/10.1126/science.1167000; PMID: 19150808
- Guse A, Mishima M, Glotzer M. Phosphorylation of ZEN-4/MKLP1 by aurora B regulates completion of cytokinesis. Curr Biol 2005; 15:778 - 86; http://dx.doi.org/10.1016/j.cub.2005.03.041; PMID: 15854913
- Steigemann P, Wurzenberger C, Schmitz MH, Held M, Guizetti J, Maar S, et al. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 2009; 136:473 - 84; http://dx.doi.org/10.1016/j.cell.2008.12.020; PMID: 19203582
- Song J, Salek-Ardakani S, So T, Croft M. The kinases aurora B and mTOR regulate the G1-S cell cycle progression of T lymphocytes. Nat Immunol 2007; 8:64 - 73; http://dx.doi.org/10.1038/ni1413; PMID: 17128276
- Sabbattini P, Canzonetta C, Sjoberg M, Nikic S, Georgiou A, Kemball-Cook G, et al. A novel role for the Aurora B kinase in epigenetic marking of silent chromatin in differentiated postmitotic cells. EMBO J 2007; 26:4657 - 69; http://dx.doi.org/10.1038/sj.emboj.7601875; PMID: 17948062
- Wu L, Ma CA, Zhao Y, Jain A. Aurora B interacts with NIR-p53, leading to p53 phosphorylation in its DNA-binding domain and subsequent functional suppression. J Biol Chem 2011; 286:2236 - 44; http://dx.doi.org/10.1074/jbc.M110.174755; PMID: 20959462
- Gully CP, Velazquez-Torres G, Shin JH, Fuentes-Mattei E, Wang E, Carlock C, et al. Aurora B kinase phosphorylates and instigates degradation of p53. Proc Natl Acad Sci USA 2012; 109:E1513 - 22; http://dx.doi.org/10.1073/pnas.1110287109; PMID: 22611192
- Nair JS, Ho AL, Tse AN, Coward J, Cheema H, Ambrosini G, et al. Aurora B kinase regulates the postmitotic endoreduplication checkpoint via phosphorylation of the retinoblastoma protein at serine 780. Mol Biol Cell 2009; 20:2218 - 28; http://dx.doi.org/10.1091/mbc.E08-08-0885; PMID: 19225156
- Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, et al. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol 2003; 161:267 - 80; http://dx.doi.org/10.1083/jcb.200208091; PMID: 12719470
- Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol 2003; 161:281 - 94; http://dx.doi.org/10.1083/jcb.200208092; PMID: 12707311
- Vader G, Cruijsen CW, van Harn T, Vromans MJ, Medema RH, Lens SM. The chromosomal passenger complex controls spindle checkpoint function independent from its role in correcting microtubule kinetochore interactions. Mol Biol Cell 2007; 18:4553 - 64; http://dx.doi.org/10.1091/mbc.E07-04-0328; PMID: 17699588
- Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol 2006; 7:644 - 56; http://dx.doi.org/10.1038/nrm1988; PMID: 16896351
- Manchado E, Guillamot M, de Cárcer G, Eguren M, Trickey M, García-Higuera I, et al. Targeting mitotic exit leads to tumor regression in vivo: Modulation by Cdk1, Mastl, and the PP2A/B55α,δ phosphatase. Cancer Cell 2010; 18:641 - 54; http://dx.doi.org/10.1016/j.ccr.2010.10.028; PMID: 21156286
- Pavletich NP. Mechanisms of cyclin-dependent kinase regulation: structures of Cdks, their cyclin activators, and Cip and INK4 inhibitors. J Mol Biol 1999; 287:821 - 8; http://dx.doi.org/10.1006/jmbi.1999.2640; PMID: 10222191
- Stewart S, Fang G. Destruction box-dependent degradation of aurora B is mediated by the anaphase-promoting complex/cyclosome and Cdh1. Cancer Res 2005; 65:8730 - 5; http://dx.doi.org/10.1158/0008-5472.CAN-05-1500; PMID: 16204042
- Malumbres M, Sotillo R, Santamaría D, Galán J, Cerezo A, Ortega S, et al. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 2004; 118:493 - 504; http://dx.doi.org/10.1016/j.cell.2004.08.002; PMID: 15315761
- Barrière C, Santamaría D, Cerqueira A, Galán J, Martín A, Ortega S, et al. Mice thrive without Cdk4 and Cdk2. Mol Oncol 2007; 1:72 - 83; http://dx.doi.org/10.1016/j.molonc.2007.03.001; PMID: 19383288
- Vazquez-Martin A, Sauri-Nadal T, Menendez OJ, Oliveras-Ferraros C, Cufí S, Corominas-Faja B, et al. Ser2481-autophosphorylated mTOR colocalizes with chromosomal passenger proteins during mammalian cell cytokinesis. Cell Cycle 2012; 11:4211 - 21; http://dx.doi.org/10.4161/cc.22551; PMID: 23095638
- Suzuki A, Ito T, Kawano H, Hayashida M, Hayasaki Y, Tsutomi Y, et al. Survivin initiates procaspase 3/p21 complex formation as a result of interaction with Cdk4 to resist Fas-mediated cell death. Oncogene 2000; 19:1346 - 53; http://dx.doi.org/10.1038/sj.onc.1203429; PMID: 10713676
- Wu L, Ma CA, Jain A. When Aurora B met p53: newly revealed regulatory phosphorylation in an old protein. Cell Cycle 2011; 10:171 - 2; http://dx.doi.org/10.4161/cc.10.2.14349; PMID: 21212738
- Teng CL, Hsieh YC, Phan L, Shin J, Gully C, Velazquez-Torres G, et al. FBXW7 is involved in Aurora B degradation. Cell Cycle 2012; 11:4059 - 68; http://dx.doi.org/10.4161/cc.22381; PMID: 23095493
- Wang Y, Zhou BP. FBW7-Aurora B-p53 feedback loop regulates mitosis and cell growth. Cell Cycle 2012; 11:4113 - 4; http://dx.doi.org/10.4161/cc.22607; PMID: 23099923
- Wu CC, Yang TY, Yu CT, Phan L, Ivan C, Sood AK, et al. p53 negatively regulates Aurora A via both transcriptional and posttranslational regulation. Cell Cycle 2012; 11:3433 - 42; http://dx.doi.org/10.4161/cc.21732; PMID: 22894933
- Chiang CM. p53-Aurora A mitotic feedback loop regulates cell cycle progression and genomic stability. Cell Cycle 2012; 11:3719 - 20; http://dx.doi.org/10.4161/cc.22113; PMID: 22982999
- Nair JS, Ho AL, Schwartz GK. The induction of polyploidy or apoptosis by the Aurora A kinase inhibitor MK8745 is p53-dependent. Cell Cycle 2012; 11:807 - 17; http://dx.doi.org/10.4161/cc.11.4.19323; PMID: 22293494
- Hsueh KW, Fu SL, Chang CB, Chang YL, Lin CH. A novel Aurora-A-mediated phosphorylation of p53 inhibits its interaction with MDM2. Biochim Biophys Acta 2012; PMID: 23201157
- Katayama H, Sen S. Functional significance of Aurora kinase A regulatory interactions with p53-ERα complex in human breast cancer cells. Horm Cancer 2011; 2:117 - 24; http://dx.doi.org/10.1007/s12672-011-0070-x; PMID: 21761334
- Yang D, Welm A, Bishop JM. Cell division and cell survival in the absence of survivin. Proc Natl Acad Sci USA 2004; 101:15100 - 5; http://dx.doi.org/10.1073/pnas.0406665101; PMID: 15477601
- Okada H, Bakal C, Shahinian A, Elia A, Wakeham A, Suh WK, et al. Survivin loss in thymocytes triggers p53-mediated growth arrest and p53-independent cell death. J Exp Med 2004; 199:399 - 410; http://dx.doi.org/10.1084/jem.20032092; PMID: 14757745
- Miyauchi K, Zhu X, Foong C, Hosoya H, Murata-Hori M. Aurora B kinase activity is required to prevent polar cortical ingression during cytokinesis. Cell Cycle 2007; 6:2549 - 53; http://dx.doi.org/10.4161/cc.6.20.4817; PMID: 17726378
- Nezi L, Musacchio A. Sister chromatid tension and the spindle assembly checkpoint. Curr Opin Cell Biol 2009; 21:785 - 95; http://dx.doi.org/10.1016/j.ceb.2009.09.007; PMID: 19846287
- Santaguida S, Vernieri C, Villa F, Ciliberto A, Musacchio A. Evidence that Aurora B is implicated in spindle checkpoint signalling independently of error correction. EMBO J 2011; 30:1508 - 19; http://dx.doi.org/10.1038/emboj.2011.70; PMID: 21407176
- Brito DA, Rieder CL. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol 2006; 16:1194 - 200; http://dx.doi.org/10.1016/j.cub.2006.04.043; PMID: 16782009
- Lee J, Kim JA, Margolis RL, Fotedar R. Substrate degradation by the anaphase promoting complex occurs during mitotic slippage. Cell Cycle 2010; 9:1792 - 801; http://dx.doi.org/10.4161/cc.9.9.11519; PMID: 20436289
- Baus F, Gire V, Fisher D, Piette J, Dulić V. Permanent cell cycle exit in G2 phase after DNA damage in normal human fibroblasts. EMBO J 2003; 22:3992 - 4002; http://dx.doi.org/10.1093/emboj/cdg387; PMID: 12881433
- Charrier-Savournin FB, Château MT, Gire V, Sedivy J, Piette J, Dulic V. p21-Mediated nuclear retention of cyclin B1-Cdk1 in response to genotoxic stress. Mol Biol Cell 2004; 15:3965 - 76; http://dx.doi.org/10.1091/mbc.E03-12-0871; PMID: 15181148
- Davoli T, de Lange T. The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol 2011; 27:585 - 610; http://dx.doi.org/10.1146/annurev-cellbio-092910-154234; PMID: 21801013
- Keen N, Taylor S. Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer 2004; 4:927 - 36; http://dx.doi.org/10.1038/nrc1502; PMID: 15573114
- Pérez de Castro I, de Cárcer G, Montoya G, Malumbres M. Emerging cancer therapeutic opportunities by inhibiting mitotic kinases. Curr Opin Pharmacol 2008; 8:375 - 83; http://dx.doi.org/10.1016/j.coph.2008.06.013; PMID: 18644252
- Giet R, Petretti C, Prigent C. Aurora kinases, aneuploidy and cancer, a coincidence or a real link?. Trends Cell Biol 2005; 15:241 - 50; http://dx.doi.org/10.1016/j.tcb.2005.03.004; PMID: 15866028
- Girdler F, Gascoigne KE, Eyers PA, Hartmuth S, Crafter C, Foote KM, et al. Validating Aurora B as an anti-cancer drug target. J Cell Sci 2006; 119:3664 - 75; http://dx.doi.org/10.1242/jcs.03145; PMID: 16912073
- Girdler F, Sessa F, Patercoli S, Villa F, Musacchio A, Taylor S. Molecular basis of drug resistance in aurora kinases. Chem Biol 2008; 15:552 - 62; http://dx.doi.org/10.1016/j.chembiol.2008.04.013; PMID: 18559266
- Curry J, Angove H, Fazal L, Lyons J, Reule M, Thompson N, et al. Aurora B kinase inhibition in mitosis: strategies for optimising the use of aurora kinase inhibitors such as AT9283. Cell Cycle 2009; 8:1921 - 9; http://dx.doi.org/10.4161/cc.8.12.8741; PMID: 19440047
- Marxer M, Foucar CE, Man WY, Chen Y, Ma HT, Poon RY. Tetraploidization increases sensitivity to Aurora B kinase inhibition. Cell Cycle 2012; 11:2567 - 77; http://dx.doi.org/10.4161/cc.20947; PMID: 22722494
- Rao B, van Leeuwen IM, Higgins M, Campbel J, Thompson AM, Lane DP, et al. Evaluation of an Actinomycin D/VX-680 aurora kinase inhibitor combination in p53-based cyclotherapy. Oncotarget 2010; 1:639 - 50; PMID: 21317459
- Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 1995; 377:552 - 7; http://dx.doi.org/10.1038/377552a0; PMID: 7566157
- García-Higuera I, Manchado E, Dubus P, Cañamero M, Méndez J, Moreno S, et al. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat Cell Biol 2008; 10:802 - 11; http://dx.doi.org/10.1038/ncb1742; PMID: 18552834
- Tiscornia G, Singer O, Verma IM. Design and cloning of lentiviral vectors expressing small interfering RNAs. Nat Protoc 2006; 1:234 - 40; http://dx.doi.org/10.1038/nprot.2006.36; PMID: 17406238
- Yang Z, Kenny AE, Brito DA, Rieder CL. Cells satisfy the mitotic checkpoint in Taxol, and do so faster in concentrations that stabilize syntelic attachments. J Cell Biol 2009; 186:675 - 84; http://dx.doi.org/10.1083/jcb.200906150; PMID: 19720871
- Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet 2004; 36:744 - 9; http://dx.doi.org/10.1038/ng1382; PMID: 15208629
- Mitchell C, Willenbring H. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat Protoc 2008; 3:1167 - 70; http://dx.doi.org/10.1038/nprot.2008.80; PMID: 18600221