Abstract
The antineoplastic agent cis-diammineplatinum(II) dichloride (cisplatin, CDDP) is part of the poorly effective standard treatment of non-small cell lung carcinoma (NSCLC). Here, we report a novel strategy to improve the efficacy of CDDP. In conditions in which CDDP alone or either of two PARP inhibitors, PJ34 hydrochloride hydrate or CEP 8983, used as standalone treatments were inefficient in killing NSCLC cells, the combination of CDDP plus PJ34 or that of CDDP plus CEP 8983 were found to kill a substantial fraction of the cells. This cytotoxic synergy could be recapitulated by combining CDDP and the siRNA-mediated depletion of the principal PARP isoform, PARP1, indicating that it is mediated by on-target effects of PJ34 or CEP 8983. CDDP and PARP inhibitors synergized in inducing DNA damage foci, mitochondrial membrane permeabilization leading to cytochrome c release, and dissipation of the inner transmembrane potential, caspase activation, plasma membrane rupture and loss of clonogenic potential in NSCLC cells. Collectively, our results indicate that CDDP can be advantageously combined with PARP inhibitors to kill several NSCLC cell lines, independently from their p53 status. Combined treatment with CDDP and PARP inhibitors elicits the intrinsic pathway of apoptosis.
Introduction
The antineoplastic agent cis-diammineplatinum(II) dichloride (cisplatin, CDDP) is best known for its capacity to form intra- and interstrand DNA adducts, leading to the activation of a (frequently p53-dependent) DNA damage response that ignites cell senescence or (mostly apoptotic) cell death.Citation1-Citation5 In addition, CDDP can activate hitherto poorly characterized cytoplasmic signaling pathways that result in the apoptotic or necrotic demise of cancer cells characterized by an oxidative/metabolic burst, and, at least in some cases, the activation of pro-apoptotic proteins including the BCL-2 family members BAX and BAK, which act on mitochondria to activate the intrinsic cell death pathway.Citation6-Citation8
CDDP was approved by the US Food and Drug Administration for use in cancer patients as early as 1978.Citation9 Since then, this platinum derivative has routinely been employed for the treatment of a large panel of solid neoplasms, including head and neck, lung, esophageal, gastric, ovarian, uterin, cervical, bladder and germ cell tumors.Citation9 Nevertheless, with the notable exception of testicular germ cell cancer, a clinical setting in which CDDP induces durable complete remissions in > 80% of patients,Citation10 the therapeutic efficacy of CDDP is limited.Citation11,Citation12 On one hand, the use of CDDP has been shown to promote non-negligible adverse effects, including grade I–II nephrotoxicity (in > 30% of patients), ototoxicity and neurotoxicity (both in 10–30% of patients).Citation9 On the other hand, most neoplasms become insensitive to the cytotoxic effects of CDDP in the course of therapy.Citation11,Citation12 Thus, chemoresistance—be it innate or acquired—currently represents the most consistent impediment to the clinical use of CDDP.Citation11,Citation13,Citation14
We have recently undertaken a systems biology study, involving both in vitro and in vivo models, to get further insights into the signaling pathways activated by CDDP in non-small cell lung carcinoma (NSCLC), a relatively frequent and aggressive variant of lung cancer that is responsible for > 1 million deaths worldwide annually.Citation15,Citation16 In this context, we have identified and characterized several CDDP response modifiers, that is, factors that limit or aggravate the cytotoxic response of NSCLC cells to CDDP,Citation17-Citation21 More recently, we have demonstrated that cancer cells often (but not always) develop acquired CDDP resistance in vitro while hyperactivating poly(ADP-ribose) (PAR) polymerases (PARPs), hence becoming sensitive to the cytotoxic effects of PARP inhibitors.Citation22
As they are intimately involved in a multitude of cellular functions, including transcriptional regulation and DNA repair,Citation23,Citation24 PARPs have attracted great attention as targets for the development of novel antineoplastic agents.Citation23,Citation25,Citation26 Thus, no less than nine chemically distinct PARP inhibitors are currently being investigated in clinical trials (www.clinicaltrials.gov),Citation26-Citation28 most often for the treatment of tumors bearing loss-of-function mutations of BRCA1 and BRCA2.Citation29,Citation30 By interfering with the base-excision repair (BER) pathway, PARP inhibitors facilitate indeed the accumulation of double-strand DNA breaks, which are not conveniently repaired in BRCA-deficient cells.Citation31 PARP inhibitors have also been tested for their ability to exacerbate the antineoplastic effects of various DNA-damaging agents, including (but not limited to) ionizing irradiation, temozolomide (an alkylating agent), camptothecin (a topoisomerase inhibitor) as well as CDDP and carboplatin (another platinum derivative).Citation32-Citation35
Here, we demonstrate that PARP inhibitors and CDDP synergize in the killing of NSCLC cell lines, independent of their p53 status. Our findings indicate that a combination of PARP inhibitors and CDDP can stimulate the induction of the mitochondrial pathway of apoptosis.
Results and Discussion
PARP inhibitors and cisplatin synergize in the killing of wild-type A549 cells
By means of a high-throughput screening (HTS) based on the fluorescence microscopy-assisted detection of nuclear shrinkage (as a marker of apoptotic cell death),Citation36,Citation37 we have identified a few drugs (among 1040 FDA-approved chemicals) that significantly aggravate the lethal response of NSCLC A549 cells to the commercial PARP inhibitor PJ34 hydrochloride hydrate (PJ). These compounds included the multiple DNA-damaging agents, such as thiotepa, dacarbazine, mitomycin C and CDDP,Citation22 prompting us to investigate the possibility that PARP inhibitors may be employed to exacerbate the cytotoxic potential of CDDP against NSCLC cells.
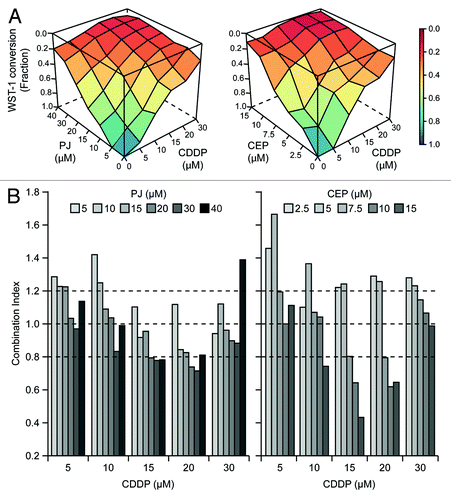
To obtain precise quantitative insights into the interaction between PARP inhibitors and CDDP, we exposed A549 cells to increasing concentrations of CDDP and either PJ or another PARP-targeting agent that is under clinical development (www.clinicaltrials.gov), CEP 8983 (CEP), for 48 h, followed by the assessment of residual cellular viability using a colorimetric test based on the mitochondrial oxidation of WST-1. Visual inspection of combination matrices () as well as combination indexes (CIs), calculated following Harbron’s methodCitation38 (; Tables S1 and 2), revealed that CDDP and PARP inhibitors can synergize in the killing of A549 cells (CI < 0.8). CEP interacted with CDDP much more efficiently than PJ, as demonstrated by the comparatively lower CIs observed at CDDP concentrations ranging from 10 to 20 μM (; Tables S1 and 2).
Figure 1. Hyperadditive interactions of PARP inhibitors and cisplatin in NSCLC A549 cells. (A and B) Human non-small cell lung carcinoma (NSCLC) A549 cells were cultured in control conditions or exposed for 48 h to PJ34 hydrochloride hydrate (PJ), CEP 8983 (CEP) or cisplatin (CDDP), alone or in the indicated combinations, followed by the colorimetric assessment of residual proliferation by means of a WST-1 conversion-based assay. In panel (A), color-coded surfaces illustrate residual WST-1-converting activity. In panel (B), combination indexes (CIs), estimated according to the Harbron’s method starting from a data set built on n = 3 independent assessments, are depicted. CI < 0.8 is generally viewed as an indicator of bona fide synergistic interactions. See also Tables S1 and S2.
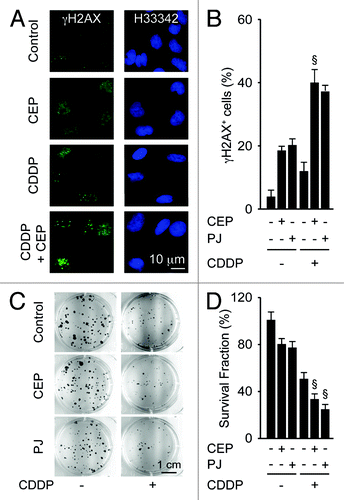
In the chronology of the response to CDDP, one of the first events (detectable within hours) is the induction of a DNA damage response that is characterized by the appearance of DNA damage foci (or γH2AX foci), which can be detected as nuclear puncta positive for phosphorylated histone H2AX.Citation39 In contrast, one of the latest events (detectable within weeks) is the loss of the clonogenic potential of cells exposed to CDDP.Citation40 The induction of γH2AX foci by CDDP was exacerbated by simultaneous treatment with CEP (). Moreover, the loss of the clonogenic survival of A549 cells induced by CDDP was increased by CEP or PJ, in conditions in which neither of the two PARP inhibitors affected clonogenic potential when used as single agents (). These results support the synergistic interaction between CDDP and PARP inhibitors, lasting from the first events mediated by CDDP, DNA damage, to its ultimate consequence, namely the loss of clonogenic potential, which constitutes a measurement for both cell death and senescence.
Figure 2. Synergistic effects of PARP inhibitors and cisplatin on DNA damage responses and clonogenic survival. (A and B) Effects of cisplatin (CDDP, 20 µM), PJ34 hydrochloride hydrate (PJ, 30 µM) or CEP 8983 (CEP, 10 µM), alone or in combination on DNA damage foci. Six hours after exposure to the indicated drug combinations, A549 cells were fixed, permeabilized and stained for the visualization of phosphorylated histone H2AX (γH2AX) in the presence of 2 µM Hoechst 33342 (H33342), which was used for nuclear counterstaining. Representative fluorescence microphotographs are shown in (A), and their quantitation (as the percentage of cells bearing > 5 γH2AX foci per nucleus) are shown in (B). (C and D). Effect of CDDP, PJ and/or CEP on clonogenic survival. After incubation with the indicated agents (1 µM CDDP, 5 µM PJ, 1 µM CEP) for 48 h, cells were cultured in drug-free medium, and colonies were rendered visible 10 d later by crystal violet staining. Representative photographs are shown in (C), and the frequency of colonies (referring to the untreated control as 100% value) is shown in (D). Results are means of triplicates ± SD. §p < 0.05 (Student’s t-test), compared with the sum of the cytotoxic effects caused by each agent alone.
Altogether, these results indicate that CDDP and PARP inhibitors engage in synergistic interactions, allowing for improved killing of NSCLC cells.
Mechanisms of the synergistic interaction between CDDP and PARP inhibitors
To explore the mechanisms of the interaction between CDDP and PARP inhibitors, we treated A549 cells with sub-apoptotic doses of PJ (30 µM), CEP (10 µM) and CDDP (20 µM)—alone or in combination—for 48 h, followed by the simultaneous cytofluorometric assessment of mitochondrial transmembrane potential (Δψm) and plasma membrane permeabilization after staining with the Δψm-sensitive fluorochrome 3,3′-dihexyloxacarbocyanine iodide [DiOC6(3)] and the exclusion dye propidium iodide (PI).Citation37,Citation41 Indeed, whereas neither PJ, neither CEP, nor CDDP per se increased the percentage of dying [DiOC6(3)lowPI−] and dead (PI+) A549 cells to more than 20%, the simultaneous administration of either PARP inhibitor and CDDP did induce consistent lethal effects (). Since pharmacological inhibitors may have off-target effects, we investigated whether the combination of CDDP and the genetic inhibition of PARP1 would exhibit synergistic effects as well. We found that the knockdown of PARP1 with three distinct, non-overlapping siRNAs was able to increase the response to a suboptimal dose (10 µM) of CDDP (), suggesting that the observed effects can indeed be attributed to PARP1 inhibition.
Figure 3. Cytotoxic response of NSCLC cells to PARP inhibitors and cisplatin. (A and B) Human non-small cell lung carcinoma (NSCLC) A549 cells were maintained in control conditions or exposed for 48 h to 30 µM PJ34 hydrochloride hydrate (PJ), 10 µM CEP 8983 (CEP) or the indicated concentration of CDDP, alone or combined, followed by the cytofluorometric assessment of apoptosis-related parameters. Panel (A) depicts representative dot plots. Numbers indicate the percentage of cells found in the corresponding gate. In panel (B), black and white columns report the percentage of dead (PI+) and dying [DiOC6(3)low PI−] cells, respectively (means ± SEM, n = 3). §p < 0.001 (Student’s t-test), compared with the sum of the cytotoxic effects caused by each agent alone. (C) Synergy between CDDP and PARP depletion. A549 cells were left untransfected (control) or were transfected with three distinct siRNAs targeting PARP1 or an unrelated (UNR) control siRNA. Twenty-four hours later, cells were treated for 48 h with the indicated CDDP concentration, followed by staining with PI and DiOC6(3). §p < 0.05 (Student’s t-test), compared with the sum of the cytotoxic effects caused by each agent alone. (D) Effects of CDDP and PARP inhibitors on additional human NSCLC cell lines. H226, H358 and H522 cells were maintained in control conditions or treated with the indicated concentrations of cisplatin (CDDP), alone or combined with increasing concentrations of CEP 8983 (CEP, 10 or 15 µM, 10 or 15 µM, and 5 or 10 µM in H522, H226 and H358 cells, respectively) or PJ34 hydrochloride hydrate (PJ, 40 or 60 µM) for 48 h, followed by the cytofluorometric assessment of apoptosis-related parameters as above. §p < 0.05 (Student’s t-test), compared with the sum of the cytotoxic effects caused by each agent alone.
![Figure 3. Cytotoxic response of NSCLC cells to PARP inhibitors and cisplatin. (A and B) Human non-small cell lung carcinoma (NSCLC) A549 cells were maintained in control conditions or exposed for 48 h to 30 µM PJ34 hydrochloride hydrate (PJ), 10 µM CEP 8983 (CEP) or the indicated concentration of CDDP, alone or combined, followed by the cytofluorometric assessment of apoptosis-related parameters. Panel (A) depicts representative dot plots. Numbers indicate the percentage of cells found in the corresponding gate. In panel (B), black and white columns report the percentage of dead (PI+) and dying [DiOC6(3)low PI−] cells, respectively (means ± SEM, n = 3). §p < 0.001 (Student’s t-test), compared with the sum of the cytotoxic effects caused by each agent alone. (C) Synergy between CDDP and PARP depletion. A549 cells were left untransfected (control) or were transfected with three distinct siRNAs targeting PARP1 or an unrelated (UNR) control siRNA. Twenty-four hours later, cells were treated for 48 h with the indicated CDDP concentration, followed by staining with PI and DiOC6(3). §p < 0.05 (Student’s t-test), compared with the sum of the cytotoxic effects caused by each agent alone. (D) Effects of CDDP and PARP inhibitors on additional human NSCLC cell lines. H226, H358 and H522 cells were maintained in control conditions or treated with the indicated concentrations of cisplatin (CDDP), alone or combined with increasing concentrations of CEP 8983 (CEP, 10 or 15 µM, 10 or 15 µM, and 5 or 10 µM in H522, H226 and H358 cells, respectively) or PJ34 hydrochloride hydrate (PJ, 40 or 60 µM) for 48 h, followed by the cytofluorometric assessment of apoptosis-related parameters as above. §p < 0.05 (Student’s t-test), compared with the sum of the cytotoxic effects caused by each agent alone.](/cms/asset/89eae081-d744-49ec-8f1b-62fa85f0ace4/kccy_a_10924034_f0003.gif)
To extend our observations to other in vitro models of NSCLC, we selected three additional human NSCLC cell lines that differ from each other (and from A549 cells, which harbor an intact p53 system)Citation42 with respect to their p53 status: (1) H358 cells lacking both copies of the TP53 gene; (2) H522 cells bearing a frame-shift mutation, resulting in the synthesis of a loss-of-function mutant form of p53 truncated at residue 191; and (3) H226 cells, whose p53 status remains controversial.Citation42-Citation44 Similar to A549 cells, H358, H522 and H226 cells were relatively insensitive to low doses of PJ, CEP and CDDP, each used alone, yet died in response to combinations of PARP inhibitors and CDDP (). These results indicate that the p53 status does not affect the cooperative cytotoxicity of CDDP and PARP inhibitors.
Based on the observation that co-treatment with PARP inhibitors and CDDP induced early Δψm dissipation (), we investigated the activation of the mitochondrial pathway of apoptosis in response to this cell death-inducing regimen. Immunofluorescence staining revealed that synergistic killing by PARP inhibitors and CDDP was preceded by the mitochondrial release of cytochrome c (which revealed a diffuse, cytosolic rather than punctuate, mitochondrial distribution) followed by the activation of caspase-3 (which became detectable by an antibody only recognizing the proteolytically mature form of the enzyme). Hence, the combination of CDDP and PJ or CEP induced mitochondrial cytochrome c release and caspase activation in a higher percentage of cells than did each of the treatments alone (). The addition of the pan-caspase inhibitor, carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone (Z-VAD-fmk) largely suppressed the loss of the viability induced by the combination treatment with CDDP plus PARP inhibitors (), indicating that cell death is indeed driven by caspases in this system.
Figure 4. Mechanisms of the synergistic action of cisplatin and PARP inhibitors. (A and B) Effects of cisplatin (CDDP), CEP 8983 (CEP) and PJ34 hydrochloride hydrate (PJ) on the subcellular location of cytochrome c and the activation of caspase-3. Upon treatment with CDDP (20 µM), CEP (10 µM) and/or PJ (30 µM) for 24 h, cells were fixed, permeabilized and stained by immunofluorescence to visualize cytochrome c (green fluorescence) and activated caspase 3a (C3a, red fluorescence). Representative microphotographs are shown in (A), and quantitative results (means ± SD of triplicate determinations) are reported in (B). (C) Effect of caspase inhibition on the cytotoxic effects of CDDP and PARP inhibitors. The experiment was performed by combining CDDP (20 µM), CEP (10 µM), PJ (30 µM) and/or Z-VAD-fmk (Z-VAD, 50 µM), as indicated, for 48 h, followed by staining with PI and DiOC6(3) and cytofluorometric determination of the percentage of dead (PI+) and dying [DiOC6(3)low PI−] cells. *p < 0.05 (Student’s t-test), compared with Z-VAD untreated cells. §p < 0.05 (Student’s t-test), compared with the sum of the cytotoxic effects caused by each agent alone.
![Figure 4. Mechanisms of the synergistic action of cisplatin and PARP inhibitors. (A and B) Effects of cisplatin (CDDP), CEP 8983 (CEP) and PJ34 hydrochloride hydrate (PJ) on the subcellular location of cytochrome c and the activation of caspase-3. Upon treatment with CDDP (20 µM), CEP (10 µM) and/or PJ (30 µM) for 24 h, cells were fixed, permeabilized and stained by immunofluorescence to visualize cytochrome c (green fluorescence) and activated caspase 3a (C3a, red fluorescence). Representative microphotographs are shown in (A), and quantitative results (means ± SD of triplicate determinations) are reported in (B). (C) Effect of caspase inhibition on the cytotoxic effects of CDDP and PARP inhibitors. The experiment was performed by combining CDDP (20 µM), CEP (10 µM), PJ (30 µM) and/or Z-VAD-fmk (Z-VAD, 50 µM), as indicated, for 48 h, followed by staining with PI and DiOC6(3) and cytofluorometric determination of the percentage of dead (PI+) and dying [DiOC6(3)low PI−] cells. *p < 0.05 (Student’s t-test), compared with Z-VAD untreated cells. §p < 0.05 (Student’s t-test), compared with the sum of the cytotoxic effects caused by each agent alone.](/cms/asset/5beed6c2-cee1-452b-afef-dd94897242a8/kccy_a_10924034_f0004.gif)
In conclusion, it appears that the synergistic action of CDDP and PARP inhibitors involves the p53-independent activation of the intrinsic pathway of apoptosis.
Concluding remarks
Most NSCLCs become resistant to the antineoplastic effects of CDDP in the course of therapy.Citation11,Citation12 The fact that CDDP resistance is often multifactorial, stemming from the simultaneous activation of non-redundant signaling pathways,Citation11 complicates the implementation of clinically useful strategies for increasing the antineoplastic potential of CDDP. In this context, we have recently demonstrated that NSCLC cells often, but not always, become resistant to CDDP along with the hyperactivation of PARP, rendering them sensitive to PARP inhibitors employed as standalone interventions in vitro and in vivo.Citation22
Here, we demonstrate that PARP inhibitors and CDDP synergize in the killing of NSCLC cells independently of their p53 status. This is an important notion, because the p53 pathway is often perturbed in NSCLC. PARP inhibitors and CDDP synergize at multiple levels, in particular in inducing DNA damage, in triggering mitochondrial membrane permeabilization with cytochrome c release and Δψm dissipation, in causal lethal caspase activation, post-apoptotic plasma membrane permeabilization and loss of the clonogenic potential. Our findings indicate that PARP inhibitors may constitute a valuable tool to sensitize NSCLC cells to the antineoplastic effects of CDDP.
Materials and Methods
Chemicals and cell cultures
Unless otherwise indicated, chemicals were purchased from Sigma-Aldrich, cell culture media and supplements from Gibco-Life Technologies and plasticware from Corning. CEP 8983 was obtained from Cephalon. Human NSCLC A549 cells were cultured in Glutamax®-containing DMEM/F12 medium supplemented with 10% fetal calf serum (FCS), 10 mM HEPES buffer, 100 units/mL penicillin G sodium and 100 µg/mL streptomycin sulfate. Human NSCLC H226, H358 and H522 cells were grown in RPMI1640 medium, supplemented as above. Cells were grown at 37°C in humidified incubator generating a 5% CO2 atmosphere. Cells were plated into appropriate supports and allowed to adapt and recover normal growth rates for at least 24 h before experimental procedures.
RNA interference
PARP-1 depletion was performed with previously validated siRNAs (PARP1.a, sense 5′-CAAACUGGAACAGAUGCCGdTdT-3′; PARP1.b, sense 5′-GCCUCCGCUCCUGAACAA UdTdT-3′; PARP1.c, sense 5′-GAUAGAGCGUGAAGGCGAAdTdT-3′).Citation22 Transfection with one control siRNA (UNR, sense 5′-GCCGGUAUGCCGGUUAAGUdTdT-3′) provided negative control conditions. A549 cells in 12-well plates were transfected with siRNAs at 30–40% confluence by means of the HiPerFect® transfection reagent (Qiagen), as previously describedCitation45 and used for experiments no earlier than 24 h after transfection.
Cytofluorometry
For the simultaneous assessment of Δψm and plasma membrane integrity, cells were collected, washed and co-stained with 40 nM 3,3′-dihexyloxacarbocyanine iodide [DiOC6(3), a Δψm-sensitive dye] and 1 µg/mL propidium iodide (PI, which only accumulates into dead cells) (Molecular Probes-Life Technologies), following established protocols.Citation37 Cytofluorometric acquisitions were performed on a FACSCalibur or FACScan cytofluorometer (BD Biosciences), and first-line data analyses were performed by using the proprietary CellQuest™ software (BD Biosciences) upon gating on events exhibiting normal light scatter (FSC and SSC) parameters.
Colorimetric assays
In vitro assessments of the interaction between PARP inhibitors and CDDP were performed by means of a colorimetric assay based on the reduction of the colorless tetrazolium salt 4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate (WST-1, from Roche) to formazan (which exhibit an absorbance peak around 450 nm), following conventional procedures.Citation18,Citation36 Absorbance at 450 nm was measured on a FLUOstar OPTIMA microplate reader (BMG Labtech) and—following background subtraction—WST-1 conversion data were normalized to the readings of DMSO-treated cells included in the same test plate.
Clonogenic survival assays
To evaluate clonogenic survival, cells were seeded at different concentrations (from 0.2 to 4 × 103 per well) in triplicate well of 6-well plates and treated for 48 h with CDDP and PARP inhibitors. Cells were cultured in drug-free medium for up to 10 d under normal conditions. Colonies were washed 1–2 times in 0.9% (w/v) NaCl in PBS then stained with an aqueous solution containing 0.25% (w/v) crystal violet, 70% (v/v) methanol and 3% (v/v) formaldehyde (Carlo Erba Reagents) for 20 min at room temperature.Citation45 Finally, stained colonies were washed again 2–3 times in water and counted.Citation40 Only colonies made of ≥ 30 cells were included in the quantification. For each treatment, the surviving fraction (SF) was estimated according to the formula SF = (number of colonies formed/ umber of cells seeded).
Immunofluorescence microscopy
Immunofluorescence microscopy assessments were performed as previously described.Citation45,Citation46 Briefly, cells were fixed in 4% (w/v) paraformaldehyde in PBS, permeabilized with 0.1% SDS and immunostained with antibodies specific for γH2AX (JBW301) (Millipore-Chemicon International), cleaved caspase-3 (Asp175) (Cell Signaling Technology Inc.) or cytochrome c (6H2.B4) (BD Biosciences). Slides were then incubated with the appropriate Alexa Fluor® conjugates (Molecular Probes-Invitrogen) in the presence of 2 µM Hoechst 33342, which was used for nuclear counterstaining. Images were captured using a DMIRE2 microscope (Leica Microsystems) equipped with a Cool Snap Ez camera (Roper Scientific, Princeton Instruments).
Common statistical procedures
Unless otherwise specified, experiments were performed in triplicate parallel instances and repeated at least twice, yielding comparable results. Data, which are generally presented as means ± SEM or SD, were analyzed with Microsoft Excel (Microsoft Co.). The interaction between PARP inhibitors and CDDP was evaluated by computing combination indexes following Harbron’s method.Citation38 Statistical significance was determined by means of two-tailed unpaired Student’s t-tests. Unless otherwise indicated, p values < 0.05 were considered statistically significant.
| Abbreviations: | ||
| CDDP | = | cisplatin |
| CEP | = | CEP 8983 |
| DiOC6(3) | = | 3′-dihexyloxacarbocyanine iodide |
| Δψm | = | mitochondrial transmembrane potential |
| GAPDH | = | glyceraldehyde-3-phosphate dehydrogenase |
| NSCLC | = | non-small cell lung carcinoma |
| PARP | = | poly(ADP-ribose) polymerase |
| PI | = | propidium iodide |
| PJ | = | PJ34 hydrochloride hydrate |
| siRNA | = | small-interfering RNA |
Additional material
Download Zip (100.3 KB)Acknowledgments
The authors would like to thank Lorenzo Galluzzi (Université Paris Descartes, Paris, France) for assistance with manuscript preparation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Grant Supports
J.M. is supported by the French Ministry of Science and Action Lions “Vaincre le Cancer” (Luxembourg). G.K. is supported by the European Commission (ArtForce); Agence National de la Recherche (ANR); Ligue contre le Cancer (Equipe labelisée); Fondation pour la Recherche Médicale (FRM); Institut National du Cancer (INCa); LabEx Immuno-Oncologie; Fondation de France; Fondation Bettencourt-Schueller; AXA Chair for Longevity Research; Cancéropôle Ile-de-France and Paris Alliance of Cancer Research Institutes (PACRI).
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/24034
References
- Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8:729 - 40; http://dx.doi.org/10.1038/nrm2233; PMID: 17667954
- Carrassa L, Broggini M, Erba E, Damia G. Chk1, but not Chk2, is involved in the cellular response to DNA damaging agents: differential activity in cells expressing or not p53. Cell Cycle 2004; 3:1177 - 81; http://dx.doi.org/10.4161/cc.3.9.1080; PMID: 15326376
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012; 19:107 - 20; http://dx.doi.org/10.1038/cdd.2011.96; PMID: 21760595
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007; 87:99 - 163; http://dx.doi.org/10.1152/physrev.00013.2006; PMID: 17237344
- Nardella C, Clohessy JG, Alimonti A, Pandolfi PP. Pro-senescence therapy for cancer treatment. Nat Rev Cancer 2011; 11:503 - 11; http://dx.doi.org/10.1038/nrc3057; PMID: 21701512
- Mandic A, Hansson J, Linder S, Shoshan MC. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J Biol Chem 2003; 278:9100 - 6; http://dx.doi.org/10.1074/jbc.M210284200; PMID: 12509415
- Berndtsson M, Hägg M, Panaretakis T, Havelka AM, Shoshan MC, Linder S. Acute apoptosis by cisplatin requires induction of reactive oxygen species but is not associated with damage to nuclear DNA. Int J Cancer 2007; 120:175 - 80; http://dx.doi.org/10.1002/ijc.22132; PMID: 17044026
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 2010; 11:700 - 14; http://dx.doi.org/10.1038/nrm2970; PMID: 20823910
- Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer 2007; 7:573 - 84; http://dx.doi.org/10.1038/nrc2167; PMID: 17625587
- Winter C, Albers P. Testicular germ cell tumors: pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 2011; 7:43 - 53; http://dx.doi.org/10.1038/nrendo.2010.196; PMID: 21116298
- Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene 2012; 31:1869 - 83; http://dx.doi.org/10.1038/onc.2011.384; PMID: 21892204
- Köberle B, Tomicic MT, Usanova S, Kaina B. Cisplatin resistance: preclinical findings and clinical implications. Biochim Biophys Acta 2010; 1806:172 - 82; PMID: 20647037
- Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 2003; 22:7265 - 79; http://dx.doi.org/10.1038/sj.onc.1206933; PMID: 14576837
- Borst P, Rottenberg S, Jonkers J. How do real tumors become resistant to cisplatin?. Cell Cycle 2008; 7:1353 - 9; http://dx.doi.org/10.4161/cc.7.10.5930; PMID: 18418074
- Beers MH. Lung carcinoma. In: Porter RS, Jones TV, eds. The Merck manual of diagnosis and therapy. Rahway: Merck & Co., Inc., 2008:2992.
- Jemal A, Thun MJ, Ries LA, Howe HL, Weir HK, Center MM, et al. Annual report to the nation on the status of cancer, 1975-2005, featuring trends in lung cancer, tobacco use, and tobacco control. J Natl Cancer Inst 2008; 100:1672 - 94; http://dx.doi.org/10.1093/jnci/djn389; PMID: 19033571
- Panaretakis T. Cisplatin-induced apoptosis and development of resistance are transcriptionally distinct processes. Cell Cycle 2012; 11:3723; http://dx.doi.org/10.4161/cc.22114; PMID: 22983001
- Galluzzi L, Vitale I, Senovilla L, Olaussen KA, Pinna G, Eisenberg T, et al. Prognostic impact of vitamin B6 metabolism in lung cancer. Cell Rep 2012; 2:257 - 69; http://dx.doi.org/10.1016/j.celrep.2012.06.017; PMID: 22854025
- Galluzzi L, Vitale I, Senovilla L, Eisenberg T, Carmona-Gutierrez D, Vacchelli E, et al. Independent transcriptional reprogramming and apoptosis induction by cisplatin. Cell Cycle 2012; 11:3472 - 80; http://dx.doi.org/10.4161/cc.21789; PMID: 22918244
- Yao Z, Szabadkai G. Transcriptional profiling of apoptosis: cell death classification moves toward the systems era. Cell Cycle 2012; 11:3721 - 2; http://dx.doi.org/10.4161/cc.22116; PMID: 22982998
- Galluzzi L, Marsili S, Vitale I, Senovilla L, Michels J, Garcia P, et al. Vitamin B6 metabolism influences the intracellular accumulation of cisplatin. Cell Cycle 2013; 12:417 - 21; http://dx.doi.org/10.4161/cc.23275; PMID: 23287530
- Michels J, Vitale I, Galluzzi L, Adam J, Olaussen KA, Kepp O, et al. Cisplatin resistance associated with PARP hyperactivation. Cancer Res 2013; In press
- Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 2012; 13:411 - 24; http://dx.doi.org/10.1038/nrm3376; PMID: 22713970
- Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 2006; 7:517 - 28; http://dx.doi.org/10.1038/nrm1963; PMID: 16829982
- Jagtap P, Szabó C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov 2005; 4:421 - 40; http://dx.doi.org/10.1038/nrd1718; PMID: 15864271
- Villanueva T. Anticancer drugs: Expanding the horizons of PARP inhibitors. Nat Rev Drug Discov 2010; 9:919; http://dx.doi.org/10.1038/nrd3335; PMID: 21119729
- Annunziata CM, O’Shaughnessy J. Poly (ADP-ribose) polymerase as a novel therapeutic target in cancer. Clin Cancer Res 2010; 16:4517 - 26; http://dx.doi.org/10.1158/1078-0432.CCR-10-0526; PMID: 20823142
- Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer 2010; 10:293 - 301; http://dx.doi.org/10.1038/nrc2812; PMID: 20200537
- Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet 2010; 376:245 - 51; http://dx.doi.org/10.1016/S0140-6736(10)60893-8; PMID: 20609468
- Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 2010; 376:235 - 44; http://dx.doi.org/10.1016/S0140-6736(10)60892-6; PMID: 20609467
- McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 2006; 66:8109 - 15; http://dx.doi.org/10.1158/0008-5472.CAN-06-0140; PMID: 16912188
- Delaney CA, Wang LZ, Kyle S, White AW, Calvert AH, Curtin NJ, et al. Potentiation of temozolomide and topotecan growth inhibition and cytotoxicity by novel poly(adenosine diphosphoribose) polymerase inhibitors in a panel of human tumor cell lines. Clin Cancer Res 2000; 6:2860 - 7; PMID: 10914735
- Clark CC, Weitzel JN, O’Connor TR. Enhancement of synthetic lethality via combinations of ABT-888, a PARP inhibitor, and carboplatin in vitro and in vivo using BRCA1 and BRCA2 isogenic models. Mol Cancer Ther 2012; 11:1948 - 58; http://dx.doi.org/10.1158/1535-7163.MCT-11-0597; PMID: 22778154
- Postel-Vinay S, Vanhecke E, Olaussen KA, Lord CJ, Ashworth A, Soria JC. The potential of exploiting DNA-repair defects for optimizing lung cancer treatment. Nat Rev Clin Oncol 2012; 9:144 - 55; http://dx.doi.org/10.1038/nrclinonc.2012.3; PMID: 22330686
- Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med 2012; 366:1382 - 92; http://dx.doi.org/10.1056/NEJMoa1105535; PMID: 22452356
- Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 2009; 16:1093 - 107; http://dx.doi.org/10.1038/cdd.2009.44; PMID: 19373242
- Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G. Cell death assays for drug discovery. Nat Rev Drug Discov 2011; 10:221 - 37; http://dx.doi.org/10.1038/nrd3373; PMID: 21358741
- Harbron C. A flexible unified approach to the analysis of pre-clinical combination studies. Stat Med 2010; 29:1746 - 56; http://dx.doi.org/10.1002/sim.3916; PMID: 20572122
- Banáth JP, Olive PL. Expression of phosphorylated histone H2AX as a surrogate of cell killing by drugs that create DNA double-strand breaks. Cancer Res 2003; 63:4347 - 50; PMID: 12907603
- Zhang P, Castedo M, Tao Y, Violot D, Métivier D, Deutsch E, et al. Caspase independence of radio-induced cell death. Oncogene 2006; 25:7758 - 70; http://dx.doi.org/10.1038/sj.onc.1209744; PMID: 16862186
- Galluzzi L, Zamzami N, de La Motte Rouge T, Lemaire C, Brenner C, Kroemer G. Methods for the assessment of mitochondrial membrane permeabilization in apoptosis. Apoptosis 2007; 12:803 - 13; http://dx.doi.org/10.1007/s10495-007-0720-1; PMID: 17294081
- Berglind H, Pawitan Y, Kato S, Ishioka C, Soussi T. Analysis of p53 mutation status in human cancer cell lines: a paradigm for cell line cross-contamination. Cancer Biol Ther 2008; 7:699 - 708; http://dx.doi.org/10.4161/cbt.7.5.5712; PMID: 18277095
- Hainaut P, Hernandez T, Robinson A, Rodriguez-Tome P, Flores T, Hollstein M, et al. IARC Database of p53 gene mutations in human tumors and cell lines: updated compilation, revised formats and new visualisation tools. Nucleic Acids Res 1998; 26:205 - 13; http://dx.doi.org/10.1093/nar/26.1.205; PMID: 9399837
- Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat 2007; 28:622 - 9; http://dx.doi.org/10.1002/humu.20495; PMID: 17311302
- Vitale I, Senovilla L, Jemaà M, Michaud M, Galluzzi L, Kepp O, et al. Multipolar mitosis of tetraploid cells: inhibition by p53 and dependency on Mos. EMBO J 2010; 29:1272 - 84; http://dx.doi.org/10.1038/emboj.2010.11; PMID: 20186124
- Mullen P. PARP cleavage as a means of assessing apoptosis. Methods Mol Med 2004; 88:171 - 81; PMID: 14634228