Abstract
Urothelial cell carcinoma (UCC) is the second most common genitourinary malignant disease in the USA, and tobacco smoking is the major known risk factor for UCC development. Exposure to carcinogens, such as those contained in tobacco smoke, is known to directly or indirectly damage DNA, causing mutations, chromosomal deletion events and epigenetic alterations in UCC. Molecular studies have shown that chromosome 9 alterations and P53, RAS, RB and PTEN mutations are among the most frequent events in UCC. Recent studies suggested that continuous tobacco carcinogen exposure drives and enhances the selection of epigenetically altered cells in UCC, predominantly in the invasive form of the disease. However, the sequence of molecular events that leads to UCC after exposure to tobacco smoke is not well understood.
To elucidate molecular events that lead to UCC oncogenesis and progression after tobacco exposure, we developed an in vitro cellular model for smoking-induced UCC. SV-40 immortalized normal HUC1 human bladder epithelial cells were continuously exposed to 0.1% cigarette smoke extract (CSE) until transformation occurred. Morphological alterations and increased cell proliferation of non-malignant urothelial cells were observed after 4 months (mo) of treatment with CSE. Anchorage-independent growth assessed by soft agar assay and increase in the migratory and invasive potential was observed in urothelial cells after 6 mo of CSE treatment. By performing a PCR mRNA expression array specific to the PI3K-AKT pathway, we found that 26 genes were upregulated and 22 genes were downregulated after 6 mo of CSE exposure of HUC1 cells. Among the altered genes, PTEN, FOXO1, MAPK1 and PDK1 were downregulated in the transformed cells, while AKT1, AKT2, HRAS, RAC1 were upregulated. Validation by RT-PCR and western blot analysis was then performed. Furthermore, genome-wide methylation analysis revealed MCAM, DCC and HIC1 are hypermethylated in CSE-treated urothelial cells when compared with non-CSE exposed cells. The methylation status of these genes was validated using quantitative methylation-specific PCR (QMSP), confirming an increase in methylation of CSE-treated urothelial cells compared to untreated controls. Therefore, our findings suggest that a tobacco signature could emerge from distinctive patterns of genetic and epigenetic alterations and can be identified using an in vitro cellular model for the development of smoking-induced cancer.
Introduction
Urothelial cell cancer (UCC) is the second most common urological malignancy in the United States, with an estimated 74,000 new cases and with 15,000 deaths in 2012.Citation1 One-third to half of all UCC are believed to be caused by smoking, although the mechanisms of action are less clearly understood.Citation2 Smokers have a three to five times higher risk of developing UCC than non-smokers.Citation3 The higher risk can be attributed to tobacco-related carcinogens that enter the body after inhalation, are absorbed into the bloodstream and eventually reach the bladder. The bladder epithelium is then exposed to these carcinogens for several hours before they are expelled. Several environmental agents such as cigarette smoke, arsenic and nicotine have been well studied and have been shown to affect gene expression through genetic and epigenetic changes.Citation4,Citation5 Recent evidence indicates that methylation and allelic deletions occur more frequently in tumors from smokers than non-smokers.Citation6 Inactivation of tumor suppressor genes (TSGs) and activation of oncogenes that show altered methylation patterns are present in 17% of human UCC.Citation7 PTEN mutations are present in approximately 35% of invasive UCC and are reported to regulate the PI3K pathway, which, in turn, regulates cell proliferation, cell survival and downstream AKT.Citation8
The pathogenic mechanisms of tobacco smoke contributing to the development of UCC have long been studied, though they remain incompletely characterized. Tobacco smoke is known to generate genotoxic reactive oxygen species that are capable of inducing DNA damage,Citation9,Citation10 leading to genetic and epigenetic alterations.Citation11,Citation12 In addition, the rapidly emerging literature indicates that silencing of TSGs via promoter methylation occurs in bladder cancer.Citation13-Citation16 However, the precise mechanisms underlying the induction of TSG methylation and the factors that influence tumor-specific methylation profiles are incompletely understood in UCC as well as other cancer types. Exposure to carcinogens has been associated with TSG methylation silencing. Initial studies demonstrated that there is a dose response for methylation silencing of CDKN2A by tobacco smoke in lung cancer.Citation17,Citation18 Moreover, methylation of TSGs CDKN2A and APC were also significantly associated with exposure to tobacco smoke in lung and UCC.Citation15,Citation19 Marsit and colleagues recently measured promoter hypermethylation of 16 different genes in UCC and found a correlation between overall methylation and age, gender and smoking history.Citation20
Taken together, the above facts strongly suggest that tobacco exposure may act to induce molecular alterations, including methylation silencing of TSGs. However, it remains unclear if this is a direct or indirect selection for TSG inactivation across phenotypically important pathways, and whether the process is stochastic and less phenotypically driven, or whether a dose and time response exists between exposure and molecular alterations. Although there is unequivocal association between cigarette smoking and UCC, the molecular mechanisms by which tobacco smoke initiates and promotes urothelium carcinogenesis have not been fully delineated. In particular, epigenetic events associated with initiation of tobacco-induced UCCs have not yet been comprehensively elucidated. The aim of this study was to characterize epigenomic alterations in cultured human immortalized urothelial cells treated by cigarette smoke and to develop an in vitro model to investigate the biological and molecular alterations involved in the development of UCC. Furthermore, key signaling pathways that are related to UCC, such as the PI3K-AKT/mTOR, were also assessed.
Results
There are three different methods for exposure of cells to cigarette smoke (CS). Cells can be exposed to CS total particulate matter (TPM), aqueous CSE containing components of the particulate and vapor phase or whole smoke. TPM is prepared by trapping the particles from the mainstream smoke on a Cambridge filter pad, a glass-fiber filter that retains 99% of particles larger than 0.1 μm, and extracting the particles with dimethyl sulphoxide (DMSO).Citation21 CSE is prepared by bubbling smoke from one cigarette into culture medium.Citation22 Cells cultured on Transwells at the air-liquid interface are exposed to freshly generated whole smoke diluted with air in a Perspex chamber.Citation23 To molecularly understand how exposure to CSE initiates and promotes the development of UCC, we sought to develop an in vitro model in which CSE exposure acts as an initiator and promoter of malignant transformation of a SV-40 immortalized normal uroepithelial cell line (SV40-HUC-1).
Cigarette smoke extract induces transformation of simian virus 40 (SV40) immortalized normal human urinary tract epithelial cells (SV40-HUC-1)
Morphological changes due to CSE exposure
The SV40-HUC-1 cell line is an appropriate model for in vitro studies as it is non-tumorigenic and remained non-tumorigenic as determined by the inability to form tumors in athymic nude mice.Citation24,Citation25 In the present study, we examined the transformation and growth characteristics of the SV-40-HUC-1 cells after treatment with CSE. SV40-HUC-1 urothelial cell line was chronically exposed to 0.1% of CSE for 6 months (mo). After each month of CSE treatment, cells were stock for future studies. The concentration of 0.1% was optimal in long-term exposure of these non-tumorgenic cell lines, because it exhibited greater than 80% viability and promoted enhanced proliferation after 24 h (data not shown). To understand and identify the early changes in cellular transformation, we extracted protein, RNA and DNA every month of exposure and monitored for any morphological changes that would indicate transformation. depicts the morphological changes associated with bi-monthly CSE exposure. After 2 mo of exposure, CSE-treated cells appeared similar to the untreated (UT) normal cells (flat morphology); however, after 4 and 6 mo of exposure, they showed significant changes in morphology, including rounding and clumping of the cells.
Figure 1. Biotransformation of SV40-HUC-1 cells with long-term CSE exposure. (A) Morphological changes: Morphologic differences were monitored over the entire treatment schedule, using a light microscope (20×). Cells were plated on 6-cm dishes at a density of 200,000 per plate. After 6 mo (M) of CSE treatment, HUC1 became more rounded and had a tendency to pile on to one another. The distribution of nuclear and cytoplasmic compartments was also altered. (B) MTT was performed for each month of treatment in order to determine if changes in cell proliferation occurred because of CSE treatment. This figure depicts 2, 4 and 6 mo of CSE-treated and mock-treated human immortalized normal uro-epithelial cells (HUC). The average for each sample was performed to get a standard deviation and mean value. Student’s t-test was also performed to determine the p value. All data represent +/− SD, p value < 0.05. (C) BRD-U incorporation assay shows that cells treated for 4 mo begin to show higher cell proliferation vs. controls cells. All data represent +/− SD, p value < 0.05. (D) Invasion assay: graphical representation for the invasion assays performed at months 2, 4 and 6 (left panel). All values were significant using Student’s t-test (p < 0.05). Invaded cells were counted using a light microscope at 10 different fields and a 20× objective. Pictures were taken at random. UT, untreated; M, month. Right panel shows invaded cells. (E) Soft agar assay: Anchorage-independent growth was evaluated using a soft agar assay. Cell were embedded in agar in triplicate and allowed to grow for 2 wk. Cell colonies were counted using a light microscope in four different fields and averaged. Cell colonies were then stained with 0.05% ethidium bromide for 24 h. UV transilluminator was used to determine the number of colonies per well greater than 50 μM in size. Student’s t-test was used to determine p values. Pictures were taken at random using a digital camera attached to a high resolution light microscope. Left panel shows the bar graphs of colonies of 6-mo CSE-treated and untreated SV40-HUC-1 cells. Right panel shows a photograph of the representative area (magnification, ×100). M, months; UT, untreated.
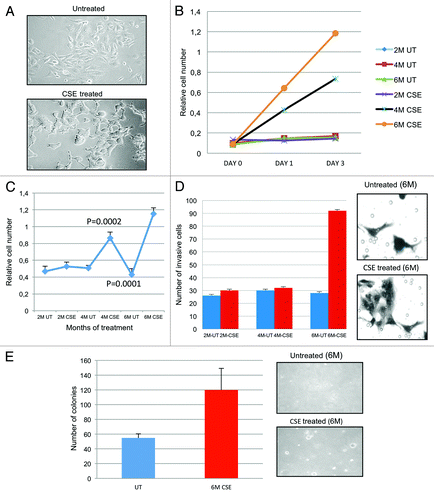
CSE increases SV40-HUC-1 cell proliferation in vitro
Cancer cells often display an uncontrolled proliferation and/or diminished apoptosis. Every month cells were tested for cell proliferation differences via MTT assay. After 4 mo of CSE exposure, we found that the growth of the SV40-HUC-1 cells was increased in a time-dependent manner. A 4-fold increase in cell doubling time was observed in cells treated for 4 mo, while cell treated for 6 mo indicated a 5–6-fold increase (). The average for each sample was performed to get a standard deviation and mean value. Student’s t-test was also performed to determine the p value. The cell’s increase in proliferation at months 4 and 6 were both statistically significant (p < 0.05).
To determine if the increase in cell proliferation after CSE exposure was a result of increases in DNA synthesis, Brdu incorporation was assayed by colorimetric changes. We examine untreated and treated cell lines at various time points (2 mo, 4 mo and 6 mo). Brdu assay was performed 24 h after seeding the untreated and CSE-treated cells. As indicated in , 4 mo and 6 mo CSE-treated cells were two to three times more proliferative, respectively, than untreated and 2 mo CSE-treated cell lines.
Invasiveness and anchorage-independent growth of CSE-treated SV40-HUC-1 cells
The aggressiveness of a malignant cell is determined by its potential to invade the extracellular matrix and metastasize. Therefore, we determined the in vitro invasion potential of CSE-treated cell lines by Matrigel chamber assays. SV40-HUC-1 cells exposed for 2, 4 and 6 mo were compared with untreated cells. No differences in the number of invading cells were observed for untreated and 2 and 4 mo exposures; however, 6 mo of treatment showed a significant (p value 0.004) increase (3-fold) in the cells’ potential to invade the Matrigel ().
Oncogenic transformation confers the ability of a cell to grow in an anchorage-independent manner. Therefore, we performed experiments to observe the anchorage-independent growth of CSE-treated cells. Briefly, SV40-HUC-1 untreated and 6 mo CSE-treated cell were plated in soft agar for 2 wk. Colony formation was present in both cell lines; however, cells treated for 6 mo with CSE formed more colonies that were also larger in size. SV40-HUC-1 cells are immortalized cells and have been shown to moderately grow in soft agar;Citation24 however, the 2-fold increase in colony formation after 6 mo of CSE treatment indicated that cells acquired more anchorage-independent growth properties associated with malignant transformation (). We chose to only test the 6-mo-exposed cells, since they were shown to be the only ones that acquired enhanced invasive properties. The soft agar assays indicated that cells exposed to CSE acquired anchorage independence after 6 mo of treatment.
CSE-treated SV40-HUC-1 transformed cells show similar molecular alterations seen in human UCC
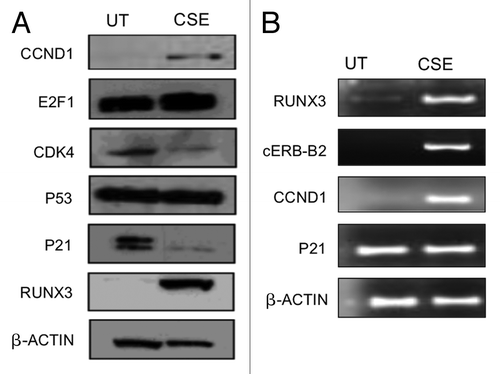
In most types of cancers, including UCC, the transformation mechanism of a normal cell into a transformed cell involves a multistep process, including the alteration of different signaling pathways and genes (oncogenes, tumor-suppressor genes, cell cycle genes and DNA-repair genes). To understand the related molecular alterations associated with CSE, we analyzed a panel of genes that are reported to be altered in smoking-related cancers, like lung, head and neck, esophageal and bladder. This panel included known TSGs, such as PTEN, p53, p21, and reported oncogenes, such as RUNX3, CCND1, E2F1 and cERB-b2. PTEN and p53 are commonly altered or mutated genes in UCC.Citation26,Citation27 We performed western blot and/or mRNA analysis to identify molecular alterations in CSE-treated urothelial cells (). Six-months-treated cell lines showed enhanced Cyclin D1 and RUNX3 protein expression and lower p21 expression compared with CSE untreated control. Interestingly, RUNX3 is reported to be methylated and silenced in urothelial cell carcinoma;Citation28 however, we observed RUNX3 protein and mRNA overexpression after CSE treatment. Translational and transcriptional levels of P53 remained unchanged,
Figure 2. CSE induces molecular changes in SV40-HUC-1 cells that are commonly found in UCC. (A) Western blot analysis of a panel of anti-human cyclin D1, E2F1, PTEN, Cdk4, p53 and p21 antibodies used to determine the expression levels of these genes in untreated (UT) and 6-mo CSE-treated SV40-HUC-1 cells. As expected, CCND1 and E2F1 were upregulated after CSE treatment, while CDK4 and p21 were downregulated. (B) RT-PCR analysis for RUNX3, Cerb-b2, Cyclin D1, p21 expression in untreated and 6-mo CSE-treated SV40-HUC-1 cells. The transcript expression level of all four genes is consistent with their translational level ().
PI3K -AKT/mTOR Pathway alterations in CSE treated SV40-HUC-1 cells analyzed by PCR array for PI3K-AKT pathway
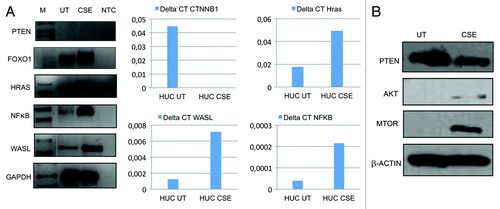
Since we observed numerous phenotypic changes after 6 mo CSE treatment, to determine the overall changes in PI3K-AKT pathway, a known deregulated pathway in UCC,Citation29 we examined the mRNA expression profile using a PCR array for 84 genes involved in the PI3K-AKT pathway in CSE-treated and untreated SV40-HUC-1 cells. By array analysis, we found that PTEN, FOXO1, MAPK1 and PDK1 were downregulated (≥ 5-fold) in the transformed cells, while WASL, NFκB, AKT 1, AKT 2, HRAS and RAC1 were upregulated (≥ 5-fold)().
Table 2. Gene expression alterations of AKT pathway associated with CSE exposure
We confirmed the array findings using semi-quantitative and/or quantitative RT-PCR and validated HRAS, NFκB and WASL to have increased mRNA levels in treated vs. untreated cell lines (); western blot analysis confirmed enhanced protein expression for AKT and mTOR in the CSE-treated cell line (). We found discordant values for FOXO1, which was found to be upregulated in the qRT-PCR while downregulated in the microarray.
Figure 3. Validation of PCR array for PI3K-AKT pathway. (A) Left panel: Expression of representative genes analyzed by semi-quantitaive RT-PCR on the SV40-HUC-1 cells untreated (UT) and exposed for 6 mo to CSE (CSE). M, molecular marker; NTC, non template control (water). GAPDH was used as an internal control. PTEN, HRAS, NFκB and WASL expression are consistent with PCR-array data while FOXO1 expression is not consistent with PCR-array data (center and right panels). Downregulation of CTNNB1 and overexpression of Hras, WASL and NFκB was observed after 6 mo CSE treatment, analyzed by real-time quantitative RT-PCR (Q-RT-PCR). Primer sequences for Q-RT-PCR are same as semi-quantitative RT-PCR and were done using SYBR green. Relative fold was calculated by the expression of target mRNA to 18S rRNA (an internal control). Experiments were performed in triplicate. Differences above a 3-fold difference were considered as significant. This result validates the finding in the AKT pathway array. (B) Western blot analysis of three AKT pathway genes: consistent with AKT pathway PCR array data, PTEN is downregulated after CSE treatment while AKT and mTOR are upregulated.
Immunohistochemistry for FOXO1
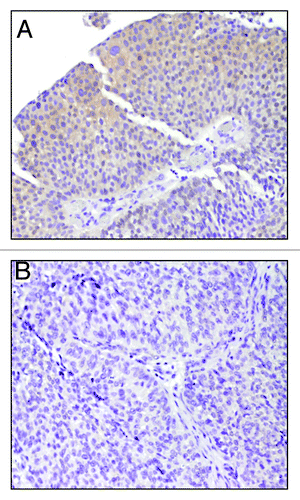
Immunohistochemistry results are summarized in . Six patients were smokers, while seven were non-smokers; 10 patients underwent trans-urethral resection while three had cystoprostatectomy. All FOXO1-positive cases showed a cytoplasmic pattern of staining; in one case nuclear staining was also observed. Of six patients who were smokers, five showed positive FOXO1 staining [5/6(83%)] while one out of seven [1/7(14%)] non-smoker patients showed positive staining for FOXO1 (). So, our RT-PCR data using cell line and pilot immunohistochemistry data using primary tumors from smokers and non-smokers indicate that FOXO1 expression is associated with smoking.
Table 3. FOXO1 immunohistochemistry
Figure 4. Immunohistochemistry for FOXO1. Immunohistochemistry for FOXO1 using FOXO1-specific monoclonal antibody. Representative examples of UCC samples are shown. (A) Positive staining for FOXO1 shows a cytoplasmic pattern of FOXO1 staining (magnification 10×) (B) Negative staining for FOXO1 (magnification 20×).
Differential methylation patterns were observed after CSE exposure
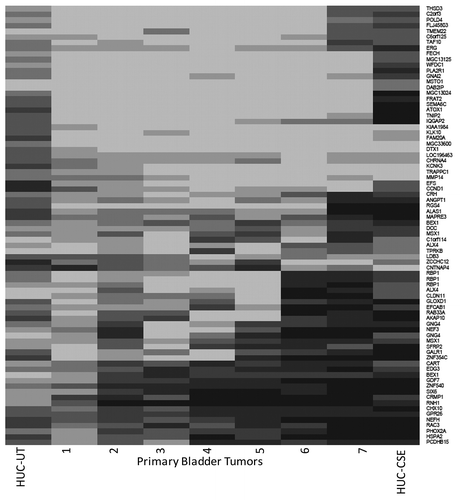
Epigenetic alterations such as DNA hypermethylation and altered histone acetylation have been observed in bladder cancer.Citation7 We hypothesized that chronic treatment of normal urothelial cells could be used as a tool to identify the epigenetic changes associated with smoking-induced transformation of urothelial cells. We performed methylation profiling using the Illumina Infinium HumanMethylation27 BeadChip Kit. Untreated and 6-mo CSE-treated (0.1% CSE) samples were compared with each other. In addition, we analyzed seven primary UCC to achieve an overall methylation profile of UCC. For the analysis, we first removed Illumina methylation probes having ≤ 3 CpG dinucleotides. From the remaining probes, using the normal urothelial cells, we identified all those for which |beta_untreated-beta_treated| resulted in a net value of > 0.25 (468 probes). These initial analyses identified a subset of genes (n = 78) that agreed well between the cell line and primary tumor samples. An overview of potentially differential methylated genes in CSE-treated SV40-HUC-1 cells is shown in . To validate our methylation array data, we performed quantitative Methylation-Specific PCR (QMSP) on 6-mo CSE-treated and untreated SV40-HUC1 cell lines DNA for DCC, HIC1 and MCAM, and found higher methylation values for all three genes (100%) in CSE-treated cells compared with untreated cells (data not shown).
Figure 5. Methylation array. Whole-genome methylation (Illumina Infinium assay) pattern in SV40-HUC-1 cell lines treated with CSE for 6 mo and untreated controls and primary UCC samples. Genes listed on the right are showing more methylation in CSE-treated cells in comparison with CSE-untreated cells on the left. Dark colors indicate methylation.
Discussion
Since cigarette condensate is a complex agent, composed of over 400 different carcinogenic compounds, it remains to be determined which chemical plays the major role in the development of UCC. A number of studies have been performed to examine the effects of individual components of tobacco smoke, such as nicotine or 4-[methylnitrosamino]-1-[3-pyridyl]-1-butanone (NNK) in cultured cells.Citation30-Citation36 Although informative, these studies are inconclusive, as the effects of single tobacco components are unlikely to represent those mediated by the complex mixture of organic as well as inorganic carcinogens detected in tobacco smoke. So our model developed in this study is more representative of the global exposure seen when an individual smokes.
We developed an in vitro model for smoking-induced transformation in normal urothelial cells that can be used to identify molecular events that lead to urothelial carcinogenesis and progression after tobacco exposure. It has been suggested that chronic sub-cytotoxic doses induce a rapid proliferation response or “wound response” in the urothelium, which may be more representative of the exposure of human bladder cells to various toxins.Citation37 Here we show that chronic CSE exposure (0.1%) can promote cell proliferation of normal bladder epithelial cells within 4 mo. However, we did not see any significant changes in invasiveness and anchorage-independent growth at this time point. After 6 mo of CSE treatment, cell proliferation was more robust, and the cells acquired additional transformative properties, like anchorage independence and invasion potential. These findings support the data suggesting a strong link between smoking and the risk of developing UCCCitation38 and the stepwise acquired transformative properties.
We identified molecular changes in 6-mo CSE-treated SV40-HUC-1 cells that are commonly seen in UCC tissue, including increased expression of cERB-b2, Cyclin D1, AKT, m-TOR and RUNX3. It has been reported that overexpression of AKT plays a key role in the tumorigenesis and angiogenesis of urothelial cells.Citation39 PTEN is one of the most frequently mutated genes in human cancer.Citation40,Citation41 PTEN tumor-suppressive activity works to regulate cell growth, proliferationCitation42,Citation43 and genomic stability.Citation44 We found a slight decrease in the expression of PTEN in our CSE-treated cell line. Whether this decrease is due to loss of heterozygosity (LOH) will be further explored. RUNX3 is a Runt domain transcription factor 3 localized to 1p36, which is a region commonly deleted in many tumors, including breast and lung cancer.Citation45 RUNX3 was reported to be hypermethylated in early-stage UCC and is associated with smoking.Citation28 It was also reported that RUNX3 promoter methylation is inversely correlated with expression.Citation46 We found an increase in RUNX3 expression after CSE treatment; therefore, it would be of great interest to find how RUNX3 expression is deregulated after CSE treatment in this cell line.
Translational and transcriptional levels of p53 were unchanged, suggesting that a longer period of exposure is needed to alter the p53 pathway. p53 TSG is located on chromosome 17p13.1 and encodes for p53 protein, the primary player of the p53 pathway, which has been classically associated with the genesis of invasive urothelial carcinoma.Citation47 Loss of heterozygosity on chromosome 17p appears to occur during the later stages of UCC and is usually associated with a more aggressive phenotype;Citation48 moreover, in a large cohort, p53 expression was found altered in 50% of patients with advanced bladder cancer.Citation49
It has been reported that overexpression of AKT plays a key role in the tumorgenesis and angiogenesis of uroepithelial cells.Citation39 We also showed that long-term exposure to CSE enhanced AKT signaling, suggesting that AKT may play a key role in smoking-induced UCC. It remains to be seen whether these transformed cells can grow faster in vivo. An in vivo model would allow us to explore the effects of inhibition of mTOR signaling after induction of AKT-mTOR signaling by CSE, a pathway that is currently being explored in clinical trials.
Aberrant activation of phosphatidylinositol 3-kinase (PI3K) signal transduction pathway has been identified in a wide range of cancers.Citation50,Citation51 Several pathway components, including AKT, PI3K and mTOR, represent potential therapeutic targets, and many small-molecule inhibitors are under development or early clinical trials. We found significant differences in the pattern of this pathway between CSE-treated and untreated SV40-HUC-1 cells. Although recent molecular studies have implicated some genes of PI3K pathway altered in UCC of all grades and stages, it is not yet well demonstrated that all the genes related to this pathway have any association with smoking. Our in vitro study data from the PCR array need to be validated in primary tumors of different phenotypes, including smokers and non-smokers, and eventually these findings will have implications for personalized therapy development and prevention strategies. Several genes involved in PI3K pathway showed over 250-fold increased expression in CSE-treated cells. These include PRKCA, NFκB, PTK2, WASL, TIRAP and ILK. PRKCA is one member of a family of protein kinase C (PKC) that comprises at least 11 serine-threonine kinases. Increased expression of PRKCA has been reported in proliferative areas of UCC when compared with normal epithelium.Citation52 Integrin-linked kinase (ILK) is an ubiquitously expressed protein serine-threonine kinase that was initially identified by Hannigan et al.Citation53 and is a component of the PI3K pathway located upstream of protein kinase B and has been shown to play a fundamental role in the regulation of cell survival, proliferation an and migration.Citation54 ILK overexpression has been shown to be involved in a variety of epithelial cancer types, including prostate,Citation55 colonCitation56 and non-small cell lung cancer (NSCLC).Citation57 Also, recent studies have investigated the role of ILK in mediating epithelial-mesenchymal transition (EMT) in UCC and lung squamous cell carcinoma.Citation58,Citation59 More recently, it has been demonstrated that overexpression of ILK protein in primary NSCLC samples correlated with the TNM stage and lymph node metastasis. Moreover, ectopic overexpression of ILK in lung cancer cells promoted cell migration and invasion in vitro.Citation60 It will be interesting to further explore the relationship of ILK expression with smoking. WASL, also known as N-WASP is a member of the WASP family of proteins that are key regulators of reorganization of the actin cytoskeleton in several signaling pathways.Citation61,Citation62 WASL was previously shown to be an essential component of invadopodia proteolytic membrane protrusions formed by highly invasive cancer cells, proposed to have roles in cancer invasion and metastasis.Citation63 Recently, it has been reported that WASL-mediated invadopodium formation is essential in breast cancer invasion, intravasation and lung metastasis.Citation64 However, its relationship with CSE exposure needs to be explored.
Among genes found to be downregulated by PCR-array analysis in CSE-treated cells are PTEN, MAPK1, PDK1 and FOXO1. Forkhead box protein O1 (FOXO1) is a member of the subfamily of mammalian FOXO transcription factors; it is known to be a direct phosphorylation target of the protein kinase AKT.Citation65 The activation of AKT with subsequent functional loss of FOXO family of transcription factors has been observed to promote tumorigenesis.Citation11 In contrast with previous reports, Kim and coworkers showed that FOXO1 expression in cancer tissues was higher than in normal mucosa.Citation14 In our study, the RT-PCR validation experiment showed discordant results for FOXO1 compared with the PCR array. This might be due to an amplification bias, mispriming or the hybridization inefficiency on the array. To further confirm our RT-PCR data at protein level, we analyzed 13 primary UCC tissues from smokers and non-smokers by immunohistochemistry. Interestingly, immunohistochemistry for FOXO1 performed on UCC showed high level and frequency of expression in the smoker group (5/6) while one out of seven non-smokers showed FOXO1 expression. In our view, this might reflect the fact that FOXO1 in smokers is subject to an enhanced phosphorylation by AKT with consequent cytoplasmic translocation and accumulation prior to degradation. However, our sample size is too small to make any conclusion; studies with a larger cohort of patients might clarify this difference, and further elucidation of the mechanistic relationship between AKT and FOXO1 needs to be performed.
We performed methylation profiling using the Illumina Infinium HumanMethylation27 BeadChip Kit in both the primary UCC tissue as well as CSE-treated cell line and also validated the genes DCC, HIC1 and MCAM with quantitative methylation-specific PCR (QMSP) on 6 mo CSE-treated and untreated SV40-HUC1 cell lines DNA. DCC has been demonstrated to be subject to specific epigenetic silencing in colon cancer cell lines.Citation66 Moreover, it has recently been shown that hypermethylation of HIC1 promoter in chronic pancreatitis may contribute to the aberrant expression of HIC1/SIRT1 pathway, leading to pancreatic carcinogenesis.Citation67 Although further validation and functional studies are needed, our results open a new avenue for understanding the epigenetic alteration due to smoking at the basis of the development of UCC.
A limitation of our study is the immortalization process that may lead to substantial molecular alterations; however, we compared our molecular analysis in the same cell lines treated with or without CSE. Although SV40-HUC-1 cell are non-tumorgenic, they express oncogenic proteins that affect major TSGs, including p53 and Rb.Citation68 These alterations may facilitate the development of cells carrying chromosomal and karyotypic abnormities, which could promote the transformed phenotype after cigarette exposure.Citation37 Moreover, we did not validate all of the deregulated genes in two groups of samples by complementary approach and did not functionally characterize any of the molecules. Future functional studies of specific molecules (like FOXO1) need to be performed to understand the in depth relationship of smoking and alterations of these genes. Another limitation of our study is using only one cell line, which may generate biased information. However, our overall purpose is to identify related molecules that are altered by smoking. We will take lead from in vitro model data reported in this paper and ultimately will test all the relevant molecules in human samples (smokers and non-smokers). Here, as a pilot study we found that frequency of FOXO1 expression is higher in smokers than non-smokers. In future studies, we will validate all our findings in human primary UCC tissues obtained from smokers and non-smokers.
In summary, we established an in vitro model to study the stepwise initiation and progression of UCC by cigarette smoke (CS). Our findings confirmed some of the previous results and identified several novel genes that are altered due to CS exposure, and the model we developed will serve as the foundation for future studies on the genetic and epigenetic basis for smoke-induced UCC. Accumulating evidence strongly indicates that UCC is driven by the accumulation of genetic and epigenetic aberrations, and it is well known that smoke induces these alterations. However, the number of genes that could be correlated with smoking among normal and tumor tissue samples was limited. In this pilot study we characterize our in vitro model by testing some key genes that are genetically altered in UCC, and we also performed PCR array for PI3K-AKT/mTOR pathways that are reported to be altered in UCC. We found some novel genes within this pathway that were not previously reported to be altered in UCC. Furthermore, by a comprehensive array-based approach; we identified differentially methylated genes in CSE-treated cells and validated some methylated genes by QMSP. We previously reported the identification of a cancer-specific methylated panel of genes in UCC that could be used in the detection and determination of prognosis of UCC.Citation16,Citation69 Further analysis of genetically and epigenetically altered genes published in smoke-associated solid tumors will be used to further validate our in vitro model and to understand the roles of identified molecules in UCC initiation, progression and detection. We feel that this in vitro model will help to identify “driver genes” that are responsible for initiation and progression of UCC. Early molecular changes that will be determined by this model have potential to facilitate the ability for early detection, treatment and follow up of superficial lesions before the patient develops advanced cancer.
Materials and Methods
Cell lines and reagents
A simian virus 40 (SV-40) immortalized human normal uroepithelial cell line (SV40-HUC1-1) was obtained from American Type Culture Collection (ATCC). SV40-HUC-1 cells were cultured in F12K medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin solution.
Mainstream smoke collection
Mainstream cigarette smoke (CSE) extract was generated using type-2R1 research cigarette packs from Tobacco Research and Development Center using a smoking machine set at 35 mL puff every 2 sec and collected by bubbling into 20 mL of phosphate buffered solution (PBS). This was regarded as a 100% extract solution (20 cigarettes into 20 mL PBS). The solution’s pH was adjusted to 7.0 using 1 mo NaOH or 3 mo HCL and filtered through a 2-μm syringe filter. After preparations, CSE stocks were aliquoted and stored at −80°C. For smoke exposure experiments, cells were cultured in a 25-cm flask. Medium and CSE was changed every 2 d. Cells were sub-cultured as necessary and frozen down each month for future studies.
Cell proliferation assay
CSE-treated and untreated cells were counted on a hemacytometer and seeded at a density of 1,000 cells per well in 96-well plates, and cellular viability was measured by the 3-(4, 5-dimethyl thiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) proliferation assay kit (ATCC) according to the manufacturer's instructions. Briefly, equal numbers of cells were cultured in 5% FBS for 24, 48 and 72 h. At the end of each time point, 10 μl of MTT labeling reagent (5 mg/ml MTT) was added to the culture media without fetal bovine serum (FBS), which was then incubated in the dark for a further 4 h at 37°C. This step was followed by cell lysis with the addition of 1 ml of a SDS-based detergent reagent from the MTT kit. The plates were incubated for 2 h at 37°C to dissolve formazan crystals. Spectrophotometric readings (A570nm−A650nm) were obtained on a Spectra Max 250 96-well plate reader (Molecular Devices). Each assay was performed in triplicate, and each experiment was repeated at least three times. Data are represented as the extent of cellular survival expressed as a percentage of control.
To measure DNA synthesis, cell proliferation was also determined with the BrdU cell proliferation enzyme-linked immunosorbent assay kit (Roche Molecular Biochemicals). Briefly CSE-treated and untreated SV40-HUC-1 cells were seeded in a 96-well culture plate at a density of 2.0 × 103 cells per well. Cells were labeled for 2 h with BrdU at 24, 48 and 72 h after seeding. BrdU incorporated into cellular DNA was quantified as instructed by the manufacturer.
Soft agar assays
Soft agar plates were made with Agar Select (Invitrogen) at 0.8% (bottom layer) and 0.3% (top layer). Five thousand cells were counted and seeded in the top layer of agar mixed with F12K medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin solution in 6-well plates and incubated at 37°C. Complete medium was added every 2 d (500 μl/well). The cells were allowed to grow for 2 wk, and colonies were photographed and counted using a light microscope. Each experiment was performed in triplicate wells and repeated 3×.
Invasion assays
Invasion assay were performed using BD Biosciences individual inserts coated with Matrigel (#354480) as directed by the manufacturer. Briefly, 24-well tissue culture plate inserts coated with Matrigel were re-hydrated for 2 h in 37°C with media containing 5% FBS. Media (0.6 ml) containing 5% FBS was added to each well as a chemo-attractant, and 0.2 ml (1 × 104 cells) of cell suspension was added to each insert. CSE-treated and CSE-untreated cells were allowed to attach and grow for 48 h. Invasion of cells to the underside of the Matrigel-coated membrane was detected by staining the cells with crystal violet solution and visualizing the cells under a microscope. After staining, cells were counted under a microscope in ten randomly selected fields (magnification ×100) per well and averaged. To normalize for growth differences between CSE-treated and CSE-untreated cells, each of the cell lines was also grown on an uncoated insert. The number of invaded cells was divided by the number of cell counted on the uncoated inserts, and results were expressed in the form of a bar graph. Assays were performed three times for each condition.
RT-PCR array
The Human PI3K-AKT Signaling Pathway RTCitation2 Profiler™ PCR Array (PAHS-058) profiles the expression of 84 genes involved in PI3K-AKT signaling. This array contains members of the AKT (Protein Kinase B) and PI3K families and their regulators. PI3K-AKT Signaling Pathway RTCitation2 Profiler™ PCR Array was obtained from SABiosciences.
The spreadsheets, gene tables and template formulas included with the PCR array package were used to calculate relative changes in gene expression as described previously.Citation70
Methylation array analysis
The Infinium Human Methylation27 BeadChip assay measures DNA methylation quantitatively at 27,578 CpG dinucleotides spanning 14,495 genes. Only 1 µg of genomic DNA is required. With the ability to provide genome-wide coverage for 12 samples concurrently, the Infinium platform is easily capable of high-throughput analyses. In a pilot study, we compared methylation differences between CSE treated and untreated SV40-HUC-1 cell lines. We also analyzed seven primary bladder cancer samples to determine genome-wide cancer specific methylation and then correlated this data with that from the CSE-treated cells. The Illumina Infinium HumanMethylation27 BeadChip Kit assays were performed in accordance with the manufacturer’s instructions and were performed at the Johns Hopkins Microarray Core Facility.
After bisulfite treatment, unmethylated C is converted to T, whereas methylation protects C from conversion. A pair of bead-bound probes is used to detect the presence of T or C by hybridization followed by single-base extension with a labeled nucleotide. The assay generates DNA methylation data for 27,578 CpG dinucleotide spanning 14,473 well-annotated, unique gene promoter and/or 50 gene regions (from −1,500 to + 1,500 from the transcription start site). The assay information is available at www.illumina.com. Selected genes determined by array analysis were validated by quantitative methylation-specific PCR (QMSP) as described previously.Citation16
Reverse transcription-PCR and real-time reverse transcription-PCR
RNA for RT-PCR was extracted from CSE-treated and untreated SV40-HUC-1 cell lines using TRizol Reagent (Invitrogen) and reverse transcribed in 20 μl reactions using SuperScript RNA Amplification System (Invitrogen), in accordance with the manufacturer’s instructions. The 260:280 and 260:230 nm ratios were calculated by the NanoDrop spectrophotometer and used to evaluate the RNA purity. Four micrograms of total RNA were reverse transcribed with Superscript II reverse transcriptase (Invitrogen), and then cDNA was amplified by PCR. Respective primer sequences are shown in . Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. The PCR products were separated by agarose gel electrophoresis and stained with ethidium bromide. For real-time RT-PCR, 1 μL of each cDNA was used and was performed using QuantiFast SYBR Green PCR kit (Promega). Amplifications were performed in 384-well plates in a 7900 Sequence Detector System (Perkin-Elmer Applied Biosystems). Expression of genes relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was calculated based on the threshold cycle (Ct) as 2−Δ (ΔCt), where Δ Ct = Ct GENE − Ct, GAPDH and Δ(ΔCt) = ΔCt, UT − ΔCt,T (UT, CSE untreated; T, CSE treated). Detailed PCR conditions are available upon request.
Table 1. RT-PCR primers
Western blot analysis
Total cell lysates of untreated and CSE exposed SV40-HUC-1 cells were used to conduct western blot analysis for p21, cyclin D1, Rb, E2F1, cdk4, p53, mTOR, AKT and β-actin (cell signaling). Briefly, cells were lysed in RIPA buffer containing a cocktail of protease inhibitors. Equal amounts of protein (20–45 μg depending on protein) solubilized in sample buffer was separated using a 4–12% gradient Bis-Tris PAGE (Invitrogen) and transferred onto a nitrocellulose membrane, followed by incubation with primary and secondary antibodies. The signals were detected using ECL-plus western detection solution (Amersham).
Patients and samples
For methylation analysis, DNA extracted from fresh-frozen seven UCC specimens were obtained from the IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo FG, Italy. An additional 13 FFPE UCC specimens from patients with known smoking history and stage were retrieved from Johns Hopkins Medical Institutions for FOXO1 immunohistochemistry. IRB approval was obtained; the tissue material has been anonymized, and the results obtained are not retraceable to the individuals concerned.
Immunohistochemistry for FOXO1
Immunohistochemistry for FOXO1 protein was performed on FFPE tissue sections from 13 UCC cases. Briefly, sections were deparaffinized in xylene, rehydrated in ethanol and subjected to heat-induced antigen retrieval with Retrievagen A (pH 6.0) (BD PharMingen) using a microwave. Endogenous peroxidase was blocked by incubation with 0.3% hydrogen peroxidase for 30 min. Sections were incubated with the primary rabbit monoclonal antibody specific to FOXO1 (Cell Signaling, dilution 1:50). After the application of a biotinylated anti-rabbit IgG (Vector Laboratories Inc., dilution 1:100), the reaction was visualized using the Vectastain ABC kit (Vector Laboratories) and counterstained with hematoxylin. FFPE cell line tissue of IGROV and SKOV3 and ttonsil tissue were used as positive controls. Expression of FOXO1 was examined according to the staining pattern (nuclear and/or cytoplasmic) and staining intensity by professional pathologists (NGR and GN) without knowledge of patient’s smoking history. Intensity was graded as follows: 0, negative; 1, weak; 2, moderate; 3, strong. Staining in more than 5% of cells was considered as positive.
Statistical analysis
Difference between values obtained and characteristic was analyzed by Student’s test or Fisher exact test; differences were considered statistical significant at p < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Grant Support
Flight Attendant Medical Research Institute Young Clinical Scientist Award, Career Development award from SPORE in Cervical Cancer Grants P50 CA098252 (M.H.) and 1R01CA163594-01(D.S. and M.H.).
Related Research Data
References
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62:10 - 29; http://dx.doi.org/10.3322/caac.20138; PMID: 22237781
- Strope SA, Montie JE. The causal role of cigarette smoking in bladder cancer initiation and progression, and the role of urologists in smoking cessation. J Urol 2008; 180:31 - 7, discussion 37; http://dx.doi.org/10.1016/j.juro.2008.03.045; PMID: 18485400
- Pelucchi C, Bosetti C, Negri E, Malvezzi M, La Vecchia C. Mechanisms of disease: The epidemiology of bladder cancer. Nat Clin Pract Urol 2006; 3:327 - 40; http://dx.doi.org/10.1038/ncpuro0510; PMID: 16763645
- Wang YH, Yeh SD, Wu MM, Liu CT, Shen CH, Shen KH, et al. Comparing the joint effect of arsenic exposure, cigarette smoking and risk genotypes of vascular endothelial growth factor on upper urinary tract urothelial carcinoma and bladder cancer. J Hazard Mater 2012; http://dx.doi.org/10.1016/j.jhazmat.2012.08.056; PMID: 23009795
- Sozzi G, Pastorino U, Croce CM. MicroRNAs and lung cancer: from markers to targets. Cell Cycle 2011; 10:2045 - 6; http://dx.doi.org/10.4161/cc.10.13.15712; PMID: 21623159
- Marsit CJ, Karagas MR, Schned A, Kelsey KT. Carcinogen exposure and epigenetic silencing in bladder cancer. Ann N Y Acad Sci 2006; 1076:810 - 21; http://dx.doi.org/10.1196/annals.1371.031; PMID: 17119258
- Enokida H, Nakagawa M. Epigenetics in bladder cancer. Int J Clin Oncol 2008; 13:298 - 307; http://dx.doi.org/10.1007/s10147-008-0811-1; PMID: 18704629
- Wu X, Obata T, Khan Q, Highshaw RA, De Vere White R, Sweeney C. The phosphatidylinositol-3 kinase pathway regulates bladder cancer cell invasion. BJU Int 2004; 93:143 - 50; http://dx.doi.org/10.1111/j.1464-410X.2004.04574.x; PMID: 14678387
- Onol FF, Demir A, Temiz Y, Yüksel M, Eren F, Türkeri LN. The inhibitory effect of vitamin E on cigarette smoke-induced oxidative damage to the rat urothelium: can it prevent transitional cell carcinoma?. Urol Int 2007; 78:150 - 4; http://dx.doi.org/10.1159/000098074; PMID: 17293656
- Stern MC, Umbach DM, van Gils CH, Lunn RM, Taylor JA. DNA repair gene XRCC1 polymorphisms, smoking, and bladder cancer risk. Cancer Epidemiol Biomarkers Prev 2001; 10:125 - 31; PMID: 11219769
- Hu YC, Yang ZH, Zhong KJ, Niu LJ, Pan XJ, Wu DC, et al. Alteration of transcriptional profile in human bronchial epithelial cells induced by cigarette smoke condensate. Toxicol Lett 2009; 190:23 - 31; http://dx.doi.org/10.1016/j.toxlet.2009.06.860; PMID: 19559774
- Marsit CJ, Wiencke JK, Nelson HH, Kim DH, Hinds PW, Aldape K, et al. Alterations of 9p in squamous cell carcinoma and adenocarcinoma of the lung: association with smoking, TP53, and survival. Cancer Genet Cytogenet 2005; 162:115 - 21; http://dx.doi.org/10.1016/j.cancergencyto.2005.04.001; PMID: 16213358
- Hoque MO, Begum S, Topaloglu O, Chatterjee A, Rosenbaum E, Van Criekinge W, et al. Quantitation of promoter methylation of multiple genes in urine DNA and bladder cancer detection. J Natl Cancer Inst 2006; 98:996 - 1004; http://dx.doi.org/10.1093/jnci/djj265; PMID: 16849682
- Kim WJ, Kim YJ. Epigenetic biomarkers in urothelial bladder cancer. Expert Rev Mol Diagn 2009; 9:259 - 69; http://dx.doi.org/10.1586/erm.09.5; PMID: 19379084
- Marsit CJ, Karagas MR, Danaee H, Liu M, Andrew A, Schned A, et al. Carcinogen exposure and gene promoter hypermethylation in bladder cancer. Carcinogenesis 2006; 27:112 - 6; http://dx.doi.org/10.1093/carcin/bgi172; PMID: 15987713
- Brait M, Begum S, Carvalho AL, Dasgupta S, Vettore AL, Czerniak B, et al. Aberrant promoter methylation of multiple genes during pathogenesis of bladder cancer. Cancer Epidemiol Biomarkers Prev 2008; 17:2786 - 94; http://dx.doi.org/10.1158/1055-9965.EPI-08-0192; PMID: 18843024
- Kim DH, Nelson HH, Wiencke JK, Zheng S, Christiani DC, Wain JC, et al. p16(INK4a) and histology-specific methylation of CpG islands by exposure to tobacco smoke in non-small cell lung cancer. Cancer Res 2001; 61:3419 - 24; PMID: 11309302
- Toyooka S, Suzuki M, Tsuda T, Toyooka KO, Maruyama R, Tsukuda K, et al. Dose effect of smoking on aberrant methylation in non-small cell lung cancers. Int J Cancer 2004; 110:462 - 4; http://dx.doi.org/10.1002/ijc.20125; PMID: 15095316
- Toyooka S, Maruyama R, Toyooka KO, McLerran D, Feng Z, Fukuyama Y, et al. Smoke exposure, histologic type and geography-related differences in the methylation profiles of non-small cell lung cancer. Int J Cancer 2003; 103:153 - 60; http://dx.doi.org/10.1002/ijc.10787; PMID: 12455028
- Marsit CJ, Houseman EA, Schned AR, Karagas MR, Kelsey KT. Promoter hypermethylation is associated with current smoking, age, gender and survival in bladder cancer. Carcinogenesis 2007; 28:1745 - 51; http://dx.doi.org/10.1093/carcin/bgm116; PMID: 17522068
- Pryor WA. Cigarette smoke radicals and the role of free radicals in chemical carcinogenicity. Environ Health Perspect 1997; 105:Suppl 4 875 - 82; PMID: 9255574
- Carp H, Janoff A. Possible mechanisms of emphysema in smokers. In vitro suppression of serum elastase-inhibitory capacity by fresh cigarette smoke and its prevention by antioxidants. Am Rev Respir Dis 1978; 118:617 - 21; PMID: 101105
- Massey E, Aufderheide M, Koch W, Lodding H, Pohlmann G, Windt H, et al. Micronucleus induction in V79 cells after direct exposure to whole cigarette smoke. Mutagenesis 1998; 13:145 - 9; http://dx.doi.org/10.1093/mutage/13.2.145; PMID: 9568586
- Christian BJ, Loretz LJ, Oberley TD, Reznikoff CA. Characterization of human uroepithelial cells immortalized in vitro by simian virus 40. Cancer Res 1987; 47:6066 - 73; PMID: 2822239
- Reznikoff CA, Loretz LJ, Christian BJ, Wu SQ, Meisner LF. Neoplastic transformation of SV40-immortalized human urinary tract epithelial cells by in vitro exposure to 3-methylcholanthrene. Carcinogenesis 1988; 9:1427 - 36; http://dx.doi.org/10.1093/carcin/9.8.1427; PMID: 2841047
- Cordon-Cardo C. Molecular alterations associated with bladder cancer initiation and progression. Scand J Urol Nephrol Suppl 2008; 154 - 65; http://dx.doi.org/10.1080/03008880802291915; PMID: 18815930
- Zhang ZF, Kurtz RC, Marshall JR. Cigarette smoking and esophageal and gastric cardia adenocarcinoma. J Natl Cancer Inst 1997; 89:1247 - 9; http://dx.doi.org/10.1093/jnci/89.17.1247; PMID: 9293909
- Wolff EM, Liang G, Cortez CC, Tsai YC, Castelao JE, Cortessis VK, et al. RUNX3 methylation reveals that bladder tumors are older in patients with a history of smoking. Cancer Res 2008; 68:6208 - 14; http://dx.doi.org/10.1158/0008-5472.CAN-07-6616; PMID: 18676844
- Platt FM, Hurst CD, Taylor CF, Gregory WM, Harnden P, Knowles MA. Spectrum of phosphatidylinositol 3-kinase pathway gene alterations in bladder cancer. Clin Cancer Res 2009; 15:6008 - 17; http://dx.doi.org/10.1158/1078-0432.CCR-09-0898; PMID: 19789314
- West KA, Brognard J, Clark AS, Linnoila IR, Yang X, Swain SM, et al. Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J Clin Invest 2003; 111:81 - 90; PMID: 12511591
- Ho YS, Chen CH, Wang YJ, Pestell RG, Albanese C, Chen RJ, et al. Tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) induces cell proliferation in normal human bronchial epithelial cells through NFkappaB activation and cyclin D1 up-regulation. Toxicol Appl Pharmacol 2005; 205:133 - 48; http://dx.doi.org/10.1016/j.taap.2004.09.019; PMID: 15893541
- Jin Q, Menter DG, Mao L, Hong WK, Lee HY. Survivin expression in normal human bronchial epithelial cells: an early and critical step in tumorigenesis induced by tobacco exposure. Carcinogenesis 2008; 29:1614 - 22; http://dx.doi.org/10.1093/carcin/bgm234; PMID: 18635526
- Parsanejad R, Fields WR, Morgan WT, Bombick BR, Doolittle DJ. The time course of expression of genes involved in specific pathways in normal human bronchial epithelial cells following exposure to cigarette smoke. Exp Lung Res 2008; 34:513 - 30; http://dx.doi.org/10.1080/01902140802271826; PMID: 18850377
- Qu MY, Luo BL, Chen HM, Feng JT, Gu QH. [Effects of secretory leukocyte protease inhibitor on expression of inflammation mediators in normal human bronchial epithelial cells induced by cigarette smoke extract]. 2008; 31:352 - 5; PMID: 18953959
- Wu JP, Chang LW, Yao HT, Chang H, Tsai HT, Tsai MH, et al. Involvement of oxidative stress and activation of aryl hydrocarbon receptor in elevation of CYP1A1 expression and activity in lung cells and tissues by arsenic: an in vitro and in vivo study. Toxicol Sci 2009; 107:385 - 93; http://dx.doi.org/10.1093/toxsci/kfn239; PMID: 19033395
- Walsh KM, Amos CI, Wenzlaff AS, Gorlov IP, Sison JD, Wu X, et al. Association study of nicotinic acetylcholine receptor genes identifies a novel lung cancer susceptibility locus near CHRNA1 in African-Americans. Oncotarget 2012; 3:1428 - 38; PMID: 23232035
- Crallan RA, Georgopoulos NT, Southgate J. Experimental models of human bladder carcinogenesis. Carcinogenesis 2006; 27:374 - 81; http://dx.doi.org/10.1093/carcin/bgi266; PMID: 16287878
- Zeegers MP, Tan FE, Dorant E, van Den Brandt PA. The impact of characteristics of cigarette smoking on urinary tract cancer risk: a meta-analysis of epidemiologic studies. Cancer 2000; 89:630 - 9; http://dx.doi.org/10.1002/1097-0142(20000801)89:3<630::AID-CNCR19>3.0.CO;2-Q; PMID: 10931463
- Oka N, Tanimoto S, Taue R, Nakatsuji H, Kishimoto T, Izaki H, et al. Role of phosphatidylinositol-3 kinase/Akt pathway in bladder cancer cell apoptosis induced by tumor necrosis factor-related apoptosis-inducing ligand. Cancer Sci 2006; 97:1093 - 8; http://dx.doi.org/10.1111/j.1349-7006.2006.00294.x; PMID: 16984382
- Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA 1999; 96:4240 - 5; http://dx.doi.org/10.1073/pnas.96.8.4240; PMID: 10200246
- Simpson L, Parsons R. PTEN: life as a tumor suppressor. Exp Cell Res 2001; 264:29 - 41; http://dx.doi.org/10.1006/excr.2000.5130; PMID: 11237521
- Sun H, Lesche R, Li DM, Liliental J, Zhang H, Gao J, et al. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci USA 1999; 96:6199 - 204; http://dx.doi.org/10.1073/pnas.96.11.6199; PMID: 10339565
- Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, et al. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet 2001; 29:396 - 403; http://dx.doi.org/10.1038/ng782; PMID: 11726926
- Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007; 128:157 - 70; http://dx.doi.org/10.1016/j.cell.2006.11.042; PMID: 17218262
- Girard L, Zöchbauer-Müller S, Virmani AK, Gazdar AF, Minna JD. Genome-wide allelotyping of lung cancer identifies new regions of allelic loss, differences between small cell lung cancer and non-small cell lung cancer, and loci clustering. Cancer Res 2000; 60:4894 - 906; PMID: 10987304
- Kim WJ, Kim EJ, Jeong P, Quan C, Kim J, Li QL, et al. RUNX3 inactivation by point mutations and aberrant DNA methylation in bladder tumors. Cancer Res 2005; 65:9347 - 54; http://dx.doi.org/10.1158/0008-5472.CAN-05-1647; PMID: 16230397
- Bakkar AA, Wallerand H, Radvanyi F, Lahaye JB, Pissard S, Lecerf L, et al. FGFR3 and TP53 gene mutations define two distinct pathways in urothelial cell carcinoma of the bladder. Cancer Res 2003; 63:8108 - 12; PMID: 14678961
- Cote RJ, Chatterjee SJ. Molecular determinants of outcome in bladder cancer. Cancer J Sci Am 1999; 5:2 - 15; PMID: 10188054
- Shariat SF, Bolenz C, Karakiewicz PI, Fradet Y, Ashfaq R, Bastian PJ, et al. p53 expression in patients with advanced urothelial cancer of the urinary bladder. BJU Int 2010; 105:489 - 95; http://dx.doi.org/10.1111/j.1464-410X.2009.08742.x; PMID: 19659466
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004; 304:554; http://dx.doi.org/10.1126/science.1096502; PMID: 15016963
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 2006; 7:606 - 19; http://dx.doi.org/10.1038/nrg1879; PMID: 16847462
- Aaltonen V, Koivunen J, Laato M, Peltonen J. Heterogeneity of cellular proliferation within transitional cell carcinoma: correlation of protein kinase C alpha/betaI expression and activity. J Histochem Cytochem 2006; 54:795 - 806; http://dx.doi.org/10.1369/jhc.5A6839.2006; PMID: 16517978
- Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, Coppolino MG, Radeva G, Filmus J, et al. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature 1996; 379:91 - 6; http://dx.doi.org/10.1038/379091a0; PMID: 8538749
- McDonald PC, Fielding AB, Dedhar S. Integrin-linked kinase--essential roles in physiology and cancer biology. J Cell Sci 2008; 121:3121 - 32; http://dx.doi.org/10.1242/jcs.017996; PMID: 18799788
- Graff JR, Deddens JA, Konicek BW, Colligan BM, Hurst BM, Carter HW, et al. Integrin-linked kinase expression increases with prostate tumor grade. Clin Cancer Res 2001; 7:1987 - 91; PMID: 11448915
- Bravou V, Klironomos G, Papadaki E, Stefanou D, Varakis J. Integrin-linked kinase (ILK) expression in human colon cancer. Br J Cancer 2003; 89:2340 - 1; http://dx.doi.org/10.1038/sj.bjc.6601482; PMID: 14676816
- Takanami I. Increased expression of integrin-linked kinase is associated with shorter survival in non-small cell lung cancer. BMC Cancer 2005; 5:1; http://dx.doi.org/10.1186/1471-2407-5-1; PMID: 15631637
- Matsui Y, Assi K, Ogawa O, Raven PA, Dedhar S, Gleave ME, et al. The importance of integrin-linked kinase in the regulation of bladder cancer invasion. Int J Cancer 2012; 130:521 - 31; http://dx.doi.org/10.1002/ijc.26008; PMID: 21351095
- Cao Q, Yu J, Dhanasekaran SM, Kim JH, Mani RS, Tomlins SA, et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 2008; 27:7274 - 84; http://dx.doi.org/10.1038/onc.2008.333; PMID: 18806826
- Chen D, Zhang Y, Zhang X, Li J, Han B, Liu S, et al. Overexpression of integrin-linked kinase correlates with malignant phenotype in non-small cell lung cancer and promotes lung cancer cell invasion and migration via regulating epithelial-mesenchymal transition (EMT)-related genes. Acta Histochem 2013; 115:128 - 36; http://dx.doi.org/10.1016/j.acthis.2012.05.004; PMID: 22683084
- Miki H, Miura K, Takenawa T. N-WASP, a novel actin-depolymerizing protein, regulates the cortical cytoskeletal rearrangement in a PIP2-dependent manner downstream of tyrosine kinases. EMBO J 1996; 15:5326 - 35; PMID: 8895577
- Stradal TE, Rottner K, Disanza A, Confalonieri S, Innocenti M, Scita G. Regulation of actin dynamics by WASP and WAVE family proteins. Trends Cell Biol 2004; 14:303 - 11; http://dx.doi.org/10.1016/j.tcb.2004.04.007; PMID: 15183187
- Lorenz M, Yamaguchi H, Wang Y, Singer RH, Condeelis J. Imaging sites of N-wasp activity in lamellipodia and invadopodia of carcinoma cells. Curr Biol 2004; 14:697 - 703; http://dx.doi.org/10.1016/j.cub.2004.04.008; PMID: 15084285
- Gligorijevic B, Wyckoff J, Yamaguchi H, Wang Y, Roussos ET, Condeelis J. N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J Cell Sci 2012; 125:724 - 34; http://dx.doi.org/10.1242/jcs.092726; PMID: 22389406
- Gomes AR, Brosens JJ, Lam EW. Resist or die: FOXO transcription factors determine the cellular response to chemotherapy. Cell Cycle 2008; 7:3133 - 6; http://dx.doi.org/10.4161/cc.7.20.6920; PMID: 18927504
- Derks S, Bosch LJ, Niessen HE, Moerkerk PT, van den Bosch SM, Carvalho B, et al. Promoter CpG island hypermethylation- and H3K9me3 and H3K27me3-mediated epigenetic silencing targets the deleted in colon cancer (DCC) gene in colorectal carcinogenesis without affecting neighboring genes on chromosomal region 18q21. Carcinogenesis 2009; 30:1041 - 8; http://dx.doi.org/10.1093/carcin/bgp073; PMID: 19329758
- Zhao G, Qin Q, Zhang J, Liu Y, Deng S, Liu L, et al. Hypermethylation of HIC1 Promoter and Aberrant Expression of HIC1/SIRT1 Might Contribute to the Carcinogenesis of Pancreatic Cancer. Ann Surg Oncol 2012; http://dx.doi.org/10.1245/s10434-012-2364-9; PMID: 22552606
- Kao C, Huang J, Wu SQ, Hauser P, Reznikoff CA. Role of SV40 T antigen binding to pRB and p53 in multistep transformation in vitro of human uroepithelial cells. Carcinogenesis 1993; 14:2297 - 302; http://dx.doi.org/10.1093/carcin/14.11.2297; PMID: 8242858
- Hoque MO, Begum S, Brait M, Jeronimo C, Zahurak M, Ostrow KL, et al. Tissue inhibitor of metalloproteinases-3 promoter methylation is an independent prognostic factor for bladder cancer. J Urol 2008; 179:743 - 7; http://dx.doi.org/10.1016/j.juro.2007.09.019; PMID: 18082200
- Castillo-Pichardo L, Martínez-Montemayor MM, Martínez JE, Wall KM, Cubano LA, Dharmawardhane S. Inhibition of mammary tumor growth and metastases to bone and liver by dietary grape polyphenols. Clin Exp Metastasis 2009; 26:505 - 16; http://dx.doi.org/10.1007/s10585-009-9250-2; PMID: 19294520